Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death


 ,
,  ,
,  ,
,  ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
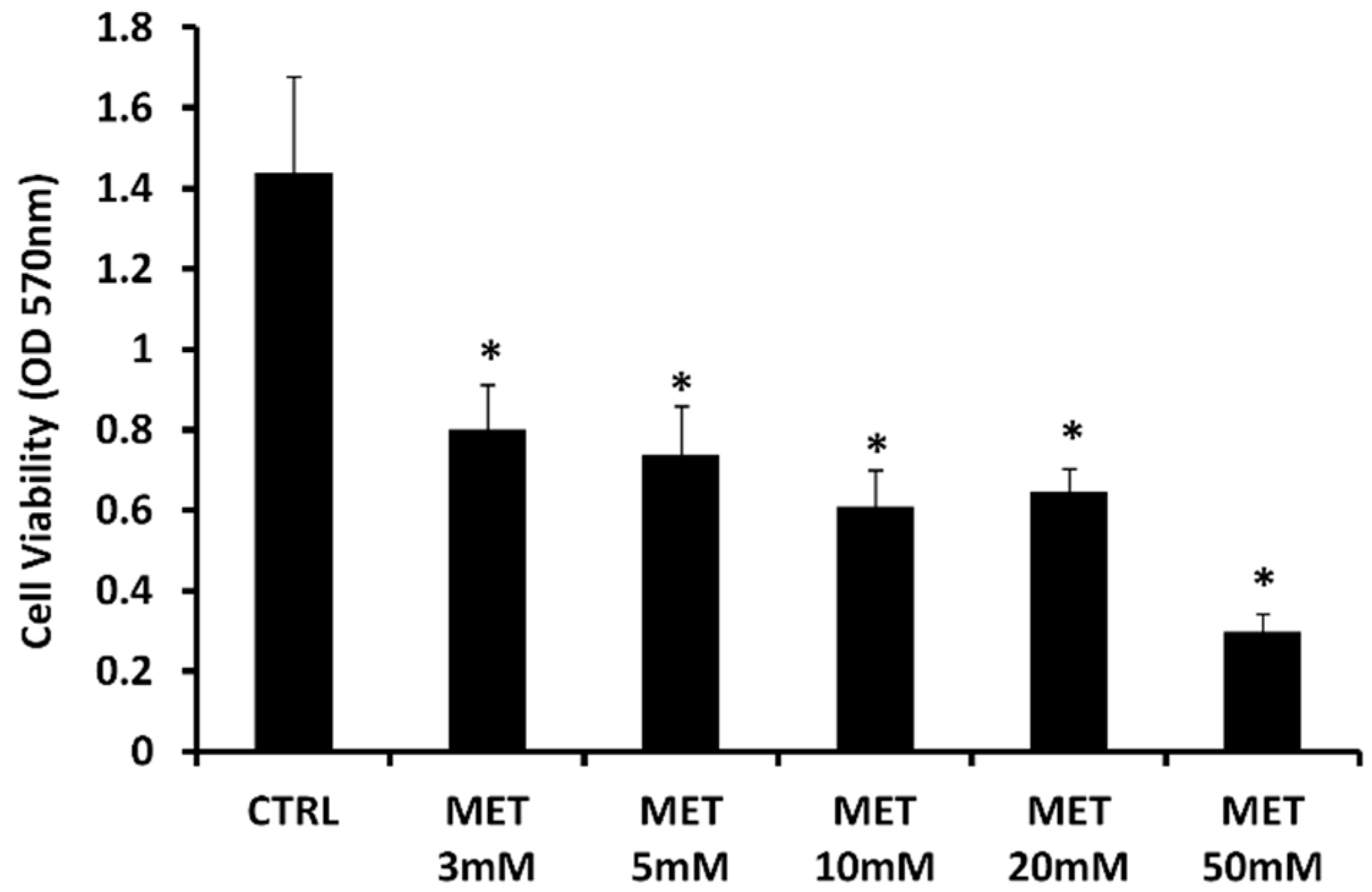
2.1. Effect of Metformin on Cell Viability
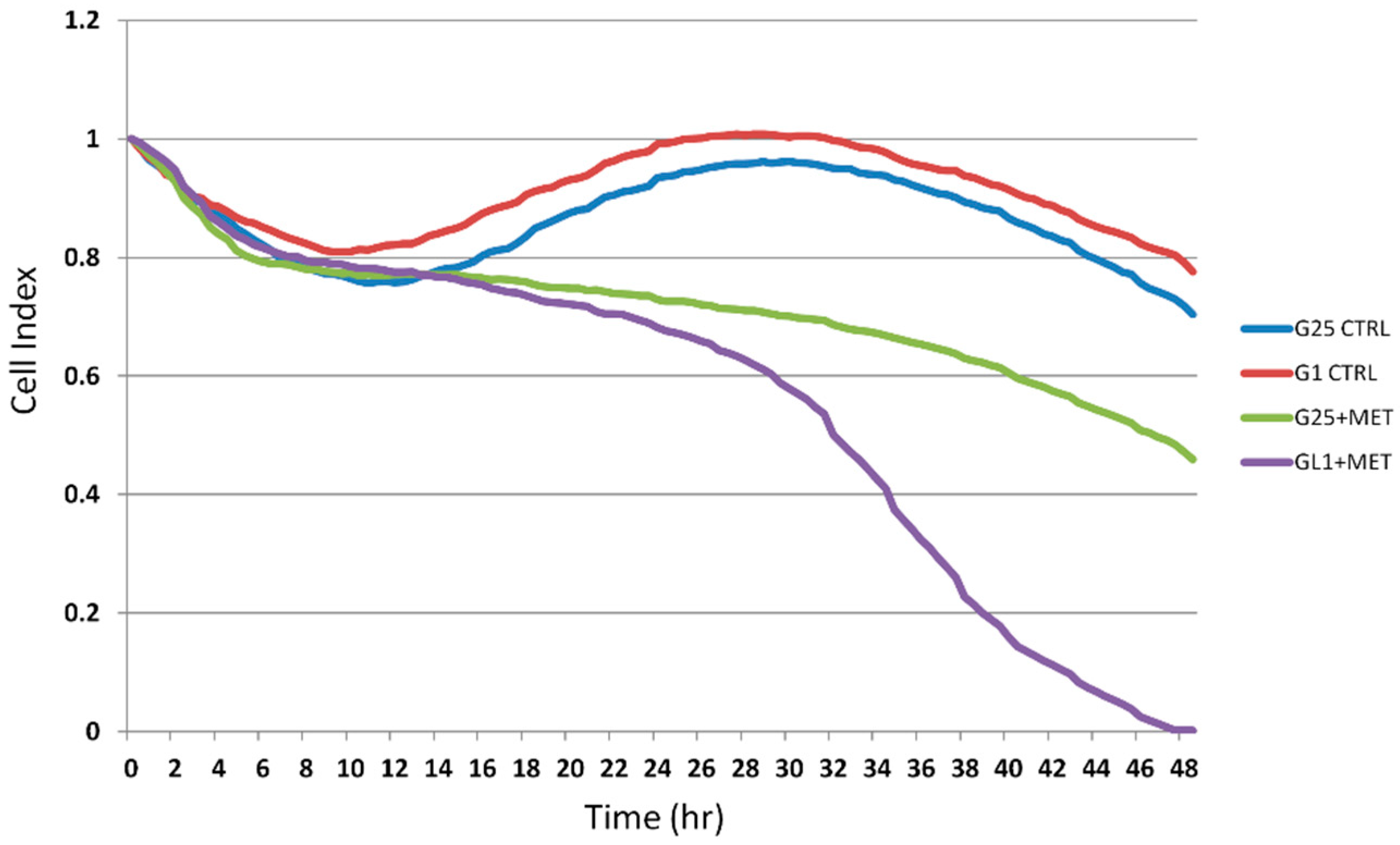
2.2. Real-Time Analysis of Cell Proliferation in Presence of Metformin and Different Glucose Concentrations
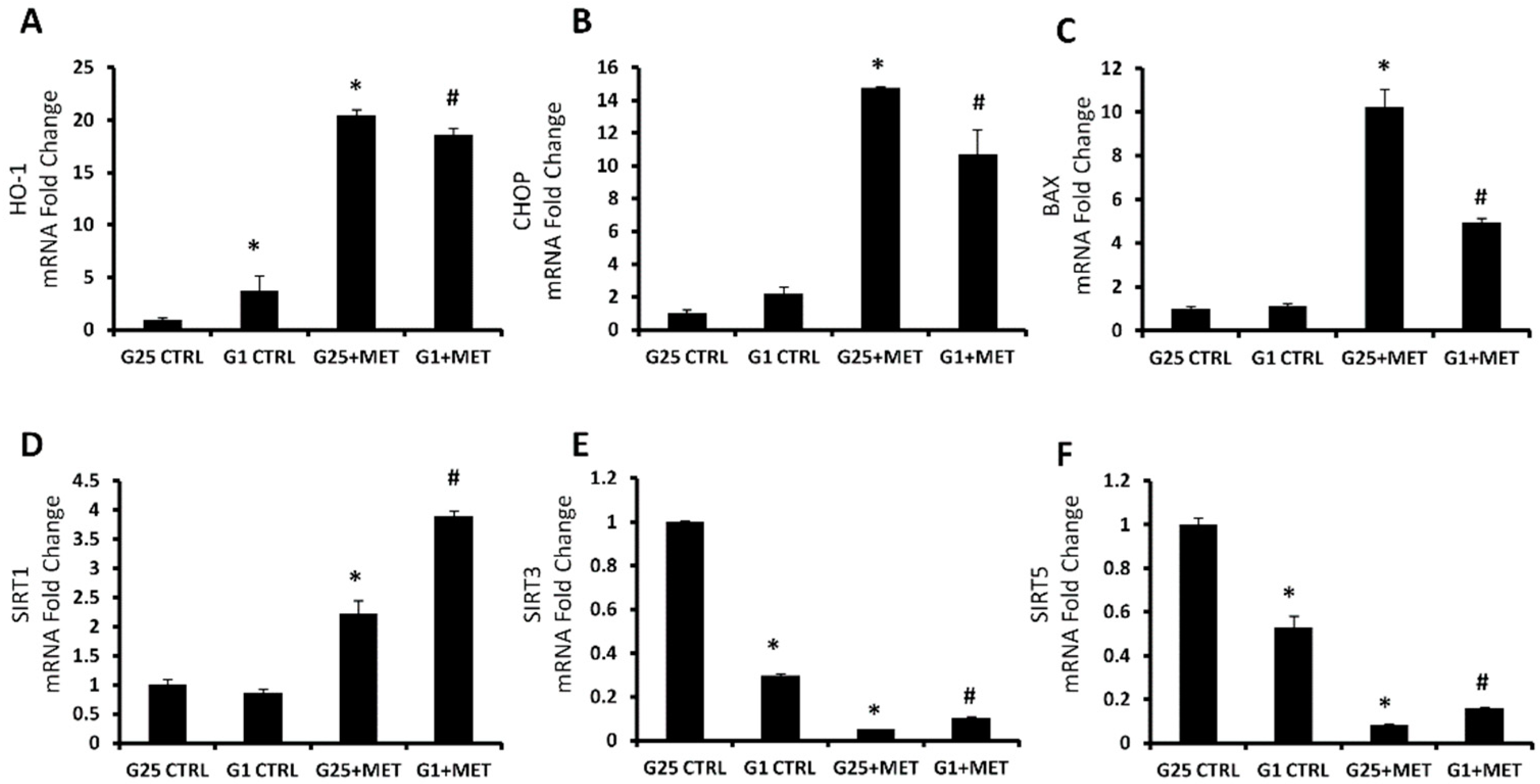
2.3. Effect of Metformin on HO-1, CHOP, BAX, and Sirtuins mRNA Expression
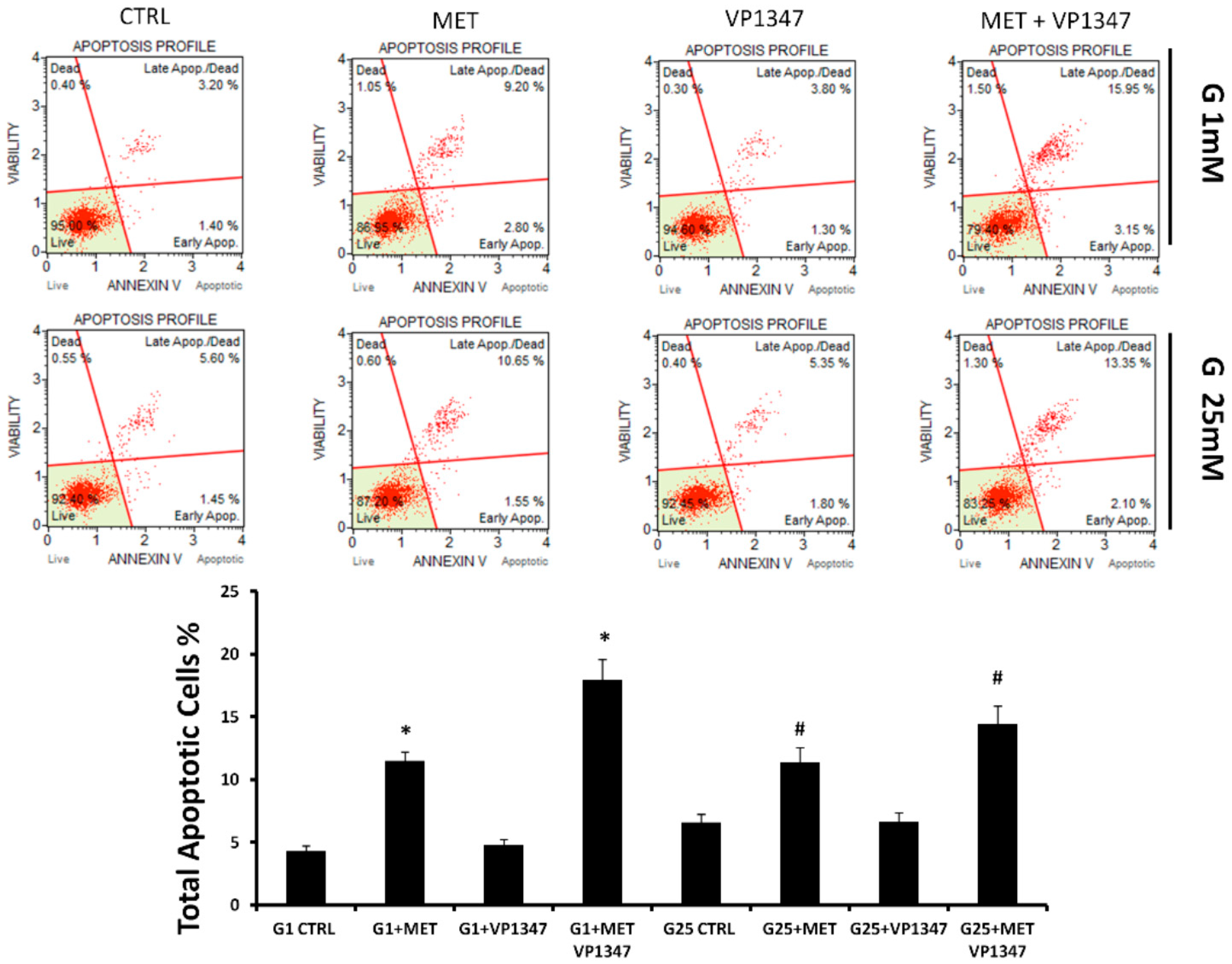
2.4. Metformin Enhances the Apoptosis Rate of DU145 in the Presence of a Selective HO-1 Activity Inhibitor
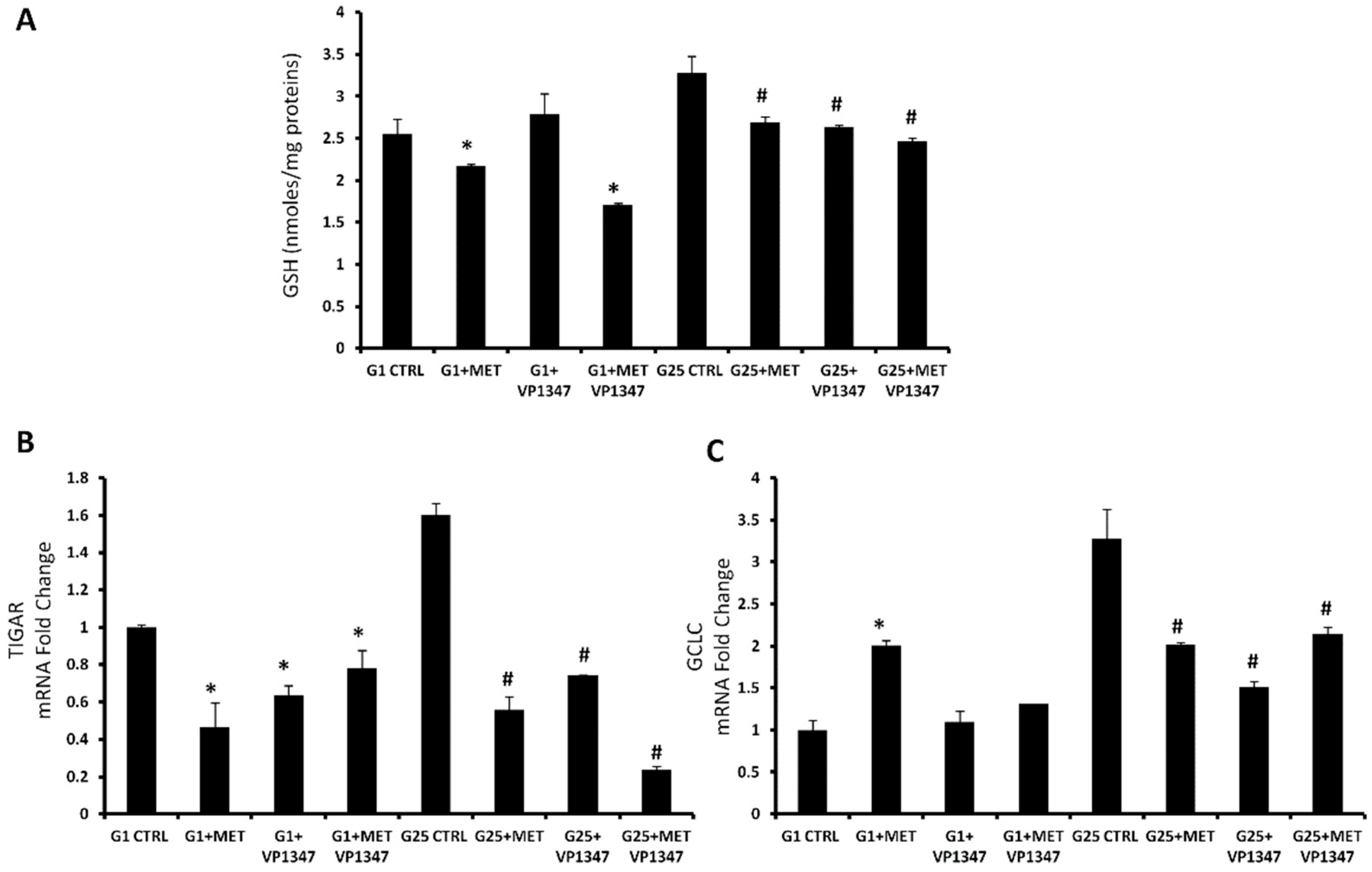
2.5. Effect of Metformin on Oxidative Stress Regulation Pathway
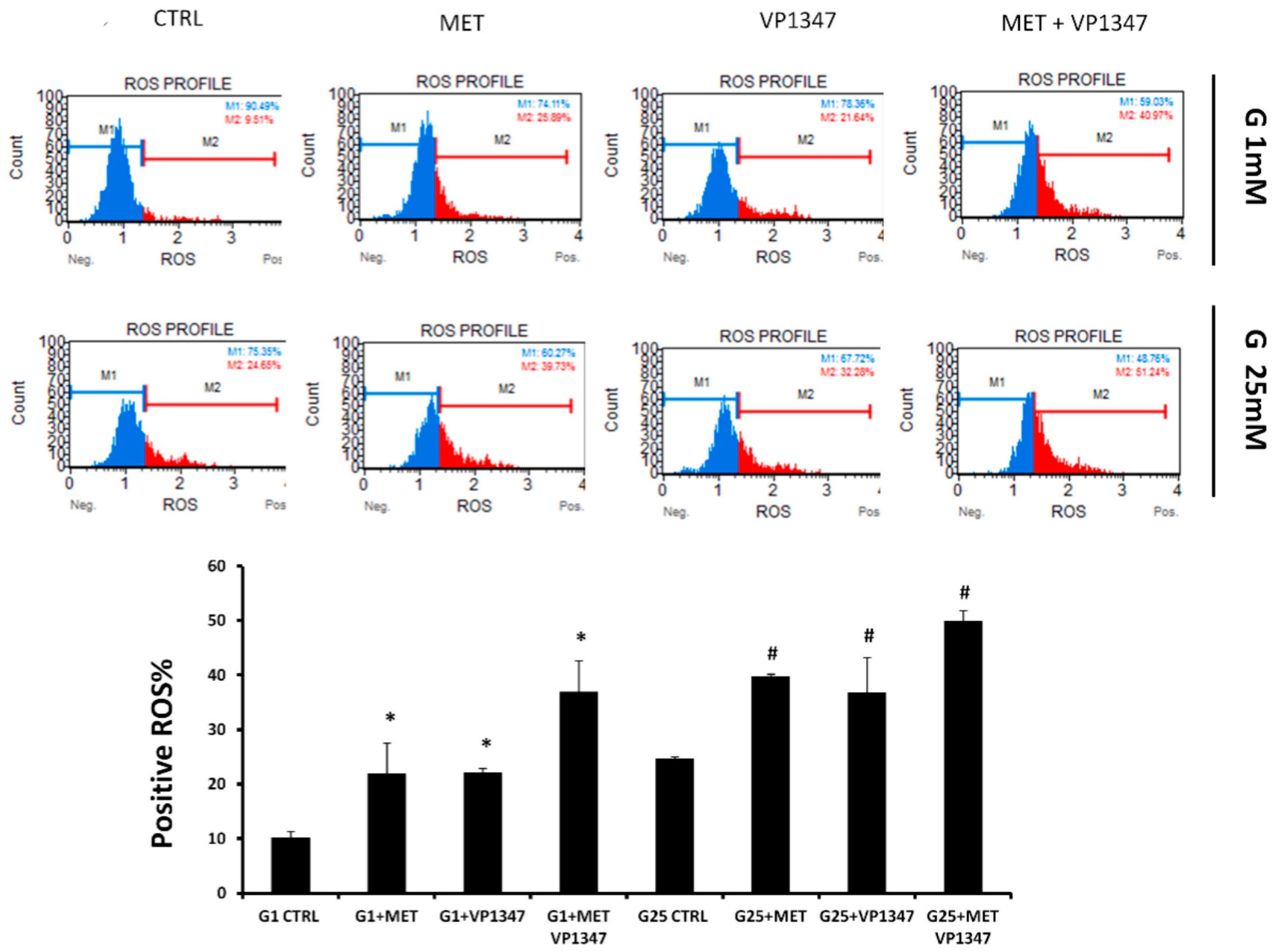
2.6. Effect of Metformin and HO-1 Activity Inhibitor on ROS Production
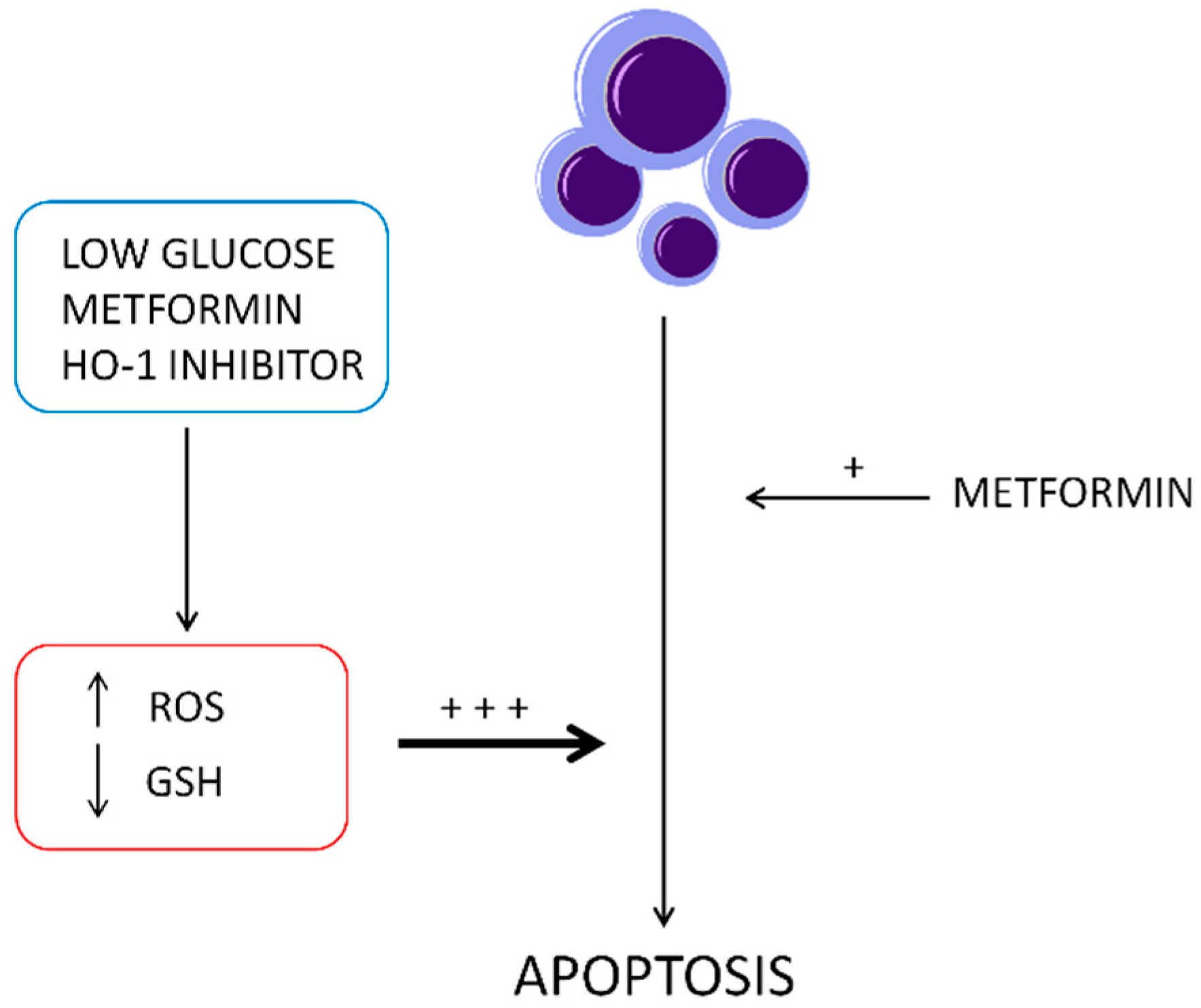
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Real-Time Monitoring of Proliferation
4.4. Oxidative Stress Assay
4.5. Annexin V Assay
4.6. RSH Evaluation
4.7. RNA Extraction and qRT-PCR
4.8. Statistical Analysis
Author Contributions
Funding
Conflicts of Interest
References
- Abraham, N.G.; Kappas, A. Pharmacological and Clinical Aspects of Heme Oxygenase. Pharmacol. Rev. 2008, 60, 79–127. [Google Scholar] [CrossRef]
- Foresti, R.; Green, C.J.; Motterlini, R. Generation of Bile Pigments by Haem Oxygenase: A Refined Cellular Strategy in Response to Stressful Insults. Biochem. Soc. Symp. 2004, 177–192. [Google Scholar] [CrossRef]
- Pittala, V.; Vanella, L.; Salerno, L.; Romeo, G.; Marrazzo, A.; di Giacomo, C.; Sorrenti, V. Effects of Polyphenolic Derivatives on Heme Oxygenase-System in Metabolic Dysfunctions. Curr. Med. Chem. 2018, 25, 1577–1595. [Google Scholar] [CrossRef]
- Vanella, L.; Barbagallo, I.; Tibullo, D.; Forte, S.; Zappala, A.; Volti, G.L. The Non-Canonical Functions of the Heme Oxygenases. Oncotarget 2016, 7, 69075–69086. [Google Scholar] [CrossRef] [PubMed]
- Converso, D.P.; Taille, C.; Carreras, M.C.; Jaitovich, A.; Poderoso, J.J.; Boczkowski, J. Ho-1 Is Located in Liver Mitochondria and Modulates Mitochondrial Heme Content and Metabolism. FASEB J. 2006, 20, 1236–1238. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.P.; Wang, X.; Galbiati, F.; Ryter, S.W.; Choi, A.M. Caveolae Compartmentalization of Heme Oxygenase-1 in Endothelial Cells. FASEB J. 2004, 18, 1080–1089. [Google Scholar] [CrossRef]
- Rodella, L.F.; Vanella, L.; Peterson, S.J.; Drummond, G.; Rezzani, R.; Falck, J.R.; Abraham, N.G. Heme Oxygenase-Derived Carbon Monoxide Restores Vascular Function in Type 1 Diabetes. Drug Metab. Lett. 2008, 2, 290–300. [Google Scholar] [CrossRef]
- Vanella, L.; Sanford, C., Jr.; Kim, D.H.; Abraham, N.G.; Ebraheim, N. Oxidative Stress and Heme Oxygenase-1 Regulated Human Mesenchymal Stem Cells Differentiation. Int. J. Hypertens. 2012, 2012, 890671. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Puri, N.; Raffaele, M.; Schragenheim, J.; Singh, S.P.; Bradbury, J.A.; Bellner, L.; Vanella, L.; Zeldin, D.C.; Cao, J.; et al. Ablation of Soluble Epoxide Hydrolase Reprogram White Fat to Beige-Like Fat through an Increase in Mitochondrial Integrity, Ho-1-Adiponectin in Vitro and in Vivo. Prostaglandins Other Lipid Mediat. 2018, 138, 1–8. [Google Scholar] [CrossRef]
- Pittala, V.; Vanella, L.; Salerno, L.; di Giacomo, C.; Acquaviva, R.; Raffaele, M.; Romeo, G.; Modica, M.N.; Prezzavento, O.; Sorrenti, V. Novel Caffeic Acid Phenethyl Ester (Cape) Analogues as Inducers of Heme Oxygenase-1. Curr. Pharm. Des. 2017, 23, 2657–2664. [Google Scholar] [CrossRef] [PubMed]
- Raffaele, M.; Volti, G.L.; Barbagallo, I.A.; Vanella, L. Therapeutic Efficacy of Stem Cells Transplantation in Diabetes: Role of Heme Oxygenase. Front. Cell Dev. Biol. 2016, 4, 80. [Google Scholar] [CrossRef]
- Stec, D.E.; John, K.; Trabbic, C.J.; Luniwal, A.; Hankins, M.W.; Baum, J.; Hinds, T.D., Jr. Bilirubin Binding to Pparalpha Inhibits Lipid Accumulation. PLoS ONE 2016, 11, e0153427. [Google Scholar] [CrossRef]
- Pittala, V.; Salerno, L.; Romeo, G.; Acquaviva, R.; di Giacomo, C.; Sorrenti, V. Therapeutic Potential of Caffeic Acid Phenethyl Ester (Cape) in Diabetes. Curr. Med. Chem. 2018, 25, 4827–4836. [Google Scholar] [CrossRef]
- Dey, S.; Sayers, C.M.; Verginadis, I.I.; Lehman, S.L.; Cheng, Y.; Cerniglia, G.J.; Tuttle, S.W.; Feldman, M.D.; Zhang, P.J.; Fuchs, S.Y.; et al. Atf4-Dependent Induction of Heme Oxygenase 1 Prevents Anoikis and Promotes Metastasis. J. Clin. Investig. 2015, 125, 2592–2608. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Zhang, X.; Liao, Z.; Liang, H.; Chu, L.; Dong, W.; Zhang, X.; Ge, Q.; Liu, Q.; Fan, P.; et al. 14-3-3zeta Inhibits Heme Oxygenase-1 (Ho-1) Degradation and Promotes Hepatocellular Carcinoma Proliferation: Involvement of Stat3 Signaling. J. Exp. Clin. Cancer Res. 2019, 38, 3. [Google Scholar] [CrossRef]
- Nitti, M.; Piras, S.; Marinari, U.M.; Moretta, L.; Pronzato, M.A.; Furfaro, A.L. Ho-1 Induction in Cancer Progression: A Matter of Cell Adaptation. Antioxidants 2017, 6, 29. [Google Scholar] [CrossRef]
- Barbagallo, I.; Giallongo, C.; Volti, G.L.; Distefano, A.; Camiolo, G.; Raffaele, M.; Salerno, L.; Pittala, V.; Sorrenti, V.; Avola, R.; et al. Heme Oxygenase Inhibition Sensitizes Neuroblastoma Cells to Carfilzomib. Mol. Neurobiol. 2019, 56, 1451–1460. [Google Scholar] [CrossRef]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; la Cava, P.; Parrinello, N.; Vanella, L.; Stagno, F.; Palumbo, G.A.; Volti, G.L.; di Raimondo, F. Nuclear Translocation of Heme Oxygenase-1 Confers Resistance to Imatinib in Chronic Myeloid Leukemia Cells. Curr. Pharm. Des. 2013, 19, 2765–2770. [Google Scholar] [CrossRef]
- Salerno, L.; Romeo, G.; Modica, M.N.; Amata, E.; Sorrenti, V.; Barbagallo, I.; Pittala, V. Heme Oxygenase-1: A New Druggable Target in the Management of Chronic and Acute Myeloid Leukemia. Eur. J. Med. Chem. 2017, 142, 163–178. [Google Scholar] [CrossRef]
- Schaefer, B.; Behrends, S. Translocation of Heme Oxygenase-1 Contributes to Imatinib Resistance in Chronic Myelogenous Leukemia. Oncotarget 2017, 8, 67406–67421. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Greish, K.F.; Salerno, L.; Al Zahrani, R.; Amata, E.; Modica, M.N.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Sorrenti, V.; Rescifina, A.; et al. Novel Structural Insight into Inhibitors of Heme Oxygenase-1 (Ho-1) by New Imidazole-Based Compounds: Biochemical and in Vitro Anticancer Activity Evaluation. Molecules 2018, 23, 1209. [Google Scholar] [CrossRef]
- Sorrenti, V.; Pittala, V.; Romeo, G.; Amata, E.; Dichiara, M.; Marrazzo, A.; Turnaturi, R.; Prezzavento, O.; Barbagallo, I.; Vanella, L.; et al. Targeting Heme Oxygenase-1 with Hybrid Compounds to Overcome Imatinib Resistance in Chronic Myeloid Leukemia Cell Lines. Eur. J. Med. Chem. 2018, 158, 937–950. [Google Scholar] [CrossRef]
- Jozkowicz, A.; Was, H.; Dulak, J. Heme Oxygenase-1 in Tumors: Is It a False Friend? Antioxid. Redox Signal. 2007, 9, 2099–2117. [Google Scholar] [CrossRef]
- Florczyk, U.; Golda, S.; Zieba, A.; Cisowski, J.; Jozkowicz, A.; Dulak, J. Overexpression of Biliverdin Reductase Enhances Resistance to Chemotherapeutics. Cancer Lett. 2011, 300, 40–47. [Google Scholar] [CrossRef]
- Banerjee, P.; Basu, A.; Wegiel, B.; Otterbein, L.E.; Mizumura, K.; Gasser, M.; Waaga-Gasser, A.M.; Choi, A.M.; Pal, S. Heme Oxygenase-1 Promotes Survival of Renal Cancer Cells through Modulation of Apoptosis- and Autophagy-Regulating Molecules. J. Biol. Chem. 2012, 287, 32113–32123. [Google Scholar] [CrossRef]
- Zingales, V.; Distefano, A.; Raffaele, M.; Zanghi, A.; Barbagallo, I.; Vanella, L. Metformin: A Bridge between Diabetes and Prostate Cancer. Front. Oncol. 2017, 7, 243. [Google Scholar] [CrossRef]
- Hankinson, S.J.; Fam, M.; Patel, N.N. A Review for Clinicians: Prostate Cancer and the Antineoplastic Properties of Metformin. Urol. Oncol. 2017, 35, 21–29. [Google Scholar] [CrossRef]
- Wright, J.L.; Stanford, J.L. Metformin Use and Prostate Cancer in Caucasian Men: Results from a Population-Based Case-Control Study. Cancer Causes Control 2009, 20, 1617–1622. [Google Scholar] [CrossRef]
- Lu, C.C.; Chiang, J.H.; Tsai, F.J.; Hsu, Y.M.; Juan, Y.N.; Yang, J.S.; Chiu, H.Y. Metformin Triggers the Intrinsic Apoptotic Response in Human Ags Gastric Adenocarcinoma Cells by Activating Ampk and Suppressing Mtor/Akt Signaling. Int. J. Oncol. 2019, 54, 1271–1281. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Gravel, S.P.; Pollak, M.; St-Pierre, J. Metformin Directly Acts on Mitochondria to Alter Cellular Bioenergetics. Cancer Metab. 2014, 2, 12. [Google Scholar] [CrossRef]
- Cantrell, L.A.; Zhou, C.; Mendivil, A.; Malloy, K.M.; Gehrig, P.A.; Bae-Jump, V.L. Metformin Is a Potent Inhibitor of Endometrial Cancer Cell Proliferation--Implications for a Novel Treatment Strategy. Gynecol. Oncol. 2010, 116, 92–98. [Google Scholar] [CrossRef]
- Ben Sahra, I.; Laurent, K.; Loubat, A.; Giorgetti-Peraldi, S.; Colosetti, P.; Auberger, P.; Tanti, J.F.; le Marchand-Brustel, Y.; Bost, F. The Antidiabetic Drug Metformin Exerts an Antitumoral Effect in Vitro and in Vivo through a Decrease of Cyclin D1 Level. Oncogene 2008, 27, 3576–3586. [Google Scholar] [CrossRef]
- Clements, A.; Gao, B.; Yeap, S.H.; Wong, M.K.; Ali, S.S.; Gurney, H. Metformin in Prostate Cancer: Two for the Price of One. Ann. Oncol. 2011, 22, 2556–2560. [Google Scholar] [CrossRef] [PubMed]
- Richards, K.A.; Liou, J.I.; Cryns, V.L.; Downs, T.M.; Abel, E.J.; Jarrard, D.F. Metformin Use Is Associated with Improved Survival for Patients with Advanced Prostate Cancer on Androgen Deprivation Therapy. J. Urol. 2018, 200, 1256–1263. [Google Scholar] [CrossRef]
- Sarmento-Cabral, A.; Fernando, L.; Gahete, M.D.; Castano, J.P.; Luque, R.M. Metformin Reduces Prostate Tumor Growth, in a Diet-Dependent Manner, by Modulating Multiple Signaling Pathways. Mol. Cancer Res. 2017, 15, 862–874. [Google Scholar] [CrossRef]
- Sharma, A.; Bandyopadhayaya, S.; Chowdhury, K.; Sharma, T.; Maheshwari, R.; Das, A.; Chakrabarti, G.; Kumar, V.; Mandal, C.C. Metformin Exhibited Anticancer Activity by Lowering Cellular Cholesterol Content in Breast Cancer Cells. PLoS ONE 2019, 14, e0209435. [Google Scholar] [CrossRef]
- Hindler, K.; Cleeland, C.S.; Rivera, E.; Collard, C.D. The Role of Statins in Cancer Therapy. Oncologist 2006, 11, 306–315. [Google Scholar] [CrossRef]
- Babcook, M.A.; Sramkoski, R.M.; Fujioka, H.; Daneshgari, F.; Almasan, A.; Shukla, S.; Nanavaty, R.R.; Gupta, S. Combination Simvastatin and Metformin Induces G1-Phase Cell Cycle Arrest and Ripk1- and Ripk3-Dependent Necrosis in C4-2b Osseous Metastatic Castration-Resistant Prostate Cancer Cells. Cell Death Dis. 2014, 5, e1536. [Google Scholar] [CrossRef]
- Wang, Z.S.; Huang, H.R.; Zhang, L.Y.; Kim, S.; He, Y.; Li, D.L.; Farischon, C.; Zhang, K.; Zheng, X.; Du, Z.Y.; et al. Mechanistic Study of Inhibitory Effects of Metformin and Atorvastatin in Combination on Prostate Cancer Cells in Vitro and in Vivo. Biol. Pharm. Bull. 2017, 40, 1247–1254. [Google Scholar] [CrossRef]
- Tran, L.N.K.; Kichenadasse, G.; Morel, K.L.; Lavranos, T.C.; Klebe, S.; Lower, K.M.; Ormsby, R.J.; Elliot, D.J.; Sykes, P.J. The Combination of Metformin and Valproic Acid Has a Greater Anti-Tumoral Effect on Prostate Cancer Growth in Vivo Than Either Drug Alone. In Vivo 2019, 33, 99–108. [Google Scholar] [CrossRef]
- Cuyas, E.; Verdura, S.; Llorach-Pares, L.; Fernandez-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct Sirt1-Activating Compound: Computational Modeling and Experimental Validation. Front. Endocrinol. (Lausanne) 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Pusapati, R.V.; Daemen, A.; Wilson, C.; Sandoval, W.; Gao, M.; Haley, B.; Baudy, A.R.; Hatzivassiliou, G.; Evangelista, M.; Settleman, J. Mtorc1-Dependent Metabolic Reprogramming Underlies Escape from Glycolysis Addiction in Cancer Cells. Cancer Cell 2016, 29, 548–562. [Google Scholar] [CrossRef] [PubMed]
- Granja, S.; Pinheiro, C.; Reis, R.M.; Martinho, O.; Baltazar, F. Glucose Addiction in Cancer Therapy: Advances and Drawbacks. Curr. Drug Metab. 2015, 16, 221–242. [Google Scholar] [CrossRef] [PubMed]
- Bikas, A.; Jensen, K.; Patel, A.; Costello, J., Jr.; McDaniel, D.; Klubo-Gwiezdzinska, J.; Larin, O.; Hoperia, V.; Burman, K.D.; Boyle, L.; et al. Glucose-Deprivation Increases Thyroid Cancer Cells Sensitivity to Metformin. Endocr. Relat. Cancer 2015, 22, 919–932. [Google Scholar] [CrossRef]
- Zordoky, B.N.; Bark, D.; Soltys, C.L.; Sung, M.M.; Dyck, J.R. The Anti-Proliferative Effect of Metformin in Triple-Negative Mda-Mb-231 Breast Cancer Cells Is Highly Dependent on Glucose Concentration: Implications for Cancer Therapy and Prevention. Biochim. Biophys. Acta 1840, 2014, 1943–1957. [Google Scholar] [CrossRef]
- Li, P.D.; Liu, Z.; Cheng, T.T.; Luo, W.G.; Yao, J.; Chen, J.; Zou, Z.W.; Chen, L.L.; Ma, C.; Dai, X.F. Redox-Dependent Modulation of Metformin Contributes to Enhanced Sensitivity of Esophageal Squamous Cell Carcinoma to Cisplatin. Oncotarget 2017, 8, 62057–62068. [Google Scholar] [CrossRef] [PubMed]
- Dodson, M.; Darley-Usmar, V.; Zhang, J. Cellular Metabolic and Autophagic Pathways: Traffic Control by Redox Signaling. Free Radic. Biol. Med. 2013, 63, 207–221. [Google Scholar] [CrossRef]
- Buzzai, M.; Jones, R.G.; Amaravadi, R.K.; Lum, J.J.; DeBerardinis, R.J.; Zhao, F.; Viollet, B.; Thompson, C.B. Systemic Treatment with the Antidiabetic Drug Metformin Selectively Impairs P53-Deficient Tumor Cell Growth. Cancer Res. 2007, 67, 6745–6752. [Google Scholar] [CrossRef]
- Gonnissen, A.; Isebaert, S.; McKee, C.M.; Muschel, R.J.; Haustermans, K. The Effect of Metformin and Gant61 Combinations on the Radiosensitivity of Prostate Cancer Cells. Int. J. Mol. Sci. 2017, 18, 399. [Google Scholar] [CrossRef]
- Knowell, A.E.; Patel, D.; Morton, D.J.; Sharma, P.; Glymph, S.; Chaudhary, J. Id4 Dependent Acetylation Restores Mutant-P53 Transcriptional Activity. Mol. Cancer 2013, 12, 161. [Google Scholar] [CrossRef]
- Salani, B.; del Rio, A.; Marini, C.; Sambuceti, G.; Cordera, R.; Maggi, D. Metformin, Cancer and Glucose Metabolism. Endocr. Relat. Cancer 2014, 21, R461–R471. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and Reduced Risk of Cancer in Diabetic Patients. BMJ 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Chatterjee, A.; Kogan, D.; Patel, D.; Foster, D.A. 5-Aminoimidazole-4-Carboxamide-1-Beta-4-Ribofuranoside (Aicar) Enhances the Efficacy of Rapamycin in Human Cancer Cells. Cell Cycle 2015, 14, 3331–3339. [Google Scholar] [CrossRef]
- Schneider, M.B.; Matsuzaki, H.; Haorah, J.; Ulrich, A.; Standop, J.; Ding, X.Z.; Adrian, T.E.; Pour, P.M. Prevention of Pancreatic Cancer Induction in Hamsters by Metformin. Gastroenterology 2001, 120, 1263–1270. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.; Fantus, I.G.; Sonenberg, N.; Pollak, M. Metformin Is an Amp Kinase-Dependent Growth Inhibitor for Breast Cancer Cells. Cancer Res. 2006, 66, 10269–10273. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Visagie, M.H.; Mqoco, T.V.; Liebenberg, L.; Mathews, E.H.; Mathews, G.E.; Joubert, A.M. Influence of Partial and Complete Glutamine-and Glucose Deprivation of Breast-and Cervical Tumorigenic Cell Lines. Cell Biosci. 2015, 5, 37. [Google Scholar] [CrossRef] [PubMed]
- Mukhopadhyay, S.; Saqcena, M.; Foster, D.A. Synthetic Lethality in Kras-Driven Cancer Cells Created by Glutamine Deprivation. Oncoscience 2015, 2, 807–808. [Google Scholar]
- Menendez, J.A.; Oliveras-Ferraros, C.; Cufi, S.; Corominas-Faja, B.; Joven, J.; Martin-Castillo, B.; Vazquez-Martin, A. Metformin Is Synthetically Lethal with Glucose Withdrawal in Cancer Cells. Cell Cycle 2012, 11, 2782–2792. [Google Scholar] [CrossRef]
- Saito, S.; Furuno, A.; Sakurai, J.; Sakamoto, A.; Park, H.R.; Shin-Ya, K.; Tsuruo, T.; Tomida, A. Chemical Genomics Identifies the Unfolded Protein Response as a Target for Selective Cancer Cell Killing During Glucose Deprivation. Cancer Res. 2009, 69, 4225–4234. [Google Scholar] [CrossRef]
- Schroder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Limonta, P.; Moretti, R.M.; Marzagalli, M.; Fontana, F.; Raimondi, M.; Marelli, M.M. Role of Endoplasmic Reticulum Stress in the Anticancer Activity of Natural Compounds. Int. J. Mol. Sci. 2019, 20, 961. [Google Scholar] [CrossRef] [PubMed]
- Haga, N.; Saito, S.; Tsukumo, Y.; Sakurai, J.; Furuno, A.; Tsuruo, T.; Tomida, A. Mitochondria Regulate the Unfolded Protein Response Leading to Cancer Cell Survival under Glucose Deprivation Conditions. Cancer Sci. 2010, 101, 1125–1132. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Bu, Y.; Chitnis, N.; Koumenis, C.; Fuchs, S.Y.; Diehl, J.A. Mir-216b Regulation of C-Jun Mediates Gadd153/Chop-Dependent Apoptosis. Nat. Commun. 2016, 7, 11422. [Google Scholar] [CrossRef] [PubMed]
- Verfaillie, T.; Garg, A.D.; Agostinis, P. Targeting Er Stress Induced Apoptosis and Inflammation in Cancer. Cancer Lett. 2013, 332, 249–264. [Google Scholar] [CrossRef] [PubMed]
- Salis, O.; Bedir, A.; Ozdemir, T.; Okuyucu, A.; Alacam, H. The Relationship between Anticancer Effect of Metformin and the Transcriptional Regulation of Certain Genes (Chop, Cav-1, Ho-1, Sgk-1 and Par-4) on Mcf-7 Cell Line. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 1602–1609. [Google Scholar]
- Yang, J.; Wei, J.; Wu, Y.; Wang, Z.; Guo, Y.; Lee, P.; Li, X. Metformin Induces Er Stress-Dependent Apoptosis through Mir-708-5p/Nnat Pathway in Prostate Cancer. Oncogenesis 2015, 4, e158. [Google Scholar] [CrossRef]
- Ma, L.; Wei, J.; Wan, J.; Wang, W.; Wang, L.; Yuan, Y.; Yang, Z.; Liu, X.; Ming, L. Low Glucose and Metformin-Induced Apoptosis of Human Ovarian Cancer Cells Is Connected to Ask1 Via Mitochondrial and Endoplasmic Reticulum Stress-Associated Pathways. J. Exp. Clin. Cancer Res. 2019, 38, 77. [Google Scholar] [CrossRef]
- Li Volti, G.; Tibullo, D.; Vanella, L.; Giallongo, C.; di Raimondo, F.; Forte, S.; di Rosa, M.; Signorelli, S.S.; Barbagallo, I. The Heme Oxygenase System in Hematological Malignancies. Antioxid. Redox Signal. 2017, 27, 363–377. [Google Scholar] [CrossRef]
- Tibullo, D.; Barbagallo, I.; Giallongo, C.; Vanella, L.; Conticello, C.; Romano, A.; Saccone, S.; Godos, J.; di Raimondo, F.; Volti, G.L. Heme Oxygenase-1 Nuclear Translocation Regulates Bortezomibinduced Cytotoxicity and Mediates Genomic Instability in Myeloma Cells. Oncotarget 2016, 7, 28868–28880. [Google Scholar] [CrossRef]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. Sirt3, a Human Sir2 Homologue, Is an Nad-Dependent Deacetylase Localized to Mitochondria. Proc. Natl. Acad. Sci. USA 2002, 99, 13653–13658. [Google Scholar] [PubMed]
- Liu, R.; Fan, M.; Candas, D.; Qin, L.; Zhang, X.; Eldridge, A.; Zou, J.X.; Zhang, T.; Juma, S.; Jin, C.; et al. Cdk1-Mediated Sirt3 Activation Enhances Mitochondrial Function and Tumor Radioresistance. Mol. Cancer Ther. 2015, 14, 2090–2102. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xu, Z.; Zeng, S.; Wang, X.; Liu, W.; Qian, L.; Wei, J.; Yang, X.; Shen, Q.; Gong, Z.; et al. Sirt5 Downregulation Is Associated with Poor Prognosis in Glioblastoma. Cancer Biomark. 2019, 24, 449–459. [Google Scholar] [CrossRef]
- Deng, C.X. Sirt1, Is It a Tumor Promoter or Tumor Suppressor? Int. J. Biol. Sci. 2009, 5, 147–152. [Google Scholar]
- Wang, C.; Chen, L.; Hou, X.; Li, Z.; Kabra, N.; Ma, Y.; Nemoto, S.; Finkel, T.; Gu, W.; Cress, W.D.; et al. Interactions between E2f1 and Sirt1 Regulate Apoptotic Response to DNA Damage. Nat. Cell Biol. 2006, 8, 1025–1031. [Google Scholar] [CrossRef]
- Liu, T.F.; McCall, C.E. Deacetylation by Sirt1 Reprograms Inflammation and Cancer. Genes Cancer 2013, 4, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Yi, J.; Luo, J. Sirt1 and P53, Effect on Cancer, Senescence and Beyond. Biochim. Biophys. Acta 1804, 2010, 1684–1689. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Cai, X.Q.; Chai, N.; Gu, Y.; Zhang, S.; Ding, M.L.; Cao, H.C.; Sha, S.M.; Yin, J.P.; Li, M.B.; et al. Sirt1 Is Reduced in Gastric Adenocarcinoma and Acts as a Potential Tumor Suppressor in Gastric Cancer. Gastrointest. Tumors 2015, 2, 109–123. [Google Scholar]
- Gerthofer, V.; Kreutz, M.; Renner, K.; Jachnik, B.; Dettmer, K.; Oefner, P.; Riemenschneider, M.J.; Proescholdt, M.; Vollmann-Zwerenz, A.; Hau, P.; et al. Combined Modulation of Tumor Metabolism by Metformin and Diclofenac in Glioma. Int. J. Mol. Sci. 2018, 19, 2586. [Google Scholar]
- Tseng, H.W.; Li, S.C.; Tsai, K.W. Metformin Treatment Suppresses Melanoma Cell Growth and Motility through Modulation of Microrna Expression. Cancers 2019, 11, 209. [Google Scholar] [CrossRef]
- Michishita, E.; Park, J.Y.; Burneskis, J.M.; Barrett, J.C.; Horikawa, I. Evolutionarily Conserved and Nonconserved Cellular Localizations and Functions of Human Sirt Proteins. Mol. Biol. Cell 2005, 16, 4623–4635. [Google Scholar] [CrossRef]
- Matsushita, N.; Yonashiro, R.; Ogata, Y.; Sugiura, A.; Nagashima, S.; Fukuda, T.; Inatome, R.; Yanagi, S. Distinct Regulation of Mitochondrial Localization and Stability of Two Human Sirt5 Isoforms. Genes Cells 2011, 16, 190–202. [Google Scholar] [CrossRef]
- Jung-Hynes, B.; Nihal, M.; Zhong, W.; Ahmad, N. Role of Sirtuin Histone Deacetylase Sirt1 in Prostate Cancer. A Target for Prostate Cancer Management Via Its Inhibition?” J. Biol. Chem. 2009, 284, 3823–3832. [Google Scholar] [CrossRef]
- Barbagallo, I.; Parenti, R.; Zappala, A.; Vanella, L.; Tibullo, D.; Pepe, F.; Onni, T.; Volti, G.L. Combined Inhibition of Hsp90 and Heme Oxygenase-1 Induces Apoptosis and Endoplasmic Reticulum Stress in Melanoma. Acta Histochem. 2015, 117, 705–711. [Google Scholar] [CrossRef]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of Cell Protection by Heme Oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef]
- Sacerdoti, D.; Singh, S.P.; Schragenheim, J.; Bellner, L.; Vanella, L.; Raffaele, M.; Meissner, A.; Grant, I.; Favero, G.; Rezzani, R.; et al. Development of Nash in Obese Mice Is Confounded by Adipose Tissue Increase in Inflammatory Nov and Oxidative Stress. Int. J. Hepatol. 2018, 2018, 3484107. [Google Scholar] [CrossRef]
- Poljsak, B.; Milisav, I. Clinical Implications of Cellular Stress Responses. Bosn J. Basic Med. Sci. 2012, 12, 122–126. [Google Scholar] [CrossRef]
- Choi, A.M.; Alam, J. Heme Oxygenase-1: Function, Regulation, and Implication of a Novel Stress-Inducible Protein in Oxidant-Induced Lung Injury. Am. J. Respir. Cell Mol. Biol. 1996, 15, 9–19. [Google Scholar] [CrossRef]
- Bahmani, P.; Hassanshahi, G.; Halabian, R.; Roushandeh, A.M.; Jahanian-Najafabadi, A.; Roudkenar, M.H. The Expression of Heme Oxygenase-1 in Human-Derived Cancer Cell Lines. Iran. J. Med. Sci. 2011, 36, 260–265. [Google Scholar]
- Chiang, S.K.; Chen, S.E.; Chang, L.C. A Dual Role of Heme Oxygenase-1 in Cancer Cells. Int. J. Mol. Sci. 2018, 20, 39. [Google Scholar] [CrossRef]
- Ryter, S.W.; Choi, A.M. Targeting Heme Oxygenase-1 and Carbon Monoxide for Therapeutic Modulation of Inflammation. Transl. Res. 2016, 167, 7–34. [Google Scholar] [CrossRef]
- Sena, P.; Mancini, S.; Benincasa, M.; Mariani, F.; Palumbo, C.; Roncucci, L. Metformin Induces Apoptosis and Alters Cellular Responses to Oxidative Stress in Ht29 Colon Cancer Cells: Preliminary Findings. Int. J. Mol. Sci. 2018, 19, 1478. [Google Scholar] [CrossRef]
- Li, Y.; Wang, M.; Zhi, P.; You, J.; Gao, J.Q. Metformin Synergistically Suppress Tumor Growth with Doxorubicin and Reverse Drug Resistance by Inhibiting the Expression and Function of P-Glycoprotein in Mcf7/Adr Cells and Xenograft Models. Oncotarget 2018, 9, 2158–2174. [Google Scholar] [CrossRef]
- Teh, J.T.; Zhu, W.L.; Newgard, C.B.; Casey, P.J.; Wang, M. Respiratory Capacity and Reserve Predict Cell Sensitivity to Mitochondria Inhibitors: Mechanism-Based Markers to Identify Metformin-Responsive Cancers. Mol. Cancer Ther. 2019, 18, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Budigner, G.S.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. Elife 2014, 3, e02242. [Google Scholar] [CrossRef]
- Wang, H.; Cheng, Q.; Li, X.; Hu, F.; Han, L.; Zhang, H.; Li, L.; Ge, J.; Ying, X.; Guo, X.; et al. Loss of Tigar Induces Oxidative Stress and Meiotic Defects in Oocytes from Obese Mice. Mol. Cell Proteom. 2018, 17, 1354–1364. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.X.; Zhang, H.Y.; Meng, X.; Guan, Y.; Wang, H.Q. Role of Oxidative Stress and Intracellular Glutathione in the Sensitivity to Apoptosis Induced by Proteasome Inhibitor in Thyroid Cancer Cells. BMC Cancer 2009, 9, 56. [Google Scholar] [CrossRef]
- Wong, E.Y.; Wong, S.C.; Chan, C.M.; Lam, E.K.; Ho, L.Y.; Lau, C.P.; Au, T.C.; Chan, A.K.; Tsang, C.M.; Tsao, S.W.; et al. Tp53-Induced Glycolysis and Apoptosis Regulator Promotes Proliferation and Invasiveness of Nasopharyngeal Carcinoma Cells. Oncol. Lett. 2015, 9, 569–574. [Google Scholar] [CrossRef]
- Bensaad, K.; Tsuruta, A.; Selak, M.A.; Vidal, M.N.; Nakano, K.; Bartrons, R.; Gottlieb, E.; Vousden, K.H. Tigar, a P53-Inducible Regulator of Glycolysis and Apoptosis. Cell 2006, 126, 107–120. [Google Scholar] [CrossRef]
- Winkler, B.S.; DeSantis, N.; Solomon, F. Multiple Nadph-Producing Pathways Control Glutathione (Gsh) Content in Retina. Exp. Eye Res. 1986, 43, 829–847. [Google Scholar] [CrossRef]
- Zhou, J.H.; Zhang, T.T.; Song, D.D.; Xia, Y.F.; Qin, Z.H.; Sheng, R. Tigar Contributes to Ischemic Tolerance Induced by Cerebral Preconditioning through Scavenging of Reactive Oxygen Species and Inhibition of Apoptosis. Sci. Rep. 2016, 6, 27096. [Google Scholar] [CrossRef] [PubMed]
- Heiss, K.; Raffaele, M.; Vanella, L.; Murabito, P.; Prezzavento, O.; Marrazzo, A.; Arico, G.; Castracani, C.C.; Barbagallo, I.; Zappala, A.; et al. (+)-Pentazocine Attenuates Sh-Sy5y Cell Death, Oxidative Stress and Microglial Migration Induced by Conditioned Medium from Activated Microglia. Neurosci. Lett. 2017, 642, 86–90. [Google Scholar] [CrossRef]
- Salerno, L.; Amata, E.; Romeo, G.; Marrazzo, A.; Prezzavento, O.; Floresta, G.; Sorrenti, V.; Barbagallo, I.; Rescifina, A.; Pittala, V. Potholing of the Hydrophobic Heme Oxygenase-1 Western Region for the Search of Potent and Selective Imidazole-Based Inhibitors. Eur. J. Med. Chem. 2018, 148, 54–62. [Google Scholar] [CrossRef] [PubMed]
- Lanteri, R.; Acquaviva, R.; di Giacomo, C.; Sorrenti, V.; Destri, G.L.; Santangelo, M.; Vanella, L.; di Cataldo, A. Rutin in Rat Liver Ischemia/Reperfusion Injury: Effect on Ddah/Nos Pathway. Microsurgery 2007, 27, 245–251. [Google Scholar] [CrossRef]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raffaele, M.; Pittalà, V.; Zingales, V.; Barbagallo, I.; Salerno, L.; Li Volti, G.; Romeo, G.; Carota, G.; Sorrenti, V.; Vanella, L. Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. Int. J. Mol. Sci. 2019, 20, 2593. https://doi.org/10.3390/ijms20102593
Raffaele M, Pittalà V, Zingales V, Barbagallo I, Salerno L, Li Volti G, Romeo G, Carota G, Sorrenti V, Vanella L. Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. International Journal of Molecular Sciences. 2019; 20(10):2593. https://doi.org/10.3390/ijms20102593
Chicago/Turabian StyleRaffaele, Marco, Valeria Pittalà, Veronica Zingales, Ignazio Barbagallo, Loredana Salerno, Giovanni Li Volti, Giuseppe Romeo, Giuseppe Carota, Valeria Sorrenti, and Luca Vanella. 2019. "Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death" International Journal of Molecular Sciences 20, no. 10: 2593. https://doi.org/10.3390/ijms20102593
APA StyleRaffaele, M., Pittalà, V., Zingales, V., Barbagallo, I., Salerno, L., Li Volti, G., Romeo, G., Carota, G., Sorrenti, V., & Vanella, L. (2019). Heme Oxygenase-1 Inhibition Sensitizes Human Prostate Cancer Cells towards Glucose Deprivation and Metformin-Mediated Cell Death. International Journal of Molecular Sciences, 20(10), 2593. https://doi.org/10.3390/ijms20102593