Abstract
Vascular cognitive impairment (VCI) is the second most common cause of cognitive deficit after Alzheimer’s disease. Since VCI patients represent an important target population for prevention, an ongoing effort has been made to elucidate the pathogenesis of this disorder. In this review, we summarize the information from animal models on the molecular changes that occur in the brain during a cerebral vascular insult and ultimately lead to cognitive deficits in VCI. Animal models cannot effectively represent the complex clinical picture of VCI in humans. Nonetheless, they allow some understanding of the important molecular mechanisms leading to cognitive deficits. VCI may be caused by various mechanisms and metabolic pathways. The pathological mechanisms, in terms of cognitive deficits, may span from oxidative stress to vascular clearance of toxic waste products (such as amyloid beta) and from neuroinflammation to impaired function of microglia, astrocytes, pericytes, and endothelial cells. Impaired production of elements of the immune response, such as cytokines, and vascular factors, such as insulin-like growth factor 1 (IGF-1), may also affect cognitive functions. No single event could be seen as being the unique cause of cognitive deficits in VCI. These events are interconnected, and may produce cascade effects resulting in cognitive impairment.
1. Vascular Cognitive Impairment (VCI)
Vascular cognitive impairment (VCI) [1] comprises both subjects with dementia and mild cognitive impairment (MCI). The symptoms of VCI include the disturbance of specific cognitive functions with various degrees of severity [2]. VCI is recognized to be the second most common cause of cognitive deficit after Alzheimer’s disease (AD). However, a comorbidity between these pathologies is frequent and characterized by synergistic processes leading to cognitive impairments [3]. This is the reason why subjects with VCI are often classified as AD, and thus VCI’s ranking as the cause of cognitive deficits may vary from second to fourth [4]. Various biomarkers have been used to increase the certainty of diagnosis underlying pathological processes [5,6,7,8,9,10].
VCI is a general term that includes a group of cognitive disorders attributable to a pathological state of the cerebral vascular system [11]. In the brain, VCI is characterized by the atrophy of gray matter and hemispheric white matter lesions [12], but may include also subcortical vascular lesions in, for example, the thalamus. The main cognitive deficits usually involve memory processes, speed of processing, and executive functions [2]. VCI types are classified according to clinical characteristics, and include vascular mild cognitive impairment (VMCI), vascular dementia (VaD), and mixed dementia (MD) associated with vascular dysfunction [13].
VCI prevalence is also age- and risk factor-related. In older people over 65 years, the prevalence of mild VCI is higher than that of VD [14]. Among other risk factors, the most relevant are those affecting the vascular system and include hypertension, hyperlipidemia, hyperuricemia, diabetes, cardiopathy, history of stroke, carotid plaque, and smoking [15].
The conceptualization of VCI has recently evolved with the adoption of VASCOG [16], VICCCS [13], and DSM-V [17] criteria. Subtypes of VCI are divided into mild VCI and major VCI (VaD) according to the level of VCI impairment, with operational criteria for the thresholds, and clinical and neuroimaging criteria to establish vascular etiology. These new criteria were adopted to overcome the limitations of those by DSM-IV and NINDS-AIREN [18] which, having memory impairment as necessary criterion for the diagnosis of VCI, often bias the diagnosis towards AD, and do not distinguish among deficits in different cognitive domains. In addition, the assessment of brain microvasculature impairment as a co-morbidity is becoming fundamental for staging cognitive decline in MCI and AD [8,19,20]. It was shown that subjects with normal functioning at the stage of MCI are more likely to progress to dementia if their Fazekas vascular score is higher [21].
Since VCI patients represent an important target population for prevention, great efforts have been made to elucidate the pathogenesis of this disorder.
2. Pathogenesis
Many studies have provided new insights into the causes of VCI. VCI is caused by decreased blood supply to the brain [11]. The affected brain regions undergo a neuronal tissue loss which compromises its structure and function and manifests as a cognitive deficit.
The causes of this reduced blood flow can be divided into three main groups: ischemic factors, hemorrhagic factors, and other factors affecting functional brain regions [22].
2.1. Ischemic Factors
Vessel obstruction can be the cause of VCI and dementia and, generally, with latency after the event. Vessel occlusions can be large, as in the case of ischemic stroke, or small, such as when caused by arteritis and arteriosclerosis of smaller vessels, where the term ‘small vessel dysfunction’ is more appropriate.
In the case of stroke, inflammatory mediators and amyloid deposition (cerebral amyloid angiopathy (CAA)) in the walls of vessels play an important role in the development of VCI [23,24]. The cognitive deficits after stroke generally occur in a shorter time frame (i.e., less than 1 year) as compared to other forms of VCI [13,25].
The damage caused by dysfunction of small vessels is slower to appear as it is the result of cortical and subcortical microinfarcts [26]. In this condition, the involved brain region is affected by a state of cerebral hypoperfusion which, in the long term, is responsible for the damage of white matter and for the insurgence of cognitive dysfunction [27,28]. These types of multiple infarctions and diffuse white matter lesions often appear in the lateral ventricle and subcortical structures, resulting in multiple cognitive domain impairment [26,27,28].
2.2. Hemorrhagic Factors
Vascular dysfunction and cognitive deficits can also occur after a cerebral hemorrhage [27,28]. Cognitive dysfunction can originate from an intracranial hemorrhage, caused by CAA [29,30] and resulting in brain edema or secondary ischemia due to mass lesion. Also, a subarachnoid hemorrhage [31], with involvement of hippocampal and frontotemporal regions [32], can result in VCI syndrome with visuospatial memory and language deficits [33]. In this case, it is believed that VCI is attributed to the impact of the subdural membrane on dural lymphatic drainage [34].
2.3. Other Factors
VCI can appear in comorbidity with other diseases, such as AD, synucleinopathy, or tauopathy, which often results from both vascular disorders and structural changes in the protein of brain tissue [22]. In addition, hypertension [35] and diabetes [3], which represent independent risk factors for AD, can be concomitant with VCI.
Lifestyle factors may also contribute to VCI, including smoking [36], vitamin deficits (vitamin E) [37], poor physical activity [38], and a low level of education. However, the contribution of these factors to cognitive deficits in VCI is, at present, controversial [36].
An improved understanding of the contribution of vascular burden to cognitive decline further originates from investigations on translational animal models [39].
3. Animal Models of VCI
Animal models for studying VCI comprise different models based on the pathogenic factors [40] exposed above (Table 1).

Table 1.
Main rodent models to study vascular cognitive impairment.
The most-used species are rodents [41], although other species can be also used once the efficacy of a particular treatment has been established in rodents. Thus, models using rabbits, monkeys, and dogs were also investigated before translation to clinical trials [41].
Stroke is usually mimicked by a transient (tMCAO) or permanent (pMCAO) middle cerebral artery occlusion [42]. Small vessel occlusion is also mimicked by tMCAO [44]. A condition of global hypoperfusion can be induced by bilateral carotid artery occlusion (BCAO) or bilateral carotid artery stenosis (BCAS) [43]. These rodents present with hypoxia and hypoperfusion of white matter, making this a reasonable model for VCI [45].
A cerebral hemorrhage can be induced by intracerebral (double or single) injection of blood or collagenase [46], or induced by a microballoon inserted into the brain [47].
A subarachnoid hemorrhage may also created by the injection of blood [50]. In addition, microhemorrhages can be induced with high spatiotemporal precision in rodent cortex by directly disrupting the walls of cortical microvessels using focused lasers [48].
VCI-relevant animal models can also result from genetically manipulated animals. The stroke-prone spontaneously hypertensive rats (SHR/SP) animal model is the most relevant model of hypertension in rodents, and was created by inbred strain manipulation [43].
Another genetic model in mice was obtained by knocking out the Notch3 gene [41,43]. This genetic manipulation resembles the hereditary form of VD [51], where the cerebrovascular alterations are similar to those observed in human autosomal dominant arteriopathy with subcortical infarcts and leukoencephalopathy (CADASIL) [52]. Other approaches to induce VCI in animals include mimicking risk factors such as diabetes, or by diet exposure to create risky conditions, such as hyperhomocysteinemia [49].
We review the current evidence obtained, from these models, for the pathogenesis of VCI.
4. Molecular Mechanism of VCI
Animal models may serve to investigate the molecular changes that occur in the brain during a cerebral vascular insult and that ultimately lead to the cognitive deficits of VCI.
Cerebral circulation assures an adequate delivery of blood to the brain. Hypoxia or chronic cerebral hypoperfusion may lead to an altered balance between the delivery of energy substrates and the clearance of metabolic waste in the neurovascular unit. The pathological mechanisms, in terms of cognitive deficits, may span from oxidative stress to vascular clearance of toxic waste products (such as Aβ), and from neuroinflammation to impaired function of microglia, astrocytes, and endothelial cells. Impaired production of elements of the immune response, such as cytokines, and vascular factors, such as insulin-like growth factor 1 (IGF-1), may also affect cognitive functions. At a molecular level, there are several lines of investigations depending on the supposed cause of the VCI.
4.1. Oxidative Stress
Oxidative stress is a condition of imbalance between free radicals and antioxidants [53,54] that can originate from vascular-related pathological states, including hypertension, diabetes, and arteriosclerosis [53]. In VCI, oxidative stress is considered to be a major contributing factor to the pathogenesis of cognitive deficits [55]. Moreover, it has been repetitively demonstrated that the excessive oxidation of proteins is also a common phenomenon in neurodegenerative disease and is correlated to cognitive deficit [56,57].
The molecular mechanism of oxidative stress-induced cognitive deficits has been investigated in animal models. In occlusion rat models (BCAO), it was shown that oxidative stress is characterized by the increased production of reactive oxygen species (ROS) [58,59] which are responsible for both cardiovascular pathophysiology and neurodegeneration [60,61]. In fact, ROS and reduced antioxidant defense may directly affect synaptic activity and neurotransmission in neurons, leading to cognitive dysfunction [62]. In the same models, it was also shown that the negative effects of ROS on memory processes could be counteracted by substances able to increase the gene expression of antioxidant proteins, such as nuclear factor erythroid 2 like 2 (NFE2L2), alcohol dehydrogenase 7 (ADH7), and glutathione peroxidase 2 (GPX2) and 3 (GPX3) [58,59].
Excessive ROS production during VCI is caused by the enzyme nicotinamide adenine dinucleotide phosphate (NADPH) oxidase [62]. NADPH oxidase is a multiunit enzyme that was discovered in neutrophils and is also present in vessel cells, particularly in cerebral blood vessels [62,63]. ROS derived from the enzyme NADPH oxidase are key pathogenic effectors of cerebrovascular dysregulation [63]. This might lead, in turn, to cognitive impairment via cellular dysfunction and cell death [62].
Supporting this notion, it has been shown that inhibition of NADPH oxidase activity can reduce cognitive impairment in BCAO rodent models of VCI [64,65]. In addition, it is likely that similar alterations may occur in human VCI. Data from subjects affected by VD demonstrated that the activity of antioxidant enzymes—such as superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase (GR), and heme oxygenase/biliverdin reductase—is decreased [55,66]. Furthermore, reduced antioxidant enzyme levels in blood samples from VD patients have also been reported [67,68].
Taken together, these data suggest that oxidative stress, through the production of ROS, may induce cognitive deficits in humans. From this perspective, the targeting of such genes is an adoptable therapy to treat ischemic-induced cognitive deficits.
4.2. Neuroinflammation and Activation of Microglia
Oxidative stress is also the cause of inflammatory processes at the neurovascular unit. Thus, it is plausible that inflammation may also be involved in the pathophysiology of VCI [53,69].
Studies in rat models of BCAO have demonstrated that a condition of hypoxia/ischemia triggers microglia to release metalloproteinases (MMPs), which can damage the blood–brain barrier (BBB) and disrupt myelinated fibers [70,71]. In addition, neuroinflammation after BCAO can induce cognitive dysfunction through the release of proinflammatory cytokines and ROS by the activated microglia [72].
Chronic microglial activation can be responsible for the pathogenesis of different forms of dementia [73]. It has been shown, in BCAO models, that inflammatory-related microglia is associated with cognitive impairment [74]. This mechanism seems to be related to the activation of receptors of advanced glycation end products (RAGE) which are present on both microglia and neurons [59,75]. RAGE activation turns on nuclear factor kappa B (NF-κB), which is a transcription factor that controls several proinflammatory genes.
The release of cytokines, such as interleukin (IL)-6 and tumor necrosis factor (TNF)-α, by microglia play important roles in the pathogenesis of dementia [73]. It has been hypothesized that this pathway is also activated in AD [76]. In animal models of stroke and in humans, IL-6 is increased in serum and cerebrospinal fluid (CSF) after stroke [77,78]. In addition, it was shown that elevated levels of IL-6 contribute to the insurgence of dementia in patients with vascular risk factors [79]. Accordingly, patients with VD also show a high level of serum IL-6 [80], which points to the inflammatory component in the development of VCI.
4.3. Astrocytes
In the past years, the role of astrocytes as fine-tune regulators of neuronal activity have been thoroughly studied. Nevertheless, recent evidence suggests that astrocytes may be key elements for the cognitive deficits induced by neurovascular dysfunction [81].
Nowadays, astrocytes are recognized as key elements for the metabolic supply to neurons by blood vessels. Astrocytes are, in fact, an element of the neurovascular unit, which regulates cerebral blood flow, BBB permeability, neuroimmune responses, and neurovascular remodeling [82]. Astrocytes can regulate cerebral blood flow through their processes by directly interacting with endothelial cells surrounding the brain vasculature. These specialized processes are called astrocytic endfeet [81].
The astrocytic endfoot is a specialized unit that functions to maintain the ionic and osmotic homeostasis of the brain. In transgenic models of cerebral amyloid angiopathy and in hyperhomocysteinemia models of VCI, it has been demonstrated that vascular alteration leads to disruption of the astrocytic endfoot [83,84]. In addition, it was shown that endfoot disruption is subsequent to microglia activation and to the release of proinflammatory cytokines [68,84]. This latter finding suggested the hypothesis that, during inflammatory processes, astrocytes and their endfeet may undergo degeneration because of the release of MMPs [85] which degrade the dystrophin–dystroglycan complex anchoring the endfoot to the basement membrane of the vasculature [65,69,81,85]. Endfeet degeneration, in turn, leads to impaired neurovascular coupling and impaired potassium homeostasis, increasing neuronal excitability and ultimately leading to cognitive deficits [86]. It should also be mentioned that astrocytes, through astrocytic CN/nuclear factor of activated T cell (NFAT), can also release inflammatory cytokines, affecting cognition [87].
Based on these data, the role of astrocytes in VCI-induced cognitive dysfunctions has been increasingly recognized [88]. Several investigations using recent genetic tools now support this notion by showing that inactivating or boosting astroglial function directly affects cognitive abilities [89].
4.4. Endothelial Cells and Nitric Oxide
Endothelial cells are part of the neurovascular unit and cover the internal surface of cerebral blood vessels [90]. Many studies have shown that endothelial cells play an important role in VCI. Endothelial cells damaged by oxidative stress contribute to cerebrovascular impairment and neurovascular uncoupling [91]. Beyond that, the link with cognitive dysfunction seems to be due to the effects on the production of nitric oxide (NO) by these cells [92].
NO is a reactive gas secreted in endothelial cells by the endothelial isoform of the enzyme NO synthase, and is tonically released to control systemic vascular tone and neuronal activity in the CNS [93]. In chronic cerebral hypoperfusion models of VCI [94] and in hypertensive rats [95], it has been observed that the bioavailability of endothelium-derived NO is reduced, and that this reduction may be the cause of cognitive deficits. Further support to this hypothesis comes from studies in AD animal models, where a reduction of NO increases the negative effects of amyloid beta (Aβ) on cognition, whereas the administration of NO has a protective effect on Aβ deposition [96].
Many studies are now concentrating on treatments aimed at restoring NO levels at normal conditions in VCI models [92,95,97].
The effects of NO on cognitive processes have been studied, and it has been seen that they are mediated by several mechanisms. NOS is postsynaptically colocalized with N-methyl-d-aspartate (NMDA) receptors. After Ca2+ influx into postsynaptic neurons, NO acts as a retrograde messenger, providing a positive feedback mechanism to maintain presynaptic glutamate release which binds to various types of postsynaptic NMDA receptors, strengthening hippocampal memory-related processes, such as long-term potentiation (LTP) [93]. NO also regulates other pathways via the post-translational modification (S-nitrosylation and 3-nitrotyrosination) of proteins involved in synaptic transmission and intracellular trafficking [98].
At the same time, however, it should be noted that excessive production of NO can have negative effects through the excessive stimulation of NMDA receptors (excitotoxicity), which in normal conditions is counteracted by the same NO through induction of cyclic guanosine monophosphate (cGMP)-mediated signaling [99]. For these reasons, a reduction in NO release by endothelial cells may actually play a very significant role in explaining the association between vascular lesions and cognitive impairment. Other studies are, however, necessary to better define the neuroprotective versus neurotoxic effects of NO in VCI.
4.5. Pericytes
Pericytes are mural cells of the neurovascular unit that surround endothelial cells [100]. The main function of pericytes is to regulate the permeability of the BBB and the clearance and phagocytosis of cellular debris [100].
Pericytes have a strong interaction with endothelial cells, forming direct cell–cell contacts, known as “peg-and-socket” contacts [101]. This interaction is essential for both types of cells. Pericytes are essential for the differentiation and survival of endothelial cells. At the same time, the proliferation and migration of pericytes depend on the release of platelet-derived growth factor B (PDGF-B) by endothelial cells [102]. PDGF-B binds to the receptor located in the pericyte membrane and activates signal transduction pathways that include TGF-β [103] and Notch [104]. Therefore, a loss of pericytes can have deleterious consequences on endothelial cells and on BBB permeability [104].
In animal models of neurodegenerative diseases and in post-mortem histological studies, a loss of pericytes has been observed, in particular, in Alzheimer’s disease [105,106]. The mechanism of pericyte loss in neurodegenerative disorders has not been yet elucidated [107]. Preliminary data have suggested that vascular factors, such as hypertension and hyperglycemia, can lead to pericyte loss [108]. Concomitantly, the loss of pericytes also leads to endothelial cell death and can exacerbate vascular dysfunction, causing regression of brain microvessels and giving rise to a condition of chronic hypoxia [109].
In pericyte-deficient transgenic mice induced by manipulation of PDGF-B gene, it has been shown that pericyte degeneration leads to BBB disruption and accumulation, in the brain, of blood-derived products potentially toxic for the neurons and neurovascular unit [110], such as plasmin, thrombin, and fibrin [111]. Pericytes are also involved in Aβ clearance through receptor LRP-1, which binds and internalizes different Aβ species [112]. A loss of brain pericytes may thus lead to reduced Aβ clearance through the LRP-1 degradative pathway, promoting Aβ accumulation and neuronal death. It was also shown that vascular damage caused by pericyte loss is sufficient to induce neurodegeneration, even in absence of Aβ accumulation [105,113].
These data suggest that pericytes may have a prominent role in inducing memory deficits in VCI. Supporting this idea, it was shown in a recent study using a rat model of cerebral small vessel disease that restoration of BBB integrity by infusion of precursor cells of pericyte and endothelial cells inhibits brain atrophy and restores cognitive functions [114].
4.6. Autophagy
In eukaryotic cells, autophagy is a fundamental process that degrades and recycles cellular constituents [115]. In the nervous system, autophagy allows nerve cell survival and reparation by clearing abnormally aggregated proteins and damaged cellular organelles, including mitochondria [116]. Autophagy is thus important in a condition of neuronal insult of vascular origin, and it has been demonstrated that it could be induced by oxidative stress [117].
Nonetheless, in a condition of chronic brain hypoperfusion, excessive activation of autophagy can cause cell death (autophagic cell death) [118] through molecular mechanisms involving multiple pathways such as AMP-activated protein kinase (AMPK) or the protein called mammalian target of rapamycin (mTOR) [119].
In rat models of VD, it was shown that excessive autophagy is present in the hippocampus and aggravates neuronal injury [120]. Similarly, in rat models of chronic brain hypoperfusion, the levels of autophagy-related proteins (Beclin-1, light chain 3B, and P62) were found to be increased before the occurrence of cognitive decline [121], suggesting the involvement of autophagy in the pathogenesis of VD. This hypothesis has been confirmed by studies showing neuroprotective and beneficial effects on cognitive function, with treatment aimed at reducing autophagy in these models [122].
There are various proposed molecular mechanisms, which include inhibition of mTOR [123], suppression of autophagy-related proteins, stimulation of vascular endothelial growth factor (VEGF) pathway [124], and reduction of proinflammatory cytokines [125].
Altogether, these data suggest that the suppression of excessive autophagy could have a neuroprotective effect and could be beneficial to preventing VCI. However, it should be noted that suppression of autophagy must be tuned gently during VCI course because beneficial effects of autophagy at the beginning of the condition related to brain hypoperfusion have been reported [126].
4.7. Insulin-Like Growth Factor-1
The incidence of VCI in humans increases with age [14]. One of the most interesting endocrine mechanisms connected to age-related cerebrovascular alterations is the decline in circulating insulin-like growth factor 1 (IGF-1) levels, which appears to contribute significantly to vascular aging and age-related cerebrovascular changes [127].
IGF-1 is a single-chain polypeptide widely expressed in brain [127]. IGF-1 is essential for neuroprotection, normal growth, and development [128]. During ontogenesis, IGF-1 exerts its roles on brain development through control of neurotrophic responses and cell signaling [129].
IGF-1 expression is high at young age and declines during aging, and this reduction correlates with cognitive decline in the elderly [130]. IGF-1 regulates cognitive functions by enhancing excitatory synaptic transmission in the CA1 region of the hippocampus [131]. After binding to its receptor (IGF1R), IGF-1 activates important pathways for memory processes, such as PI3K/mTOR/AKT1 (phosphatidylinositol-3 kinase/mammalian target of rapamycin/serine–threonine-specific protein kinase AKT-PKB), and MAPK/ERK (mitogen-activated protein kinase/extracellular signal-regulated kinase) [132].
In addition, IGF-1 induces angiogenesis [133] and neurogenesis [134] in hippocampus. These latter effects are important in the context of VCI pathology. Preclinical studies established a causal link between cognitive decline and microvascular rarefaction in the hippocampus [135]. IGF-1 has important actions on the brain vessels. It has been shown that age-related decline in circulating IGF-1 levels results in functional impairment of the cerebral microvessels [136]. Knockout of the IGF-1 gene produced neurovascular uncoupling in mice [137]. In addition, when hypertension conditions were induced in the same mice, cerebrovascular autoregulatory dysfunctions [138] and microvascular rarefaction in the hippocampus and neocortex [136] were present.
In non-genetic models of VCI (BCVAO), it was shown that IGF-1 and IGF-1 mRNA were downregulated in the hippocampus [139]. Moreover, in human subjects affected by cognitive deficits, it was demonstrated that the reduction of IGF-1 serum levels represents a risk factor for VD [140] and stroke [141].
All these data indicate that IGF-1 may play an important role in maintaining cognitive function and that VCI-induced cognitive are associated with reduction of this peptide. Supporting this notion, IGF-1 has been demonstrated to have potential neuroprotective effects in treating cerebral ischemia. Treatments aimed at restoring IGF-1 brain levels in MCAO rodent models reduced the infarct volume [142] and the rate of apoptosis [143], improving cognitive deficits through the IGF-1/AKT pathway [144].
Notably, peripherally injected IGF-1 can cross the BBB and exert its effect in the CNS [145]. This fact is of considerable interest as a potential treatment for VCI [146]. However, despite the well-recognized positive effects on synaptic function and cognition, the complex role of IGF-1 in vascular and neurodegenerative diseases is still unclear and requires additional research.
5. Conclusions
In this review, we analyzed the most important factors contributing to the onset of cognitive deficits in vascular disorders in humans by exploring data from animal models. The animal models of VCI allow understanding of the important molecular mechanisms leading to the cognitive deficits observed in humans. Moreover, these models also allow testing of the various therapeutic strategies based on various experimental hypotheses.
VCI may be caused by various mechanisms and metabolic pathways. It is difficult to determine which one is the most important, and the whole picture looks more multifactorial. Certainly, these events are interconnected and may produce cascade effects resulting in cognitive impairment. Initially, a chronic condition of hypoperfusion or hypoxia/ischemia engages a series of reactions at the level of the neurovascular unit, of which the most relevant appear to be oxidative stress and the induction of a neuroinflammatory state. Downstream processes include the activation of the microglia and the release of ROS. Recently, the role of astrocytes has also been greatly emphasized as they are elements of structural and functional support of the neurovascular unit. Astrocytes support the interactions between the vascular and nervous systems through the astrocytic endfeet, which can be destroyed by the release of inflammatory mediators, such as cytokines and ROS, but also by an excessive increase in autophagy. Increased autophagy represents a functional attempt by neurons to recovery from degeneration, but in a chronic condition, it can cause neuronal death implying cognitive deficits. In addition, the activity of endothelial cells surrounding blood vessels may also be affected by oxidative stress, neuroinflammation, and autophagy. In such conditions, these cells decrease the production of NO, a gas of fundamental importance for the activity of hippocampal neurons (LTP) and synaptic plasticity. Loss of pericytes can also exacerbate endothelial cell dysfunction and contribute to generating oxidative stress and regression of brain microvessels. All these alterations can actually be interconnected, as summarized in Figure 1. Therefore, interfering one or more of these factors has led to beneficial effects, at least in laboratory animals. Nonetheless, it should be noted that animal models, especially rodents, cannot effectively represent the complex clinical picture of VCI in humans. Moreover, potential treatments, like those currently available, are aimed at reducing the symptoms but not at curing the causes of VCI.
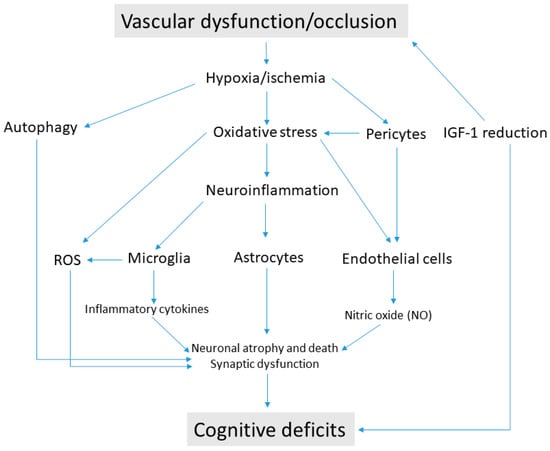
Figure 1.
Pathogenic mechanisms causing cognitive deficits in vascular cognitive impairment. Vascular dysfunction/occlusion induces a state of hypoxia/ischemia at the neurovascular unit. The consequent oxidative stress triggers the production of reactive oxygen species (ROS) and sustains the process of neuroinflammation in glial cells. Microglia activation produce proinflammatory cytokines and destruction of astrocyte endfeet contribute to disrupting cytoarchitecture in brain regions involved in cognitive function. Hypoxia/ischemia is also responsible for the loss of pericytes, which cause blood–brain barrier (BBB) and endothelial cell dysfunction. Endothelial cells damaged by oxidative stress cause reduced release of nitric oxide (NO), while excessive autophagy contributes to neuronal damage. The age-related decline of insulin-like growth factor-1 (IGF-1) may be the cause of vascular pathologies from one side, and it may impair synaptic activity from the other side, thus generating cognitive deficits.
Studies on IGF-1 factor seem very interesting for a number of reasons: First, a reduction of this factor is associated with the onset of vascular pathologies and, simultaneously, with cognitive decline. Secondly, IGF-1 appears to be able to bypass the BBB and produce beneficial effects on both the vascular and nervous systems. However, although its positive effect on synaptic function are fairly well-recognized, its potential in the prevention and treatment of VCI requires more investigation, possibly using animal species with a more evolved brain (i.e., primates).
Author Contributions
Conceptualization, F.A., J.H., K.K., and M.V.; Writing—Original Draft Preparation, F.A. and J.H.; Writing—Review & Editing, K.K. and M.V.; Funding Acquisition, J.H., K.K., and M.V. All authors read and approved the final manuscript.
Funding
This work was supported by the project no. LQ1605 from the National Program of Sustainability II (MEYS CR), the Institutional Support of Excellence 2. LF UK (Grant no. 699012) and by the Grant Agency of Charles University (Project no. 176317). The study was also supported in part by the grants from the Ministry of Education, Youth and Sports of the Czech Republic (project ERDF no. CZ.02.1.01/0.0/0.0/18_069/0010054) and of the Ministry of Health of the Czech Republic (RVO—FN HK 00179906). Also supported by the Long-term development plan UHK. Supported by MH CZ—DRO (UHHK, 00179906)- by the grant projects of the Ministry of Health of the Czech Republic (FN HK 00179906) and of the Charles University in Prague, Czech Republic (PROGRES Q40). Supported by the project: PERSONMED—Center for the Development of Personalized Medicine in Age-Related Diseases, Reg. Nr. CZ.02.1.01/0.0/0.0/17_048/0007441, co-financed by ERDF and state budget of the Czech Republic. The sponsors had no role in the design, execution, interpretation, or writing of the study.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Graff-Radford, J. Vascular Cognitive Impairment. Contin. Lifelong Learn. Neurol. 2019, 25, 147–164. [Google Scholar] [CrossRef]
- Tiel, C.; Sudo, F.K.; Alves, G.S.; Ericeira-Valente, L.; Moreira, D.M.; Laks, J.; Engelhardt, E. Neuropsychiatric symptoms in Vascular Cognitive Impairment: A systematic review. Dement. Neuropsychol. 2015, 9, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Van der Flier, W.M.; Skoog, I.; Schneider, J.A.; Pantoni, L.; Mok, V.; Chen, C.L.H.; Scheltens, P. Vascular cognitive impairment. Nat. Rev. Dis. Prim. 2018, 4, 18003. [Google Scholar] [CrossRef] [PubMed]
- Wiederkehr, S.; Simard, M.; Fortin, C.; van Reekum, R. Comparability of the Clinical Diagnostic Criteria for Vascular Dementia: A Critical Review. Part I. J. Neuropsychiatr. 2008, 20, 150–161. [Google Scholar] [CrossRef] [PubMed]
- McKhann, G.M.; Knopman, D.S.; Chertkow, H.; Hyman, B.T.; Jack, C.R.; Kawas, C.H.; Klunk, W.E.; Koroshetz, W.J.; Manly, J.J.; Mayeux, R.; et al. The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 263–269. [Google Scholar] [CrossRef]
- Jack, C.R.; Albert, M.S.; Knopman, D.S.; McKhann, G.M.; Sperling, R.A.; Carrillo, M.C.; Thies, B.; Phelps, C.H. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement. 2011, 7, 257–262. [Google Scholar] [CrossRef]
- Sorbi, S.; Hort, J.; Erkinjuntti, T.; Fladby, T.; Gainotti, G.; Gurvit, H.; Nacmias, B.; Pasquier, F.; Popescu, B.O.; Rektorova, I.; et al. EFNS-ENS Guidelines on the diagnosis and management of disorders associated with dementia. Eur. J. Neurol. 2012, 19, 1159–1179. [Google Scholar] [CrossRef]
- Tomek, A.; Urbanová, B.; Hort, J. Utility of transcranial ultrasound in predicting Alzheimer’s disease risk. J. Alzheimer’s Dis. 2014, 42, S365–S374. [Google Scholar] [CrossRef] [PubMed]
- Urbanova, B.; Tomek, A.; Mikulik, R.; Magerova, H.; Horinek, D.; Hort, J. Neurosonological examination: A non-invasive approach for the detection of cerebrovascular impairment in AD. Front. Behav. Neurosci. 2014, 8, 4. [Google Scholar] [CrossRef][Green Version]
- Hort, J.; Glosova, L.; Vyhnalek, M.; Bojar, M.; Skoda, D.; Hladikova, M. The liquor tau protein and beta amylold in Alzheimer’s disease. Ces. Slov. Neurol. Neurochir. 2007, 70, 30–36. [Google Scholar]
- Barbay, M.; Taillia, H.; Nedelec-Ciceri, C.; Arnoux, A.; Puy, L.; Wiener, E.; Canaple, S.; Lamy, C.; Godefroy, O.; Roussel, M. Vascular cognitive impairment: Advances and trends. Rev. Neurol. 2017, 173, 473–480. [Google Scholar] [CrossRef]
- Heiss, W.-D.; Rosenberg, G.A.; Thiel, A.; Berlot, R.; de Reuck, J. Neuroimaging in vascular cognitive impairment: A state-of-the-art review. BMC Med. 2016, 14, 174. [Google Scholar] [CrossRef]
- Skrobot, O.A.; Love, S.; Kehoe, P.G.; O’Brien, J.; Black, S.; Chen, C.; DeCarli, C.; Erkinjuntti, T.; Ford, G.A.; Kalaria, R.N.; et al. The Vascular Impairment of Cognition Classification Consensus Study. Alzheimer’s Dement. 2017, 13, 624–633. [Google Scholar] [CrossRef]
- Wang, T.; Xiao, S.; Chen, K.; Yang, C.; Dong, S.; Cheng, Y.; Li, X.; Wang, J.; Zhu, M.; Yang, F.; et al. Prevalence, incidence, risk and protective factors of amnestic mild cognitive impairment in the elderly in Shanghai. Curr. Alzheimer Res. 2017, 14, 460–466. [Google Scholar]
- Corrao, S.; Lo Coco, D.; Lopez, G. Cognitive impairment and stroke in elderly patients. Vasc. Health Risk Manag. 2016, 12, 105. [Google Scholar] [CrossRef] [PubMed]
- Sachdev, P.S.; Lipnicki, D.M.; Crawford, J.D.; Brodaty, H. The VASCOG criteria for vascular cognitive disorders: A validation study. Eur. J. Neurol. 2019. [Google Scholar] [CrossRef] [PubMed]
- APA American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed.; APA American Psychiatric Association: Washington, DC, USA, 2013; ISBN 9780890425541. [Google Scholar]
- Bick, K.L.; Pajeau, A.K.; Kokmen, E.; Gorelick, P.B.; Yamaguchi, T.; Scheinberg, P.; Bermejo, F.; DeCarli, C.; Fisher, M.; Masdeu, J.C.; et al. Vascular dementia: Diagnostic criteria for research studies: Report of the NINDS-AIREN International Workshop. Neurology 2012, 43, 250–260. [Google Scholar]
- Urbanova, B.S.; Schwabova, J.P.; Magerova, H.; Jansky, P.; Markova, H.; Vyhnalek, M.; Laczo, J.; Hort, J.; Tomek, A. Reduced Cerebrovascular Reserve Capacity as a Biomarker of Microangiopathy in Alzheimer’s Disease and Mild Cognitive Impairment. J. Alzheimer’s Dis. 2018, 63, 465–477. [Google Scholar] [CrossRef]
- Ressner, P. Recommendations for the diagnosis and management of Alzheimer’s disease and other disorders associated with dementia: EFNS guideline. Eur. J. Neurol. 2008, 71, 494–501. [Google Scholar]
- Inzitari, D.; Simoni, M.; Pracucci, G.; Poggesi, A.; Basile, A.M.; Chabriat, H.; Erkinjuntti, T.; Fazekas, F.; Ferro, J.M.; Hennerici, M.; et al. Risk of rapid global functional decline in elderly patients with severe cerebral age-related white matter changes: The LADIS study. Arch. Intern. Med. 2007, 167, 81–88. [Google Scholar] [CrossRef]
- Dichgans, M.; Leys, D. Vascular Cognitive Impairment. Circ. Res. 2017, 120, 573–591. [Google Scholar] [CrossRef]
- Schaapsmeerders, P.; Tuladhar, A.M.; Arntz, R.M.; Franssen, S.; Maaijwee, N.A.M.; Rutten-Jacobs, L.C.A.; Schoonderwaldt, H.C.; Dorresteijn, L.D.A.; van Dijk, E.J.; Kessels, R.P.C.; et al. Remote Lower White Matter Integrity Increases the Risk of Long-Term Cognitive Impairment After Ischemic Stroke in Young Adults. Stroke 2016, 47, 2517–2525. [Google Scholar] [CrossRef]
- Mijajlović, M.D.; Pavlović, A.; Brainin, M.; Heiss, W.-D.; Quinn, T.J.; Ihle-Hansen, H.B.; Hermann, D.M.; Assayag, E.B.; Richard, E.; Thiel, A.; et al. Post-stroke dementia—a comprehensive review. BMC Med. 2017, 15, 11. [Google Scholar] [CrossRef]
- Mandzia, J.L.; Smith, E.E.; Horton, M.; Hanly, P.; Barber, P.A.; Godzwon, C.; Donaldson, E.; Asdaghi, N.; Patel, S.; Coutts, S.B. Imaging and Baseline Predictors of Cognitive Performance in Minor Ischemic Stroke and Patients With Transient Ischemic Attack at 90 Days. Stroke 2016, 47, 726–731. [Google Scholar] [CrossRef]
- Choi, B.-R.; Kim, D.-H.; Back, D.B.; Kang, C.H.; Moon, W.-J.; Han, J.-S.; Choi, D.-H.; Kwon, K.J.; Shin, C.Y.; Kim, B.-R.; et al. Characterization of White Matter Injury in a Rat Model of Chronic Cerebral Hypoperfusion. Stroke 2016, 47, 542–547. [Google Scholar] [CrossRef]
- Moulin, S.; Labreuche, J.; Bombois, S.; Rossi, C.; Boulouis, G.; Hénon, H.; Duhamel, A.; Leys, D.; Cordonnier, C. Dementia risk after spontaneous intracerebral haemorrhage: A prospective cohort study. Lancet Neurol. 2016, 15, 820–829. [Google Scholar] [CrossRef]
- You, S.; Wang, X.; Lindley, R.I.; Robinson, T.; Anderson, C.S.; Cao, Y.; Chalmers, J. Early Cognitive Impairment after Intracerebral Hemorrhage in the INTERACT1 Study. Cerebrovasc. Dis. 2017, 44, 320–324. [Google Scholar] [CrossRef]
- Valenti, R.; Charidimou, A.; Xiong, L.; Boulouis, G.; Fotiadis, P.; Ayres, A.; Riley, G.; Kuijf, H.J.; Reijmer, Y.D.; Pantoni, L.; et al. Visuospatial Functioning in Cerebral Amyloid Angiopathy: A Pilot Study. J. Alzheimer’s Dis. 2017, 56, 1223–1227. [Google Scholar] [CrossRef]
- Banerjee, G.; Wilson, D.; Ambler, G.; Appiah, K.O.B.; Shakeshaft, C.; Lunawat, S.; Cohen, H.; Yousry, T.; Habil, M.; Lip, G.Y.H.; et al. Cognitive impairment before intracerebral hemorrhage is associated with cerebral amyloid angiopathy. Stroke 2018, 49, 40–45. [Google Scholar] [CrossRef]
- Wong, G.K.C.; Lam, S.W.; Wong, A.; Ngai, K.; Mok, V.; Poon, W.S. Early Cognitive Domain Deficits in Patients with Aneurysmal Subarachnoid Hemorrhage Correlate with Functional Status. In Acta Neurochirurgica Supplement; Springer: Cham, Switzerland, 2016; pp. 129–132. [Google Scholar]
- Da Costa, L.; Dunkley, B.T.; Bethune, A.; Robertson, A.; Keller, A.; Pang, E.W. Increased Frontal Lobe Activation After Aneurysmal Subarachnoid Hemorrhage. Stroke 2016, 47, 2503–2510. [Google Scholar] [CrossRef][Green Version]
- Su, J.; E, T.; Guo, Q.; Lei, Y.; Gu, Y. Memory Deficits After Aneurysmal Subarachnoid Hemorrhage: A Functional Magnetic Resonance Imaging Study. World Neurosurg. 2018, 111, e500–e506. [Google Scholar] [CrossRef]
- Sahyouni, R.; Goshtasbi, K.; Mahmoodi, A.; Tran, D.K.; Chen, J.W. Chronic Subdural Hematoma: A Perspective on Subdural Membranes and Dementia. World Neurosurg. 2017, 108, 954–958. [Google Scholar] [CrossRef]
- Tucsek, Z.; Noa Valcarcel-Ares, M.; Tarantini, S.; Yabluchanskiy, A.; Fülöp, G.; Gautam, T.; Orock, A.; Csiszar, A.; Deak, F.; Ungvari, Z. Hypertension-induced synapse loss and impairment in synaptic plasticity in the mouse hippocampus mimics the aging phenotype: Implications for the pathogenesis of vascular cognitive impairment. GeroScience 2017, 39, 385–406. [Google Scholar] [CrossRef]
- Dichgans, M.; Zietemann, V. Prevention of Vascular Cognitive Impairment. Stroke 2012, 43, 3137–3146. [Google Scholar] [CrossRef]
- Scarmeas, N.; Stern, Y.; Mayeux, R.; Manly, J.J.; Schupf, N.; Luchsinger, J.A. Mediterranean Diet and Mild Cognitive Impairment. Arch. Neurol. 2009, 66, 216–225. [Google Scholar] [CrossRef]
- Lautenschlager, N.T.; Cox, K.L.; Flicker, L.; Foster, J.K.; van Bockxmeer, F.M.; Xiao, J.; Greenop, K.R.; Almeida, O.P. Effect of Physical Activity on Cognitive Function in Older Adults at Risk for Alzheimer Disease. JAMA 2008, 300, 1027. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Allan, S.M.; Boltze, J.; Cunningham, C.; Farris, C.; Head, E.; Ihara, M.; Isaacs, J.D.; Kalaria, R.N.; Lesnik Oberstein, S.A.M.J.; et al. Translational models for vascular cognitive impairment: A review including larger species. BMC Med. 2017, 15, 16. [Google Scholar] [CrossRef]
- Skrobot, O.A.; Attems, J.; Esiri, M.; Hortobágyi, T.; Ironside, J.W.; Kalaria, R.N.; King, A.; Lammie, G.A.; Mann, D.; Neal, J.; et al. Vascular cognitive impairment neuropathology guidelines (VCING): The contribution of cerebrovascular pathology to cognitive impairment. Brain 2016, 139, 2957–2969. [Google Scholar] [CrossRef]
- Hietamies, T.M.; Ostrowski, C.; Pei, Z.; Feng, L.; McCabe, C.; Work, L.M.; Quinn, T.J. Variability of functional outcome measures used in animal models of stroke and vascular cognitive impairment—A review of contemporary studies. J. Cereb. Blood Flow Metab. 2018, 38, 1872–1884. [Google Scholar] [CrossRef]
- Kleinschnitz, C.; Fluri, F.; Schuhmann, M. Animal models of ischemic stroke and their application in clinical research. Drug Des. Devel. Ther. 2015, 9, 3445. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Kimura-Ohba, S.; Thompson, J.; Rosenberg, G.A. Rodent Models of Vascular Cognitive Impairment. Transl. Stroke Res. 2016, 7, 407–414. [Google Scholar] [CrossRef]
- El Amki, M.; Clavier, T.; Perzo, N.; Bernard, R.; Guichet, P.-O.; Castel, H. Hypothalamic, thalamic and hippocampal lesions in the mouse MCAO model: Potential involvement of deep cerebral arteries? J. Neurosci. Methods 2015, 254, 80–85. [Google Scholar] [CrossRef]
- Duncombe, J.; Kitamura, A.; Hase, Y.; Ihara, M.; Kalaria, R.N.; Horsburgh, K. Chronic cerebral hypoperfusion: A key mechanism leading to vascular cognitive impairment and dementia. Closing the translational gap between rodent models and human vascular cognitive impairment and dementia. Clin. Sci. 2017, 131, 2451–2468. [Google Scholar] [CrossRef]
- Klebe, D.; Iniaghe, L.; Burchell, S.; Reis, C.; Akyol, O.; Tang, J.; Zhang, J.H. Intracerebral Hemorrhage in Mice. Methods Mol. Biol. 2018, 83–91. [Google Scholar]
- Ma, Q.; Khatibi, N.H.; Chen, H.; Tang, J.; Zhang, J.H. History of Preclinical Models of Intracerebral Hemorrhage. In Intracerebral Hemorrhage Research; Springer: Vienna, Austria, 2011; pp. 3–8. [Google Scholar]
- Nishimura, N.; Schaffer, C.B.; Friedman, B.; Tsai, P.S.; Lyden, P.D.; Kleinfeld, D. Targeted insult to subsurface cortical blood vessels using ultrashort laser pulses: Three models of stroke. Nat. Methods 2006, 3, 99–108. [Google Scholar] [CrossRef]
- Jiwa, N.S.; Garrard, P.; Hainsworth, A.H. Experimental models of vascular dementia and vascular cognitive impairment: A systematic review. J. Neurochem. 2010, 115, 814–828. [Google Scholar] [CrossRef]
- Sugawara, T.; Ayer, R.; Jadhav, V.; Zhang, J.H. A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J. Neurosci. Methods 2008, 167, 327–334. [Google Scholar] [CrossRef] [PubMed]
- Muiño, E.; Gallego-Fabrega, C.; Cullell, N.; Carrera, C.; Torres, N.; Krupinski, J.; Roquer, J.; Montaner, J.; Fernández-Cadenas, I. Systematic Review of Cysteine-Sparing NOTCH3 Missense Mutations in Patients with Clinical Suspicion of CADASIL. Int. J. Mol. Sci. 2017, 18, 1964. [Google Scholar] [CrossRef] [PubMed]
- Joutel, A. Pathogenesis of CADASIL. BioEssays 2010, 33, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Grochowski, C.; Litak, J.; Kamieniak, P.; Maciejewski, R. Oxidative stress in cerebral small vessel disease. Role of reactive species. Free Radic. Res. 2017, 52, 1–13. [Google Scholar] [CrossRef]
- Skoumalová, A.; Hort, J. Blood markers of oxidative stress in Alzheimer’s disease. J. Cell. Mol. Med. 2012. [Google Scholar] [CrossRef]
- Liu, H.; Zhang, J. Cerebral Hypoperfusion and Cognitive Impairment: The Pathogenic Role of Vascular Oxidative Stress. Int. J. Neurosci. 2012, 122, 494–499. [Google Scholar] [CrossRef]
- Bennett, S.; Grant, M.M.; Aldred, S. Oxidative Stress in Vascular Dementia and Alzheimer’s Disease: A Common Pathology. J. Alzheimer’s Dis. 2008, 17, 245–257. [Google Scholar] [CrossRef]
- Gackowski, D.; Rozalski, R.; Siomek, A.; Dziaman, T.; Nicpon, K.; Klimarczyk, M.; Araszkiewicz, A.; Olinski, R. Oxidative stress and oxidative DNA damage is characteristic for mixed Alzheimer disease/vascular dementia. J. Neurol. Sci. 2008, 266, 57–62. [Google Scholar] [CrossRef]
- Liu, D.; Yuan, X.; Chu, S.; Chen, C.; Ren, Q.; Luo, P.; Lin, M.; Wang, S.; Zhu, T.; Ai, Q.; et al. CZ-7, a new derivative of Claulansine F, ameliorates 2VO-induced vascular dementia in rats through a Nrf2-mediated antioxidant responses. Acta Pharmacol. Sin. 2018, 40, 425. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Z. Gastrodin improves cognitive dysfunction and decreases oxidative stress in vascular dementia rats induced by chronic ischemia. Int. J. Clin. Exp. Pathol. 2015, 8, 14099. [Google Scholar]
- Csányi, G.; Miller, F.J., Jr. Oxidative Stress in Cardiovascular Disease. Int. J. Mol. Sci. 2014, 15, 6002–6008. [Google Scholar] [CrossRef]
- White, A.; Culmsee, C.; Beart, P. Oxidative stress and neurodegeneration. Neurochem. Int. 2013, 62, 521. [Google Scholar] [CrossRef] [PubMed]
- Tönnies, E.; Trushina, E. Oxidative Stress, Synaptic Dysfunction, and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 57, 1105–1121. [Google Scholar] [CrossRef]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov. 2011, 10, 453–471. [Google Scholar] [CrossRef]
- Choi, D.-H.; Lee, K.-H.; Kim, J.-H.; Seo, J.-H.; Kim, H.Y.; Shin, C.Y.; Han, J.-S.; Han, S.-H.; Kim, Y.-S.; Lee, J. NADPH Oxidase 1, a Novel Molecular Source of ROS in Hippocampal Neuronal Death in Vascular Dementia. Antioxid. Redox Signal. 2014, 21, 533–550. [Google Scholar] [CrossRef]
- Kim, H.A.; Miller, A.A.; Drummond, G.R.; Thrift, A.G.; Arumugam, T.V.; Phan, T.G.; Srikanth, V.K.; Sobey, C.G. Vascular cognitive impairment and Alzheimer’s disease: Role of cerebral hypoperfusion and oxidative stress. Naunyn. Schmiedebergs. Arch. Pharmacol. 2012, 385, 953–959. [Google Scholar] [CrossRef]
- Casado, Á.; Encarnación López-Fernández, M.; Concepción Casado, M.; de La Torre, R. Lipid Peroxidation and Antioxidant Enzyme Activities in Vascular and Alzheimer Dementias. Neurochem. Res. 2007, 33, 450–458. [Google Scholar] [CrossRef]
- Famulari, A.L.; Marschoff, E.R.; Llesuy, S.F.; Kohan, S.; Serra, J.A.; Dominguez, R.O.; Repetto, M.; Reides, C.; de Lustig, E.S. The antioxidant enzymatic blood profile in Alzheimer’s and vascular diseases. Their association and a possible assay to differentiate demented subjects and controls. J. Neurol. Sci. 1996, 141, 69–78. [Google Scholar] [CrossRef]
- Shi, G.-X.; Liu, C.-Z.; Wang, L.-P.; Guan, L.-P.; Li, S.-Q. Biomarkers of Oxidative Stress in Vascular Dementia Patients. Can. J. Neurol. Sci. 2012, 39, 65–68. [Google Scholar] [CrossRef]
- Wang, M.; Norman, J.E.; Srinivasan, V.J.; Rutledge, J.C. Metabolic, inflammatory, and microvascular determinants of white matter disease and cognitive decline. Am. J. Neurodegener. Dis. 2016, 5, 171. [Google Scholar]
- Sood, R.; Yang, Y.; Taheri, S.; Candelario-Jalil, E.; Estrada, E.Y.; Walker, E.J.; Thompson, J.; Rosenberg, G.A. Increased Apparent Diffusion Coefficients on MRI Linked with Matrix Metalloproteinases and Edema in White Matter after Bilateral Carotid Artery Occlusion in Rats. J. Cereb. Blood Flow Metab. 2008, 29, 308–316. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, X.-C.; Ren, H.-M.; Bao, W.-M. Effects of ischemic preconditioning on blood–brain barrier permeability and MMP-9 expression of ischemic brain. Neurol. Res. 2006, 28, 21–24. [Google Scholar] [CrossRef]
- Jiang, P.; Chen, L.; Sun, J.; Li, J.; Xu, J.; Liu, W.; Feng, F.; Qu, W. Chotosan ameliorates cognitive impairment and hippocampus neuronal loss in experimental vascular dementia via activating the Nrf2-mediated antioxidant pathway. J. Pharmacol. Sci. 2019, 139, 105–111. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, B.; Lu, K.; Deng, J.; Zhao, F.; Zhao, B.; Zhao, Y. Prevention of Hippocampal Neuronal Damage and Cognitive Function Deficits in Vascular Dementia by Dextromethorphan. Mol. Neurobiol. 2016, 53, 3494–3502. [Google Scholar] [CrossRef]
- Origlia, N.; Criscuolo, C.; Arancio, O.; Yan, S.S.; Domenici, L. RAGE Inhibition in Microglia Prevents Ischemia-Dependent Synaptic Dysfunction in an Amyloid-Enriched Environment. J. Neurosci. 2014, 34, 8749–8760. [Google Scholar] [CrossRef]
- Lue, L.-F.; Walker, D.G.; Brachova, L.; Beach, T.G.; Rogers, J.; Schmidt, A.M.; Stern, D.M.; Yan, S.D. Involvement of Microglial Receptor for Advanced Glycation Endproducts (RAGE) in Alzheimer’s Disease: Identification of a Cellular Activation Mechanism. Exp. Neurol. 2001, 171, 29–45. [Google Scholar] [CrossRef]
- Wei, J.; Sun, C.; Liu, C.; Zhang, Q. Effects of Rat Anti-mouse Interleukin-6 Receptor Antibody on the Recovery of Cognitive Function in Stroke Mice. Cell. Mol. Neurobiol. 2017, 38, 507–515. [Google Scholar] [CrossRef]
- Wytrykowska, A.; Prosba-Mackiewicz, M.; Nyka, W.M. IL-1β, TNF-α, and IL-6 levels in gingival fluid and serum of patients with ischemic stroke. J. Oral Sci. 2016, 58, 509–513. [Google Scholar] [CrossRef]
- Miwa, K.; Okazaki, S.; Sakaguchi, M.; Mochizuki, H.; Kitagawa, K. Interleukin-6, interleukin-6 receptor gene variant, small-vessel disease and incident dementia. Eur. J. Neurol. 2016, 23, 656–663. [Google Scholar] [CrossRef]
- Dukic, L.; Simundic, A.-M.; Martinic-Popovic, I.; Kackov, S.; Diamandis, A.; Begcevic, I.; Diamandis, E.P. The role of human kallikrein 6, clusterin and adiponectin as potential blood biomarkers of dementia. Clin. Biochem. 2016, 49, 213–218. [Google Scholar] [CrossRef]
- Price, B.R.; Norris, C.M.; Sompol, P.; Wilcock, D.M. An emerging role of astrocytes in vascular contributions to cognitive impairment and dementia. J. Neurochem. 2018, 144, 644–650. [Google Scholar] [CrossRef]
- Stanimirovic, D.B.; Friedman, A. Pathophysiology of the neurovascular unit: Disease cause or consequence? Blood Flow Metab. 2012, 32, 1207–1221. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, D.M.; Vitek, M.P.; Colton, C.A. Vascular amyloid alters astrocytic water and potassium channels in mouse models and humans with Alzheimer’s disease. Neuroscience 2009, 159, 1055–1069. [Google Scholar] [CrossRef]
- Sudduth, T.L.; Weekman, E.M.; Price, B.R.; Gooch, J.L.; Woolums, A.; Norris, C.M.; Wilcock, D.M. Time-course of glial changes in the hyperhomocysteinemia model of vascular cognitive impairment and dementia (VCID). Neuroscience 2017, 341, 42–51. [Google Scholar] [CrossRef] [PubMed]
- Weekman, E.M.; Wilcock, D.M. Matrix Metalloproteinase in Blood-Brain Barrier Breakdown in Dementia. J. Alzheimer’s Dis. 2015, 49, 893–903. [Google Scholar] [CrossRef]
- Tarantini, S.; Tran, C.H.T.; Gordon, G.R.; Ungvari, Z.; Csiszar, A. Impaired neurovascular coupling in aging and Alzheimer’s disease: Contribution of astrocyte dysfunction and endothelial impairment to cognitive decline. Exp. Gerontol. 2017, 94, 52–58. [Google Scholar] [CrossRef]
- Kraner, S.D.; Norris, C.M. Astrocyte Activation and the Calcineurin/in Cerebrovascular Disease. Front. Aging Neurosci. 2018, 10. [Google Scholar] [CrossRef]
- Rouach, N.; Dallérac, G. Astrocytes as new targets to improve cognitive functions. Prog. Neurobiol. 2016, 144, 48–67. [Google Scholar]
- Santello, M.; Toni, N.; Volterra, A. Astrocyte function from information processing to cognition and cognitive impairment. Nat. Neurosci. 2019, 1. [Google Scholar] [CrossRef] [PubMed]
- Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell to Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Cao, Y.; Ma, L.; Pei, H.; Rausch, W.D.; Li, H. Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Front. Aging Neurosci. 2018, 10, 367. [Google Scholar] [CrossRef]
- Stephan, B.C.M.; Harrison, S.L.; Keage, H.A.D.; Babateen, A.; Robinson, L.; Siervo, M. Cardiovascular Disease, the Nitric Oxide Pathway and Risk of Cognitive Impairment and Dementia. Curr. Cardiol. Rep. 2017, 19, 87. [Google Scholar] [CrossRef]
- Džoljić, E. Why is nitric oxide important for our brain? Funct. Neurol. 2015, 30, 159. [Google Scholar] [CrossRef]
- Ren, C.; Li, N.; Li, S.; Han, R.; Huang, Q.; Hu, J.; Jin, K.; Ji, X. Limb Ischemic Conditioning Improved Cognitive Deficits via eNOS-Dependent Augmentation of Angiogenesis after Chronic Cerebral Hypoperfusion in Rats. Aging Dis. 2018, 9, 869. [Google Scholar] [CrossRef]
- Zhang, L.; Zheng, H.; Luo, J.; Li, L.; Pan, X.; Jiang, T.; Xiao, C.; Pei, Z.; Hu, X. Inhibition of endothelial nitric oxide synthase reverses the effect of exercise on improving cognitive function in hypertensive rats. Hypertens Res. 2018, 41, 414–425. [Google Scholar] [CrossRef]
- Manukhina, E.B.; Pshennikova, M.G.; Goryacheva, A.V.; Khomenko, I.P.; Mashina, S.Y.; Pokidyshev, D.A.; Malyshev, I.Y. Role of nitric oxide in prevention of cognitive disorders in neurodegenerative brain injuries in rats. Bull. Exp. Biol. Med. 2008, 146, 391–395. [Google Scholar] [CrossRef]
- Zhao, X.; Liu, J.; Yang, S.; Song, D.; Wang, C.; Chen, C.; Li, X.; Wang, Q.; Ge, S.; Yang, R.; et al. Ling-Yang-Gou-Teng-decoction prevents vascular dementia through inhibiting oxidative stress induced neurovascular coupling dysfunction. J. Ethnopharmacol. 2018, 222, 229–238. [Google Scholar] [CrossRef]
- Ledo, A.; Barbosa, R.M.; Laranjinha, J.; Lourenço, C.F. Neurovascular-neuroenergetic coupling axis in the brain: Master regulation by nitric oxide and consequences in aging and neurodegeneration. Free Radic. Biol. Med. 2017, 108, 668–682. [Google Scholar]
- Balez, R.; Ooi, L. Getting to NO Alzheimer’s disease: Neuroprotection versus neurotoxicity mediated by nitric oxide. Oxid. Med. Cell Longev. 2016. [Google Scholar] [CrossRef]
- Cai, W.; Liu, H.; Zhao, J.; Chen, L.Y.; Chen, J.; Lu, Z.; Hu, X. Pericytes in Brain Injury and Repair After Ischemic Stroke. Transl. Stroke Res. 2017, 8, 107–121. [Google Scholar] [CrossRef]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res. 2005, 97, 512–523. [Google Scholar] [CrossRef]
- Bhowmick, S.; D’Mello, V.; Caruso, D.; Wallerstein, A.; Abdul-Muneer, P.M. Impairment of pericyte-endothelium crosstalk leads to blood-brain barrier dysfunction following traumatic brain injury. Exp. Neurol. 2019, 317, 260–270. [Google Scholar] [CrossRef]
- Takahashi, Y.; Maki, T.; Liang, A.C.; Itoh, K.; Lok, J.; Osumi, N.; Arai, K. p38 MAP kinase mediates transforming-growth factor-β1-induced upregulation of matrix metalloproteinase-9 but not -2 in human brain pericytes. Brain Res. 2014, 1593, 1–8. [Google Scholar] [CrossRef]
- Hill, J.; Rom, S.; Ramirez, S.H.; Persidsky, Y. Emerging Roles of Pericytes in the Regulation of the Neurovascular Unit in Health and Disease. J. Neuroimmune Pharmacol. 2014, 9, 591–605. [Google Scholar] [CrossRef]
- Winkler, E.A.; Sagare, A.P.; Zlokovic, B.V. The pericyte: A forgotten cell type with important implications for alzheimer’s disease? Brain Pathol. 2014, 24, 371–386. [Google Scholar] [CrossRef]
- Sengillo, J.D.; Winkler, E.A.; Walker, C.T.; Sullivan, J.S.; Johnson, M.; Zlokovic, B.V. Deficiency in mural vascular cells coincides with blood-brain barrier disruption in alzheimer’s disease. Brain Pathol. 2013, 24, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Nikolakopoulou, A.M.; Zhao, Z.; Montagne, A.; Zlokovic, B.V. Regional early and progressive loss of brain pericytes but not vascular smooth muscle cells in adult mice with disrupted platelet-derived growth factor receptor-β signaling. PLoS ONE 2017, 12, e0176225. [Google Scholar] [CrossRef] [PubMed]
- Beltramo, E.; Porta, M. Pericyte Loss in Diabetic Retinopathy: Mechanisms and Consequences. Curr. Med. Chem. 2013, 20, 3218–3225. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Rege, S.V.; Ramanathan, A.; Wang, Y.; Ahuja, A.; Lazic, D.; Tsai, P.S.; Zhao, Z.; Zhou, Y.; et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 2017, 20, 406. [Google Scholar] [CrossRef]
- Montagne, A.; Nikolakopoulou, A.M.; Zhao, Z.; Sagare, A.P.; Si, G.; Lazic, D.; Barnes, S.R.; Daianu, M.; Ramanathan, A.; Go, A.; et al. Pericyte degeneration causes white matter dysfunction in the mouse central nervous system. Nat. Med. 2018, 24, 326. [Google Scholar] [CrossRef]
- Winkler, E.A.; Bell, R.D.; Zlokovic, B.V. Pericyte-specific expression of PDGF beta receptor in mouse models with normal and deficient PDGF beta receptor signaling. Mol. Neurodegener. 2010, 5, 32. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Deane, R.; Sagare, A.P.; Bell, R.D.; Winkler, E.A. Low-density lipoprotein receptor-related protein-1: A serial clearance homeostatic mechanism controlling Alzheimer’s amyloid β-peptide elimination from the brain. J. Neurochem. 2010, 115, 1077–1089. [Google Scholar] [CrossRef]
- Sagare, A.P.; Bell, R.D.; Zhao, Z.; Ma, Q.; Winkler, E.A.; Ramanathan, A.; Zlokovic, B.V. Pericyte loss influences Alzheimer-like neurodegeneration in mice. Nat. Commun. 2013, 4, 2932. [Google Scholar] [CrossRef]
- Nakazaki, M.; Sasaki, M.; Kataoka-Sasaki, Y.; Oka, S.; Suzuki, J.; Sasaki, Y.; Nagahama, H.; Hashi, K.; Kocsis, J.D.; Honmou, O. Intravenous infusion of mesenchymal stem cells improves impaired cognitive function in a cerebral small vessel disease model. Neuroscience 2019, 408, 361–377. [Google Scholar] [CrossRef]
- Uchiyama, Y.; Shibata, M.; Koike, M.; Yoshimura, K.; Sasaki, M. Autophagy-physiology and pathophysiology. Histochem. Cell Biol. 2008, 129, 407–420. [Google Scholar] [CrossRef]
- Mariño, G.; Madeo, F.; Kroemer, G. Autophagy for tissue homeostasis and neuroprotection. Curr. Opin. Cell Biol. 2011, 23, 198–206. [Google Scholar] [CrossRef]
- Kiffin, R.; Bandyopadhyay, U.; Cuervo, A.M. Oxidative stress and autophagy. Antioxid Redox Signal. 2006, 8, 152–162. [Google Scholar] [CrossRef]
- Thornton, C.; Leaw, B.; Mallard, C.; Nair, S.; Jinnai, M.; Hagberg, H. Cell Death in the Developing Brain after Hypoxia-Ischemia. Front. Cell. Neurosci. 2017, 11, 248. [Google Scholar] [CrossRef]
- Alers, S.; Loffler, A.S.; Wesselborg, S.; Stork, B. Role of AMPK-mTOR-Ulk1/2 in the Regulation of Autophagy: Cross Talk, Shortcuts, and Feedbacks. Mol. Cell. Biol. 2011, 32, 2–11. [Google Scholar] [CrossRef]
- Liu, B.; Tang, J.; Li, S.; Zhang, J.; Yuan, M.; Wang, R. Autophagy activation aggravates neuronal injury in the hippocampus of vascular dementia rats. Neural Regen Res. 2014, 9, 1288. [Google Scholar] [CrossRef]
- Zou, W.; Song, Y.; Li, Y.; Du, Y.; Zhang, X.; Fu, J. The Role of Autophagy in the Correlation Between Neuron Damage and Cognitive Impairment in Rat Chronic Cerebral Hypoperfusion. Mol. Neurobiol. 2017, 55, 776–791. [Google Scholar] [CrossRef]
- Wang, D.P.; Yin, H.; Kang, K.; Lin, Q.; Su, S.H.; Hai, J. The potential protective effects of cannabinoid receptor agonist WIN55, on cognitive dysfunction is associated with the suppression of autophagy and inflammation in an experimental model of vascular dementia. Psychiatry Res. 2018, 267, 281–288. [Google Scholar] [CrossRef]
- Wang, D.; Lin, Q.; Su, S.; Liu, K.; Wu, Y.; Hai, J. URB597 improves cognitive impairment induced by chronic cerebral hypoperfusion by inhibiting mTOR-dependent autophagy. Neuroscience 2017, 344, 293–304. [Google Scholar] [CrossRef]
- Wang, L.; Wang, J.; Wang, F.; Liu, C.; Yang, X.; Yang, J.; Ming, D. VEGF-Mediated Cognitive and Synaptic Improvement in Chronic Cerebral Hypoperfusion Rats Involves Autophagy Process. Neuromol. Med. 2017, 19, 423–435. [Google Scholar] [CrossRef]
- Xia, D.; Sui, R.; Min, L.; Zhang, L.; Zhang, Z. Fastigial nucleus stimulation ameliorates cognitive impairment via modulating autophagy and inflammasomes activation in a rat model of vascular dementia. J. Cell. Biochem. 2018, 120, 5108–5117. [Google Scholar] [CrossRef]
- Ferrucci, M.; Biagioni, F.; Ryskalin, L.; Limanaqi, F.; Gambardella, S.; Frati, A.; Fornai, F. Ambiguous Effects of Autophagy Activation Following Hypoperfusion/Ischemia. Int. J. Mol. Sci. 2018, 19, 2756. [Google Scholar] [CrossRef]
- Sonntag, W.E.; Deak, F.; Ashpole, N.; Toth, P.; Csiszar, A.; Freeman, W.; Ungvari, Z. Insulin-like growth factor-1 in CNS and cerebrovascular aging. Front. Aging Neurosci. 2013, 5, 27. [Google Scholar] [CrossRef]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF-1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef]
- Bianchi, V.; Locatelli, V.; Rizzi, L. Neurotrophic and Neuroregenerative Effects of GH/IGF1. Int. J. Mol. Sci. 2017, 18, 2441. [Google Scholar] [CrossRef]
- Frater, J.; Lie, D.; Bartlett, P.; McGrath, J.J. Insulin-like Growth Factor 1 (IGF-1) as a marker of cognitive decline in normal ageing: A review. Ageing Res. Rev. 2018, 42, 14–27. [Google Scholar] [CrossRef]
- Ramsey, M.M.; Adams, M.M.; Ariwodola, O.J.; Sonntag, W.E.; Weiner, J.L. Functional characterization of des-IGF-1 action at excitatory synapses in the CA1 region of rat hippocampus. J. Neurophysiol. 2005, 94, 247–254. [Google Scholar] [CrossRef]
- Lopez-Lopez, C.; Dietrich, M.O.; Metzger, F.; Loetscher, H.; Torres-Aleman, I. Disturbed cross talk between insulin-like growth factor I and AMP-activated protein kinase as a possible cause of vascular dysfunction in the amyloid precursor protein/presenilin 2 mouse model of Alzheimer’s disease. J. Neurosci. 2007, 27, 824–831. [Google Scholar] [CrossRef]
- Trejo, J.L.; Carro, E.; Lopez-Lopez, C.; Torres-Aleman, I.; Suppl, A. Role of serum insulin-like growth factor I in mammalian brain aging. Growth Horm. 2004, 14, 39–43. [Google Scholar] [CrossRef]
- Åberg, M.A.I.; Åberg, N.D.; Hedbäcker, H.; Oscarsson, J.; Eriksson, P.S. Peripheral Infusion of IGF-I Selectively Induces Neurogenesis in the Adult Rat Hippocampus. J. Neurosci. 2000, 20, 2896–2903. [Google Scholar] [CrossRef]
- Tucsek, Z.; Toth, P.; Tarantini, S.; Sosnowska, D.; Gautam, T.; Warrington, J.P.; Giles, C.B.; Wren, J.D.; Koller, A.; Ballabh, P.; et al. Aging exacerbates obesity-induced cerebromicrovascular rarefaction, neurovascular uncoupling, and cognitive decline in mice. Sci. Med. Sci. 2014, 69, 1339–1352. [Google Scholar] [CrossRef]
- Tarantini, S.; Tucsek, Z.; Valcarcel-Ares, M.N.; Toth, P.; Gautam, T.; Giles, C.B.; Ballabh, P.; Wei, J.Y.; Wren, J.D.; Ashpole, N.M.; et al. Circulating IGF-1 deficiency exacerbates hypertension-induced microvascular rarefaction in the mouse hippocampus and retrosplenial cortex: Implications for cerebromicrovascular and brain aging. Age 2016, 38, 273–289. [Google Scholar] [CrossRef]
- Toth, P.; Tarantini, S.; Ashpole, N.M.; Tucsek, Z.; Milne, G.L.; Valcarcel-Ares, N.M.; Menyhart, A.; Farkas, E.; Sonntag, W.E.; Csiszar, A.; et al. IGF-1 deficiency impairs neurovascular coupling in mice: Implications for cerebromicrovascular aging. Aging Cell 2015, 14, 1034–1044. [Google Scholar] [CrossRef]
- Toth, P.; Tucsek, Z.; Tarantini, S.; Sosnowska, D.; Gautam, T.; Mitschelen, M.; Koller, A.; Sonntag, W.E.; Csiszar, A.; Ungvari, Z. IGF-1 deficiency impairs cerebral myogenic autoregulation in hypertensive mice. Blood Flow Metab. 2014, 34, 1887–1897. [Google Scholar] [CrossRef]
- Gong, X.; Ma, M.; Fan, X.; Li, M.; Liu, Q.; Liu, X.; Xu, G. Down-regulation of IGF-1/IGF-1R in hippocampus of rats with vascular dementia. Neurosci. Lett. 2012, 513, 20–24. [Google Scholar] [CrossRef]
- Quinlan, P.; Horvath, A.; Nordlund, A.; Wallin, A.; Svensson, J. Low serum insulin-like growth factor-I (IGF-I) level is associated with increased risk of vascular dementia. Psychoneuroendocrinology 2017, 86, 169–175. [Google Scholar] [CrossRef]
- Johnsen, S.P.; Hundborg, H.H.; Sørensen, H.T.; Ørskov, H.; Tjønneland, A.; Overvad, K.; Jørgensen, J.O.L. Insulin-Like Growth Factor (IGF) I, -II, and IGF Binding Protein-3 and Risk of Ischemic Stroke. J. Clin. Endocrinol. Metab. 2005, 90, 5937–5941. [Google Scholar] [CrossRef]
- Liu, X.F.; Fawcett, J.R.; Thorne, R.G.; Frey, W.H., II. Non-invasive intranasal insulin-like growth factor-I reduces infarct volume and improves neurologic function in rats following middle cerebral artery occlusion. Neurosci. Lett. 2001, 308, 91–94. [Google Scholar] [CrossRef]
- Zhu, W.; Fan, Y.; Frenzel, T.; Gasmi, M.; Bartus, R.T.; Young, W.L.; Yang, G.Y.; Chen, Y. Insulin growth factor-1 gene transfer enhances neurovascular remodeling and improves long-term stroke outcome in mice. Stroke 2008, 39, 1254–1261. [Google Scholar] [CrossRef]
- Ma, X.; Xu, W.; Zhang, Z.; Liu, N.; Yang, J.; Wang, M.; Wang, Y. Salvianolic Acid B Ameliorates Cognitive Deficits Through IGF-1/Akt Pathway in Rats with Vascular Dementia. Cell. Physiol. Biochem. 2017, 43, 1381–1391. [Google Scholar] [CrossRef]
- Riikonen, R. Insulin-like growth factor delivery across the blood-brain barrier. Chemotheraphy 2006, 52, 279–281. [Google Scholar] [CrossRef]
- Farooq, M.U.; Min, J.; Goshgarian, C.; Gorelick, P.B. Pharmacotherapy for Vascular Cognitive Impairment. CNS Drugs 2017, 31, 759–776. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).