Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart


Abstract
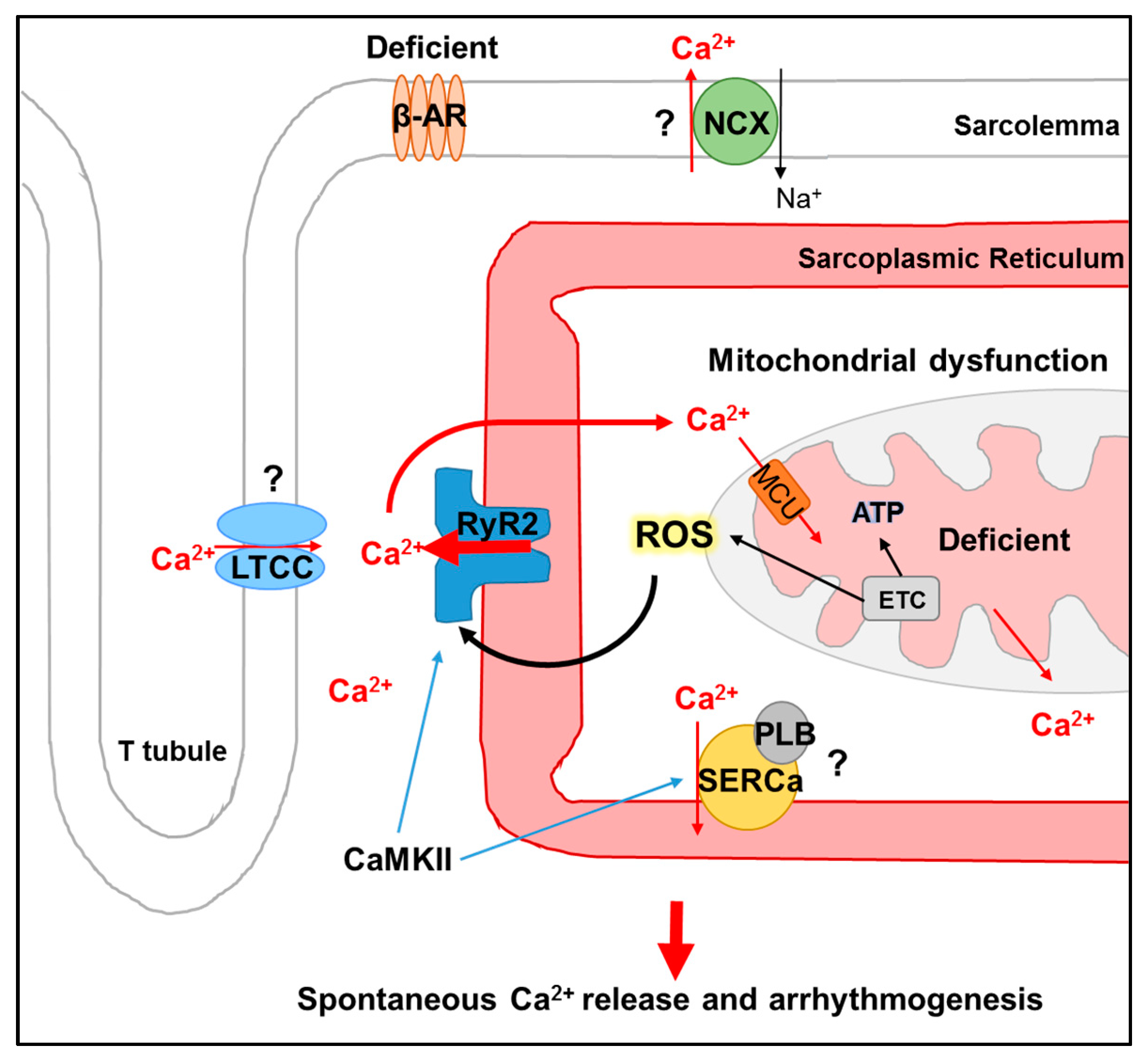
1. Introduction
2. Intracellular Ca2+ Homeostasis in the Aged Heart
2.1. Ryanodine Receptor
2.2. Sarcoplasmic Reticulum Ca2+-ATP-ase
2.3. L-Type Ca2+ Channel
2.4. Na+/Ca2+ Exchanger
2.5. Sexual Dimorphism of Intracellular Ca2+ Release
2.6. Effects of Mitochondria on Intracellular Ca2+ Release
3. Perspective
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| AF | Atrial fibrillation |
| CaM | Calmodulin |
| CaMKII | Ca2+/calmodulin-dependent protein kinase II |
| CPVT | Catecholaminergic polymorphic ventricular tachycardia |
| CSQ | Calsequestrin |
| DAD | Delayed afterdepolarization |
| EAD | Early afterdepolarization |
| LTCC | L-type Ca2+ channel |
| NCX1 | Na+/Ca2+ exchanger |
| PKA | Protein kinase A |
| PP1 | Protein phosphatase 1 |
| PP2A | Protein phosphatase 2A |
| PP2B | Protein phosphatase 2B |
| ROS | Reactive oxygen species |
| RyR2 | Ryanodine receptor, type 2 |
| SERCa2a | Sarco/endoplasmic reticulum Ca2+-ATPase, type 2a |
| VF | Ventricular fibrillation |
| VT | Ventricular tachycardia |
References
- Benjamin, E.J.; Virani, S.S.; Callaway, C.W.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Chiuve, S.E.; Cushman, M.; Delling, F.N.; Deo, R.; et al. Heart Disease and Stroke Statistics—2018 Update: A Report from the American Heart Association. Circulation 2018, 137, e67–e492. [Google Scholar] [CrossRef]
- Steenman, M.; Lande, G. Cardiac aging and heart disease in humans. Biophys. Rev. 2017, 9, 131–137. [Google Scholar] [CrossRef]
- Lakatta, E.G.; Levy, D. Arterial and cardiac aging: Major shareholders in cardiovascular disease enterprises: Part II: The aging heart in health: Links to heart disease. Circulation 2003, 107, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.P.; A Spurgeon, H.; O’Connor, F.; Lakatta, E.G. Age-associated changes in beta-adrenergic modulation on rat cardiac excitation-contraction coupling. J. Clin. Investig. 1994, 94, 2051–2059. [Google Scholar] [CrossRef] [PubMed]
- Xiao, R.P.; Tomhave, E.D.; Wang, D.J.; Ji, X.; O Boluyt, M.; Cheng, H.; Lakatta, E.G.; Koch, W.J. Age-associated reductions in cardiac beta1- and beta2-adrenergic responses without changes in inhibitory G proteins or receptor kinases. J. Clin. Investig. 1998, 101, 1273–1282. [Google Scholar] [CrossRef]
- De Lucia, C.; Eguchi, A.; Koch, W.J. New Insights in Cardiac β-Adrenergic Signaling during Heart Failure and Aging. Front. Pharmacol. 2018, 9, 904. [Google Scholar] [CrossRef] [PubMed]
- Tocchi, A.; Quarles, E.K.; Basisty, N.; Gitari, L.; Rabinovitch, P.S. Mitochondrial Dysfunction in Cardiac Ageing. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1847, 1424–1433. [Google Scholar] [CrossRef]
- Zorov, D.B. Reactive Oxygen Species (ROS)-induced ROS Release: A New Phenomenon Accompanying Induction of the Mitochondrial Permeability Transition in Cardiac Myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef]
- Cooper, L.L.; Li, W.; Lu, Y.; Centracchio, J.; Terentyeva, R.; Koren, G.; Terentyev, D. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J. Physiol. 2013, 591, 5895–5911. [Google Scholar] [CrossRef]
- Janczewski, A.M.; Lakatta, E.G. Modulation of sarcoplasmic reticulum Ca(2+) cycling in systolic and diastolic heart failure associated with aging. Heart Fail. Rev. 2010, 15, 431–445. [Google Scholar] [CrossRef]
- Feridooni, H.A.; Dibb, K.M.; Howlett, S.E. How cardiomyocyte excitation, calcium release and contraction become altered with age. J. Mol. Cell. Cardiol. 2015, 83, 62–72. [Google Scholar] [CrossRef]
- Hamilton, S.; Terentyev, D. Proarrhythmic Remodeling of Calcium Homeostasis in Cardiac Disease; Implications for Diabetes and Obesity. Front. Physiol. 2018, 9, 1517. [Google Scholar] [CrossRef] [PubMed]
- Eisner, D.A.; Caldwell, J.L.; Kistamás, K.; Trafford, A.W. Calcium and Excitation-Contraction Coupling in the Heart. Circ. Res. 2017, 121, 181–195. [Google Scholar] [CrossRef] [PubMed]
- Landstrom, A.P.; Dobrev, D.; Wehrens, X.H. Calcium Signaling and Cardiac Arrhythmias. Circ. Res. 2017, 120, 1969–1993. [Google Scholar] [CrossRef] [PubMed]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Zima, A.V.; Bovo, E.; Mazurek, S.R.; Rochira, J.A.; Li, W.; Terentyev, D. Ca handling during Excitation-Contraction Coupling in Heart Failure. Pflugers Arch. Eur. J. Physiol. 2014, 466, 1129–1137. [Google Scholar] [CrossRef]
- Fill, M.; Copello, J.A. Ryanodine Receptor Calcium Release Channels. Physiol. Rev. 2002, 82, 893–922. [Google Scholar] [CrossRef]
- Cheng, H.; Lederer, M.R.; Lederer, W.J.; Cannell, M.B. Calcium sparks and [Ca2+]i waves in cardiac myocytes. Am. J. Physiol. 1996, 270 Pt 1, C148–C159. [Google Scholar] [CrossRef]
- Fabiato, A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. J. Physiol. 1985, 85, 247–289. [Google Scholar] [CrossRef] [PubMed]
- Bassani, R.A.; Bers, D.M.; Bers, D. Rate of diastolic Ca release from the sarcoplasmic reticulum of intact rabbit and rat ventricular myocytes. Biophys. J. 1995, 68, 2015–2022. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac Sarcoplasmic Reticulum Calcium Leak: Basis and Roles in Cardiac Dysfunction. Annu. Physiol. 2014, 76, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Boyden, P.A.; Smith, G.L. Ca2+ leak-What is it? Why should we care? Can it be managed? Heart Rhythm 2018, 15, 607–614. [Google Scholar] [CrossRef]
- Shannon, T.R.; Pogwizd, S.M.; Bers, D.M. Elevated sarcoplasmic reticulum Ca2+ leak in intact ventricular myocytes from rabbits in heart failure. Circ. Res. 2003, 93, 592–594. [Google Scholar] [CrossRef] [PubMed]
- Uehara, A.; Murayama, T.; Yasukochi, M.; Fill, M.; Horie, M.; Okamoto, T.; Matsuura, Y.; Uehara, K.; Fujimoto, T.; Sakurai, T.; et al. Extensive Ca2+ leak through K4750Q cardiac ryanodine receptors caused by cytosolic and luminal Ca2+ hypersensitivity. J. Gen. Physiol. 2017, 149, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Nori, A.; Santoro, M.; Viatchenko-Karpinski, S.; Kubalova, Z.; Gyorke, I.; Terentyeva, R.; Vedamoorthyrao, S.; Blom, N.A.; Valle, G.; et al. Abnormal Interactions of Calsequestrin with the Ryanodine Receptor Calcium Release Channel Complex Linked to Exercise-Induced Sudden Cardiac Death. Circ. Res. 2006, 98, 1151–1158. [Google Scholar] [CrossRef]
- Howlett, S.E.; Grandy, S.A.; Ferrier, G.R. Calcium spark properties in ventricular myocytes are altered in aged mice. Am. J. Physiol. Circ. Physiol. 2006, 290, H1566–H1574. [Google Scholar] [CrossRef] [PubMed]
- Zhu, X.; Altschafl, B.A.; Hajjar, R.J.; Valdivia, H.H.; Schmidt, U. Altered Ca2+ sparks and gating properties of ryanodine receptors in aging cardiomyocytes. Cell Calcium 2005, 37, 583–591. [Google Scholar] [CrossRef] [PubMed]
- Domeier, T.L.; Roberts, C.J.; Gibson, A.K.; Hanft, L.M.; McDonald, K.S.; Segal, S.S. Dantrolene suppresses spontaneous Ca2+ release without altering excitation-contraction coupling in cardiomyocytes of aged mice. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H818–H829. [Google Scholar] [CrossRef] [PubMed]
- Niggli, E.; Ullrich, N.D.; Gutierrez, D.; Kyrychenko, S.; Poláková, E.; Shirokova, N. Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 866–875. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Hamilton, S. Regulation of sarcoplasmic reticulum Ca2+ release by serine-threonine phosphatases in the heart. J. Mol. Cell. Cardiol. 2016, 101, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Houser, S.R. Role of RyR2 phosphorylation in heart failure and arrhythmias: Protein kinase A-mediated hyperphosphorylation of the ryanodine receptor at serine 2808 does not alter cardiac contractility or cause heart failure and arrhythmias. Circ. Res. 2014, 114, 1320–1327. [Google Scholar] [CrossRef]
- Bovo, E.; Huke, S.; Blatter, L.A.; Zima, A.V. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J. Mol. Cell. Cardiol. 2017, 104, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Dobrev, D.; Wehrens, X.H. Role of RyR2 phosphorylation in heart failure and arrhythmias: Controversies around ryanodine receptor phosphorylation in cardiac disease. Circ. Res. 2014, 114, 1311–1319. [Google Scholar] [CrossRef] [PubMed]
- Ai, X.; Curran, J.W.; Shannon, T.R.; Bers, D.M.; Pogwizd, S.M. Ca2+/calmodulin-dependent protein kinase modulates cardiac ryanodine receptor phosphorylation and sarcoplasmic reticulum Ca2+ leak in heart failure. Circ. Res. 2005, 97, 1314–1322. [Google Scholar] [CrossRef] [PubMed]
- Curran, J.; Brown, K.H.; Santiago, D.J.; Pogwizd, S.; Bers, D.M.; Shannon, T.R. Spontaneous Ca waves in ventricular myocytes from failing hearts depend on Ca(2+)-calmodulin-dependent protein kinase II. J. Mol. Cell. Cardiol. 2010, 49, 25–32. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Nishijima, Y.; Sridhar, A.; Hamlin, R.L.; Carnes, C.A.; Györke, S. The relationship between arrhythmogenesis and impaired contractility in heart failure: Role of altered ryanodine receptor function. Cardiovasc. Res. 2011, 90, 493–502. [Google Scholar] [CrossRef]
- Mattiazzi, A.; Bassani, R.A.; Escobar, A.L.; Palomeque, J.; Valverde, C.A.; Petroff, M.V.; Bers, D.M. Chasing cardiac physiology and pathology down the CaMKII cascade. Am. J. Physiol. Circ. Physiol. 2015, 308, H1177–H1191. [Google Scholar] [CrossRef][Green Version]
- Hegyi, B.; Bers, D.M.; Bossuyt, J. CaMKII signaling in heart diseases: Emerging role in diabetic cardiomyopathy. J. Mol. Cell. Cardiol. 2019, 127, 246–259. [Google Scholar] [CrossRef]
- Yan, J.; Zhao, W.; Thomson, J.K.; Gao, X.; Demarco, D.M.; Carrillo, E.; Chen, B.; Wu, X.; Ginsburg, K.S.; Bakhos, M.; et al. Stress Signaling JNK2 Crosstalk with CaMKII Underlies Enhanced Atrial Arrhythmogenesis. Circ. Res. 2018, 122, 821–835. [Google Scholar] [CrossRef]
- Guo, X.; Yuan, S.; Liu, Z.; Fang, Q. Oxidation- and CaMKII-mediated sarcoplasmic reticulum Ca(2+) leak triggers atrial fibrillation in aging. J. Cardiovasc. Electrophysiol. 2014, 25, 645–652. [Google Scholar] [CrossRef]
- Mochizuki, M.; Yano, M.; Oda, T.; Tateishi, H.; Kobayashi, S.; Yamamoto, T.; Ikeda, Y.; Ohkusa, T.; Ikemoto, N.; Matsuzaki, M. Scavenging free radicals by low-dose carvedilol prevents redox-dependent Ca2+ leak via stabilization of ryanodine receptor in heart failure. J. Am. Coll. Cardiol. 2007, 49, 1722–1732. [Google Scholar] [CrossRef]
- Zima, A.V.; Mazurek, S.R. Functional Impact of Ryanodine Receptor Oxidation on Intracellular Calcium Regulation in the Heart. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Cham, Switzerland, 2016; Volume 171, pp. 39–62. [Google Scholar]
- Nikolaienko, R.; Bovo, E.; Zima, A.V. Redox Dependent Modifications of Ryanodine Receptor: Basic Mechanisms and Implications in Heart Diseases. Front. Physiol. 2018, 9, 1775. [Google Scholar] [CrossRef]
- Boraso, A.; Williams, A.J. Modification of the gating of the cardiac sarcoplasmic reticulum Ca(2+)-release channel by H2O2 and dithiothreitol. Am. J. Physiol. 1994, 267 Pt 2, H1010–H1016. [Google Scholar] [CrossRef]
- Terentyev, D.; Györke, I.; Belevych, A.E.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; de Blanco, E.C.; Khanna, S.; Sen, C.K.; Cardounel, A.J.; et al. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008, 103, 1466–1472. [Google Scholar] [CrossRef]
- Edwards, J.N.; Blatter, L.A. Cardiac alternans and intracellular calcium cycling. Clin. Exp. Pharmacol. Physiol. 2014, 41, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Sobie, E.A.; Song, L.S.; Lederer, W.J. Restitution of Ca(2+) release and vulnerability to arrhythmias. J. Cardiovasc. Electrophysiol. 2006, 17 (Suppl. 1), S64–S70. [Google Scholar] [CrossRef] [PubMed]
- Nivala, M.; Qu, Z. Calcium alternans in a couplon network model of ventricular myocytes: Role of sarcoplasmic reticulum load. Am. J. Physiol. Circ. Physiol. 2012, 303, H341–H352. [Google Scholar] [CrossRef][Green Version]
- Alvarez-Lacalle, E.; Cantalapiedra, I.R.; Peñaranda, A.; Cinca, J.; Hove-Madsen, L.; Echebarria, B.; Cantalapiedra, I.R. Dependency of Calcium Alternans on Ryanodine Receptor Refractoriness. PLoS ONE 2013, 8, e55042. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Viatchenko-Karpinski, S.; Terentyeva, R.; Sridhar, A.; Nishijima, Y.; Wilson, L.D.; Cardounel, A.J.; Laurita, K.R.; Carnes, C.A.; et al. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc. Res. 2009, 84, 387–395. [Google Scholar] [CrossRef]
- Belevych, A.E.; Terentyev, D.; Terentyeva, R.; Ho, H.T.; Gyorke, I.; Bonilla, I.M.; Carnes, C.A.; Billman, G.E.; Györke, S. Shortened Ca2+ signaling refractoriness underlies cellular arrhythmogenesis in a postinfarction model of sudden cardiac death. Circ. Res. 2012, 110, 569–577. [Google Scholar] [CrossRef]
- Ho, H.-T.; Stevens, S.C.W.; Terentyeva, R.; Carnes, C.A.; Terentyev, D.; Györke, S. Arrhythmogenic adverse effects of cardiac glycosides are mediated by redox modification of ryanodine receptors. J. Physiol. 2011, 589, 4697–4708. [Google Scholar] [CrossRef] [PubMed]
- Kandilci, H.B.; Tuncay, E.; Zeydanli, E.N.; Sozmen, N.N.; Turan, B. Age-related regulation of excitation–contraction coupling in rat heart. J. Physiol. Biochem. 2011, 67, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Bovo, E.; Lipsius, S.L.; Zima, A.V. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 2012, 590, 3291–3304. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, S.; Terentyeva, R.; Kim, T.Y.; Bronk, P.; Clements, R.T.; O-Uchi, J.; Csordás, G.; Choi, B.R.; Terentyev, D. Pharmacological Modulation of Mitochondrial Ca2+ Content Regulates Sarcoplasmic Reticulum Ca2+ Release via Oxidation of the Ryanodine Receptor by Mitochondria-Derived Reactive Oxygen Species. Front. Physiol. 2018, 9, 1831. [Google Scholar] [CrossRef] [PubMed]
- Pogwizd, S.M.; Qi, M.; Yuan, W.; Samarel, A.M.; Bers, D.M. Upregulation of Na(+)/Ca(2+) exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ. Res. 1999, 85, 1009–1019. [Google Scholar] [CrossRef]
- Bidasee, K.R.; Nallani, K.; Yu, Y.; Cocklin, R.R.; Zhang, Y.; Wang, M.; Dincer, Ü.D.; Besch, H.R. Chronic Diabetes Increases Advanced Glycation End Products on Cardiac Ryanodine Receptors/Calcium-Release Channels. Diabetes 2003, 52, 1825–1836. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Minguet, M.; Bou-Teen, D.; Miro-Casas, E.; Castans, C.; Castellano, J.; Bonzon-Kulichenko, E.; Igual, A.; Rodriguez-Lecoq, R.; Vazquez, J.; et al. Ryanodine Receptor Glycation Favors Mitochondrial Damage in the Senescent Heart. Circulation 2019, 139, 949–964. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Viatchenko-Karpinski, S.; Valdivia, H.H.; Escobar, A.L.; Györke, S. Luminal Ca2+ controls termination and refractory behavior of Ca2+-induced Ca2+ release in cardiac myocytes. Circ. Res. 2002, 91, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Sobie, E.A.; Song, L.S.; Lederer, W.J. Local recovery of Ca2+ release in rat ventricular myocytes. J. Physiol. 2005, 565 Pt 2, 441–447. [Google Scholar] [CrossRef]
- Xu, A.; Narayanan, N. Effects of aging on sarcoplasmic reticulum Ca2+-cycling proteins and their phosphorylation in rat myocardium. Am. J. Physiol. 1998, 275, H2087–H2094. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, U.; del Monte, F.; Miyamoto, M.I.; Matsui, T.; Gwathmey, J.K.; Rosenzweig, A.; Hajjar, R.J. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circulation 2000, 101, 790–796. [Google Scholar] [CrossRef]
- Fernandez-Sanz, C.; Ruiz-Meana, M.; Miro-Casas, E.; Nunez, E.; Castellano, J.; Loureiro, M.; Barba, I.; Poncelas, M.; Rodriguez-Sinovas, A.; Vazquez, J.; et al. Defective sarcoplasmic reticulum–mitochondria calcium exchange in aged mouse myocardium. Cell Death Dis. 2014, 5, e1573. [Google Scholar] [CrossRef]
- Gyorke, I.; Hester, N.; Jones, L.R.; Györke, S. The Role of Calsequestrin, Triadin, and Junctin in Conferring Cardiac Ryanodine Receptor Responsiveness to Luminal Calcium. Biophys. J. 2004, 86, 2121–2128. [Google Scholar] [CrossRef]
- Meissner, G. The structural basis of ryanodine receptor ion channel function. J. Gen. Physiol. 2017, 149, 1065–1089. [Google Scholar] [CrossRef] [PubMed]
- Taffet, G.E.; Tate, C.A. CaATPase content is lower in cardiac sarcoplasmic reticulum isolated from old rats. Am. J. Physiol. Circ. Physiol. 1993, 264 Pt 2, H1609–H1614. [Google Scholar] [CrossRef]
- Herraiz-Martínez, A.; Álvarez-García, J.; Llach, A.; Molina, C.E.; Fernandes, J.; Ferrero-Gregori, A.; Rodríguez, C.; Vallmitjana, A.; Benítez, R.; Padró, J.M.; et al. Ageing is associated with deterioration of calcium homeostasis in isolated human right atrial myocytes. Cardiovasc. Res. 2015, 106, 76–86. [Google Scholar] [CrossRef] [PubMed]
- Terentyev, D.; Viatchenko-Karpinski, S.; Györke, I.; Volpe, P.; Williams, S.C.; Györke, S. Calsequestrin determines the functional size and stability of cardiac intracellular calcium stores: Mechanism for hereditary arrhythmia. Proc. Natl. Acad. Sci. USA 2003, 100, 11759–11764. [Google Scholar] [CrossRef] [PubMed]
- Knollmann, B.C.; Chopra, N.; Hlaing, T.; Akin, B.; Yang, T.; Ettensohn, K.; Knollmann, B.E.; Horton, K.D.; Weissman, N.J.; Holinstat, I.; et al. Casq2 deletion causes sarcoplasmic reticulum volume increase, premature Ca2+ release, and catecholaminergic polymorphic ventricular tachycardia. J. Clin. Investig. 2006, 116, 2510–2520. [Google Scholar] [CrossRef]
- MacLennan, D.H.; Kranias, E.G. Phospholamban: A crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003, 4, 566–577. [Google Scholar] [CrossRef]
- Isenberg, G.; Borschke, B.; Rueckschloss, U. Ca2+ transients of cardiomyocytes from senescent mice peak late and decay slowly. Cell Calcium 2003, 34, 271–280. [Google Scholar] [CrossRef]
- Salameh, A.; Dhein, S.; Fleischmann, B.; Grohe, C.; Hescheler, J.; Linz, K.W.; Meyer, R. The aging heart: Changes in the pharmacodynamic electrophysiological response to verapamil in aged rabbit hearts. J. Physiol. Pharmacol. 2010, 61, 141–151. [Google Scholar] [PubMed]
- Froehlich, J. Studies of sarcoplasmic reticulum function and contraction duration in young adult and aged rat myocardium. J. Mol. Cell. Cardiol. 1978, 10, 427–438. [Google Scholar] [CrossRef]
- Kaplan, P.; Jurkovicova, D.; Babusikova, E.; Hudecova, S.; Racay, P.; Sirova, M.; Lehotsky, J.; Drgova, A.; Dobrota, D.; Krizanova, O. Effect of aging on the expression of intracellular Ca(2+) transport proteins in a rat heart. Mol. Cell. Biochem. 2007, 301, 219–226. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Kass, D.A.; Tomaselli, G.F.; Kääb, S.; Tunin, R.; Marbán, E.; O’Rourke, B.; Kaab, S.; Marban, E. Mechanisms of Altered Excitation-Contraction Coupling in Canine Tachycardia-Induced Heart Failure, I. Circ. Res. 1999, 84, 562–570. [Google Scholar] [CrossRef]
- Zhu, X.; Huq, F.; Schmidt, U.; Lebeche, D.; Guerrero, J.L.; Hajjar, R.J. In vivo gene transfer of parvalbumin improves diastolic function in aged rat hearts. Cardiovasc. Res. 2005, 66, 318–323. [Google Scholar]
- Lompre, A.M.; Lambert, F.; Lakatta, E.G.; Schwartz, K. Expression of sarcoplasmic reticulum Ca(2+)-ATPase and calsequestrin genes in rat heart during ontogenic development and aging. Circ. Res. 1991, 69, 1380–1388. [Google Scholar] [CrossRef] [PubMed]
- Cain, B.S.; Meldrum, D.R.; Joo, K.S.; Wang, J.F.; Meng, X.; Cleveland, J.C., Jr.; Banerjee, A.; Harken, A.H. Human SERCA2a levels correlate inversely with age in senescent human myocardium. J. Am. Coll. Cardiol. 1998, 32, 458–467. [Google Scholar] [CrossRef]
- Lim, C.C.; Liao, R.; Varma, N.; Apstein, C.S. Impaired lusitropy-frequency in the aging mouse: Role of Ca(2+)-handling proteins and effects of isoproterenol. Am. J. Physiol. 1999, 277, H2083–H2090. [Google Scholar] [CrossRef]
- Tian, R.; Halow, J.M.; Meyer, M.; Dillmann, W.H.; Figueredo, V.M.; Ingwall, J.S.; Camacho, S.A. Thermodynamic limitation for Ca2+ handling contributes to decreased contractile reserve in rat hearts. Am. J. Physiol. 1998, 275, H2064–H2071. [Google Scholar] [CrossRef]
- Michele, D.E.; Szatkowski, M.L.; Albayya, F.P.; Metzger, J.M. Parvalbumin gene delivery improves diastolic function in the aged myocardium in vivo. Mol. Ther. 2004, 10, 399–403. [Google Scholar] [CrossRef]
- Jiang, M.T.; Moffat, M.P.; Narayanan, N. Age-related alterations in the phosphorylation of sarcoplasmic reticulum and myofibrillar proteins and diminished contractile response to isoproterenol in intact rat ventricle. Circ. Res. 1993, 72, 102–111. [Google Scholar] [CrossRef]
- Neumann, J.; Eschenhagen, T.; Jones, L.R.; Linck, B.; Schmitz, W.; Scholz, H.; Zimmermann, N. Increased Expression of Cardiac Phosphatases in Patients with End-stage Heart Failure. J. Mol. Cell. Cardiol. 1997, 29, 265–272. [Google Scholar] [CrossRef]
- Carr, A.N.; Schmidt, A.G.; Suzuki, Y.; Del Monte, F.; Sato, Y.; Lanner, C.; Breeden, K.; Jing, S.-L.; Allen, P.B.; Greengard, P.; et al. Type 1 Phosphatase, a Negative Regulator of Cardiac Function. Mol. Cell. Biol. 2002, 22, 4124–4135. [Google Scholar] [CrossRef]
- Gupta, R.C.; Mishra, S.; Rastogi, S.; Imai, M.; Habib, O.; Sabbah, H.N. Cardiac SR-coupled PP1 activity and expression are increased and inhibitor 1 protein expression is decreased in failing hearts. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H2373–H2381. [Google Scholar] [CrossRef] [PubMed]
- Pamminger, T.; Ditz, D.; El-Armouche, A.; Zolk, O.; Eschenhagen, T. Decreased protein and phosphorylation level of the protein phosphatase inhibitor-1 in failing human hearts. Cardiovasc. Res. 2004, 61, 87–93. [Google Scholar]
- Haghighi, K.; Bidwell, P.; Kranias, E.G. Phospholamban Interactome in Cardiac Contractility and Survival: A New Vision of an OLD Friend. J. Mol. Cell. Cardiol. 2014, 77, 160–167. [Google Scholar] [CrossRef]
- Pritchard, T.J.; Kawase, Y.; Haghighi, K.; Anjak, A.; Cai, W.; Jiang, M.; Nicolaou, P.; Pylar, G.; Karakikes, I.; Rapti, K.; et al. Active Inhibitor-1 Maintains Protein Hyper-Phosphorylation in Aging Hearts and Halts Remodeling in Failing Hearts. PLoS ONE 2013, 8, e80717. [Google Scholar] [CrossRef]
- Knyushko, T.V.; Sharov, V.S.; Williams, T.D.; Schöneich, C.; Bigelow, D.J. 3-Nitrotyrosine Modification of SERCA2a in the Aging Heart: A Distinct Signature of the Cellular Redox Environment†. Biochemistry 2005, 44, 13071–13081. [Google Scholar] [CrossRef]
- Babušíková, E.; Lehotský, J.; Dobrota, D.; Račay, P.; Kaplán, P. Age-associated changes in Ca(2+)-ATPase and oxidative damage in sarcoplasmic reticulum of rat heart. Physiol. Res. 2012, 61, 453–460. [Google Scholar]
- Qin, F.; Siwik, D.A.; Lancel, S.; Zhang, J.; Kuster, G.M.; Luptak, I.; Wang, L.; Tong, X.Y.; Kang, Y.J.; Cohen, R.A.; et al. Hydrogen Peroxide–Mediated SERCA Cysteine 674 Oxidation Contributes to Impaired Cardiac Myocyte Relaxation in Senescent Mouse Heart. J. Am. Heart Assoc. 2013, 2, 000184. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.N.; Garfinkel, A.; Karagueuzian, H.S.; Chen, P.-S.; Qu, Z. Early Afterdepolarizations and Cardiac Arrhythmias. Heart Rhythm 2010, 7, 1891–1899. [Google Scholar] [CrossRef] [PubMed]
- Lakatta, E.G.; Houser, S.R.; E Walker, K. Age associated changes in membrane currents in rat ventricular myocytes. Cardiovasc. Res. 1993, 27, 1968–1977. [Google Scholar]
- Liu, S.J.; Wyeth, R.P.; Melchert, R.B.; Kennedy, R.H. Aging-associated changes in whole cell K+ and L-type Ca2+ currents in rat ventricular myocytes. Am. J. Physiol. 2000, 279, H889–H900. [Google Scholar] [CrossRef]
- Josephson, I.R.; Guia, A.; Stern, M.D.; Lakatta, E.G. Alterations in Properties of L-Type Ca Channels in Aging Rat Heart. J. Mol. Cell. Cardiol. 2002, 34, 297–308. [Google Scholar] [CrossRef]
- Grandy, S.A.; Howlett, S.E. Cardiac excitation-contraction coupling is altered in myocytes from aged male mice but not in cells from aged female mice. Am. J. Physiol. Circ. Physiol. 2006, 291, H2362–H2370. [Google Scholar] [CrossRef]
- Dun, W.; Yagi, T.; Rosen, M.R.; A Boyden, P. Calcium and potassium currents in cells from adult and aged canine right atria. Cardiovasc. Res. 2003, 58, 526–534. [Google Scholar] [CrossRef]
- Gan, T.-Y.; Qiao, W.; Xu, G.-J.; Zhou, X.-H.; Tang, B.-P.; Song, J.-G.; Li, Y.-D.; Zhang, J.; Li, F.-P.; Mao, T.; et al. Aging-associated changes in L-type calcium channels in the left atria of dogs. Exp. Ther. Med. 2013, 6, 919–924. [Google Scholar] [CrossRef]
- Xu, G.-J.; Gan, T.-Y.; Zhang, Y.; Tang, B.-P.; Chen, Z.-H.; Mahemuti, A.; Jiang, T.; Song, J.-G.; Guo, X.; Li, Y.-D.; et al. Alterations in the expression of atrial calpains in electrical and structural remodeling during aging and atrial fibrillation. Mol. Med. Rep. 2013, 8, 1343–1352. [Google Scholar] [CrossRef]
- Xu, G.-J.; Gan, T.-Y.; Tang, B.-P.; Chen, Z.-H.; Jiang, T.; Song, J.-G.; Guo, X.; Li, J.-X. Age-related changes in cellular electrophysiology and calcium handling for atrial fibrillation. J. Cell. Mol. Med. 2013, 17, 1109–1118. [Google Scholar] [CrossRef]
- Dibb, K.; Rueckschloss, U.; Eisner, D.; Isenberg, G.; Trafford, A. Mechanisms underlying enhanced cardiac excitation contraction coupling observed in the senescent sheep myocardium. J. Mol. Cell. Cardiol. 2004, 37, 1171–1181. [Google Scholar] [CrossRef]
- Clarke, J.D.; Caldwell, J.L.; Pearman, C.M.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Increased Ca buffering underpins remodelling of Ca2+ handling in old sheep atrial myocytes. J. Physiol. 2017, 595, 6263–6279. [Google Scholar] [CrossRef]
- Pearman, C.M.; Madders, G.W.; Radcliffe, E.J.; Kirkwood, G.J.; Lawless, M.; Watkins, A.; Smith, C.E.; Trafford, A.W.; Eisner, D.A.; Dibb, K.M. Increased Vulnerability to Atrial Fibrillation Is Associated with Increased Susceptibility to Alternans in Old Sheep. J. Am. Heart Assoc. 2018, 7, 009972. [Google Scholar] [CrossRef]
- Harvey, R.D.; Hell, J.W. CaV1.2 signaling complexes in the heart. J. Mol. Cell. Cardiol. 2013, 58, 143–152. [Google Scholar] [CrossRef]
- Chen, X.; Piacentino, V., 3rd; Furukawa, S.; Goldman, B.; Margulies, K.B.; Houser, S.R. L-type Ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ. Res. 2002, 91, 517–524. [Google Scholar] [CrossRef]
- Wei, J.Y.; Spurgeon, H.A.; Lakatta, E.G. Excitation-contraction in rat myocardium: Alterations with adult aging. Am. J. Physiol. Circ. Physiol. 1984, 246, H784–H791. [Google Scholar] [CrossRef]
- Crossman, D.J.; Young, A.A.; Ruygrok, P.N.; Nason, G.P.; Baddelely, D.; Soeller, C.; Cannell, M.B. t-tubule disease: Relationship between t-tubule organization and regional contractile performance in human dilated cardiomyopathy. J. Mol. Cell. Cardiol. 2015, 84, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Scardigli, M.; Ferrantini, C.; Crocini, C.; Pavone, F.S.; Sacconi, L. Interplay Between Sub-Cellular Alterations of Calcium Release and T-Tubular Defects in Cardiac Diseases. Front. Physiol. 2018, 9, 1474. [Google Scholar] [CrossRef]
- Jones, P.P.; Macquaide, N.; Louch, W.E. Dyadic Plasticity in Cardiomyocytes. Front. Physiol. 2018, 9, 1773. [Google Scholar] [CrossRef]
- Kong, C.H.T.; Bryant, S.M.; Watson, J.J.; Gadeberg, H.C.; Roth, D.M.; Patel, H.H.; Cannell, M.B.; Orchard, C.H.; James, A.F. The Effects of Aging on the Regulation of T-Tubular ICa by Caveolin in Mouse Ventricular Myocytes. J. Gerontol. A Biol. Sci. Med. Sci. 2018, 73, 711–719. [Google Scholar] [CrossRef]
- Ñeco, P.; Rose, B.; Huynh, N.; Zhang, R.; Bridge, J.H.; Philipson, K.D.; Goldhaber, J.I. Sodium-Calcium Exchange Is Essential for Effective Triggering of Calcium Release in Mouse Heart. Biophys. J. 2010, 99, 755–764. [Google Scholar] [CrossRef]
- Despa, S.; Bers, D.M. Na⁺ transport in the normal and failing heart—Remember the balance. J. Mol. Cell. Cardiol. 2013, 61, 2–10. [Google Scholar] [CrossRef]
- Ottolia, M.; Torres, N.; Bridge, J.H.B.; Philipson, K.D.; Goldhaber, J.I. Na/Ca exchange and contraction of the heart. J. Mol. Cell. Cardiol. 2013, 61, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Janapati, V.; Wu, A.; Davis, N.; Derrico, C.A.; Levengood, J.; Schummers, J.; Colvin, R.A. Post-transcriptional regulation of the Na+/Ca2+ exchanger in aging rat heart. Mech. Ageing Dev. 1995, 84, 195–208. [Google Scholar] [CrossRef]
- Assayag, P.; Charlemagne, D.; Marty, I.; De Leiris, J.; Lompré, A.M.; Boucher, F.; Valère, P.-E.; Lortet, S.; Swynghedauw, B.; Besse, S. Effects of sustained low-flow ischemia on myocardial function and calcium-regulating proteins in adult and senescent rat hearts. Cardiovasc. Res. 1998, 38, 169–180. [Google Scholar] [CrossRef]
- Mace, L.C.; Palmer, B.M.; Brown, D.A.; Jew, K.N.; Lynch, J.M.; Glunt, J.M.; Parsons, T.A.; Cheung, J.Y.; Moore, R.L. Influence of age and run training on cardiac Na+/Ca2+ exchange. J. Appl. Physiol. 2003, 95, 1994–2003. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Wu, S.; Li, S.-Y.; Lopez, F.L.; Du, M.; Kajstura, J.; Anversa, P.; Ren, J. Cardiac-specific overexpression of insulin-like growth factor 1 attenuates aging-associated cardiac diastolic contractile dysfunction and protein damage. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1398–H1403. [Google Scholar] [CrossRef]
- Ozturk, N.; Olgar, Y.; Er, H.; Kucuk, M.; Ozdemir, S. Swimming exercise reverses aging-related contractile abnormalities of female heart by improving structural alterations. Cardiol. J. 2017, 24, 85–93. [Google Scholar] [CrossRef]
- Keller, K.M.; Howlett, S.E. Sex Differences in the Biology and Pathology of the Aging Heart. Can. J. Cardiol. 2016, 32, 1065–1073. [Google Scholar] [CrossRef]
- Kane, A.E.; Howlett, S.E. Differences in Cardiovascular Aging in Men and Women. Adv. Exp. Med. Biol. 2018, 1065, 389–411. [Google Scholar] [PubMed]
- Howlett, S.E. Age-associated changes in excitation-contraction coupling are more prominent in ventricular myocytes from male rats than in myocytes from female rats. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H659–H670. [Google Scholar] [CrossRef] [PubMed]
- Farrell, S.R.; Howlett, S.E. The effects of isoproterenol on abnormal electrical and contractile activity and diastolic calcium are attenuated in myocytes from aged Fischer 344 rats. Mech. Ageing Dev. 2007, 128, 566–573. [Google Scholar] [CrossRef]
- Ross, J.L.; Howlett, S.E. Age and Ovariectomy Abolish Beneficial Effects of Female Sex on Rat Ventricular Myocytes Exposed to Simulated Ischemia and Reperfusion. PLoS ONE 2012, 7, 38425. [Google Scholar] [CrossRef] [PubMed]
- Claessens, T.E.; Rietzschel, E.R.; De Buyzere, M.L.; De Bacquer, D.; De Backer, G.; Gillebert, T.C.; Verdonck, P.R.; Segers, P.; Gillebert, T. Noninvasive assessment of left ventricular and myocardial contractility in middle-aged men and women: Disparate evolution above the age of 50? Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H856–H865. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sanz, A.; Hiona, A.; Kujoth, G.C.; Seo, A.Y.; Hofer, T.; Kouwenhoven, E.; Kalani, R.; Prolla, T.; Barja, G.; Leeuwenburgh, C.; et al. Evaluation of sex differences on mitochondrial bioenergetics and apoptosis in mice. Exp. Gerontol. 2007, 42, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Li, Y.; He, C.; Gou, B.; Song, M. Mitochondrial regulation of cardiac aging. Biochim. Biophys. Acta Mol. Basis Dis. 2018. [Google Scholar] [CrossRef]
- Chaudhary, K.R.; El-Sikhry, H.; Seubert, J.M. Mitochondria and the aging heart. J. Geriatr. Cardiol. 2011, 8, 159–167. [Google Scholar]
- Blatter, L.A.; Zima, A.V. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006, 71, 310–321. [Google Scholar]
- Kim, T.Y.; Terentyeva, R.; Roder, K.H.; Li, W.; Liu, M.; Greener, I.; Hamilton, S.; Polina, I.; Murphy, K.R.; Clements, R.T.; et al. SK channel enhancers attenuate Ca2+-dependent arrhythmia in hypertrophic hearts by regulating mito-ROS-dependent oxidation and activity of RyR. Cardiovasc. Res. 2017, 113, 343–353. [Google Scholar]
- Ying, J.; Sharov, V.; Xu, S.; Jiang, B.; Gerrity, R.; Schöneich, C.; Cohen, R.A. Cysteine-674 oxidation and degradation of sarcoplasmic reticulum Ca(2+) ATPase in diabetic pig aorta. Free Radic. Biol. Med. 2008, 45, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan, P.; Szappanos, H.C.; Ingley, E.; Hool, L.C. The cardiac L-type calcium channel alpha subunit is a target for direct redox modification during oxidative stress-the role of cysteine residues in the alpha interacting domain. Clin. Exp. Pharmacol. Physiol. 2017, 44, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; O’Rourke, B. Regulation of the Na+/Ca2+ exchanger by pyridine nucleotide redox potential in ventricular myocytes. J. Biol. Chem. 2013, 288, 31984–31992. [Google Scholar] [CrossRef]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.-X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 11427. [Google Scholar] [CrossRef]
- Yoo, S.; Aistrup, G.; Shiferaw, Y.; Ng, J.; Mohler, P.J.; Hund, T.J.; Waugh, T.; Browne, S.; Gussak, G.; Gilani, M.; et al. Oxidative stress creates a unique, CaMKII-mediated substrate for atrial fibrillation in heart failure. JCI Insight 2018, 3, 120728. [Google Scholar] [CrossRef]
- Morita, N.; Sovari, A.A.; Xie, Y.; Fishbein, M.C.; Mandel, W.J.; Garfinkel, A.; Lin, S.-F.; Chen, P.-S.; Xie, L.-H.; Chen, F.; et al. Increased susceptibility of aged hearts to ventricular fibrillation during oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1594–H1605. [Google Scholar] [CrossRef]
- Dietl, A.; Maack, C. Targeting Mitochondrial Calcium Handling and Reactive Oxygen Species in Heart Failure. Heart Fail. Rep. 2017, 14, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Escobales, N.; Nuñez, R.E.; Jang, S.; Parodi-Rullan, R.; Ayala-Peña, S.; Sacher, J.R.; Skoda, E.M.; Wipf, P.; Frontera, W.; Javadov, S. Mitochondria-targeted ROS scavenger improves post-ischemic recovery of cardiac function and attenuates mitochondrial abnormalities in aged rats. J. Mol. Cell. Cardiol. 2014, 77, 136–146. [Google Scholar] [CrossRef]
- Olgar, Y.; Degirmenci, S.; Durak, A.; Billur, D.; Can, B.; Mutlu, G.K.; Inan, E.A.; Turan, B. Aging related functional and structural changes in the heart and aorta: MitoTEMPO improves aged-cardiovascular performance. Exp. Gerontol. 2018, 110, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Dai, D.-F.; Santana, L.F.; Vermulst, M.; Tomazela, D.M.; Emond, M.J.; MacCoss, M.J.; Gollahon, K.; Martin, G.M.; Loeb, L.A.; Ladiges, W.C.; et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 2009, 119, 2789–2797. [Google Scholar] [CrossRef]
- Yao, C.; Behring, J.B.; Shao, D.; Sverdlov, A.L.; Whelan, S.A.; Elezaby, A.; Yin, X.; Siwik, D.A.; Seta, F.; Costello, C.E.; et al. Overexpression of Catalase Diminishes Oxidative Cysteine Modifications of Cardiac Proteins. PLoS ONE 2015, 10, 0144025. [Google Scholar] [CrossRef]
- Umanskaya, A.; Santulli, G.; Xie, W.; Andersson, D.C.; Reiken, S.R.; Marks, A.R. Genetically enhancing mitochondrial antioxidant activity improves muscle function in aging. Proc. Natl. Acad. Sci. USA 2014, 111, 15250–15255. [Google Scholar] [CrossRef]
- Valdés, Á.; Treuer, A.V.; Barrios, G.; Ponce, N.; Fuentealba, R.; Dulce, R.A.; González, D.R. NOX Inhibition Improves β-Adrenergic Stimulated Contractility and Intracellular Calcium Handling in the Aged Rat Heart. Int. J. Mol. Sci. 2018, 19, 2404. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Takimoto, E.; Dimaano, V.L.; DeMazumder, D.; Kettlewell, S.; Smith, G.; Sidor, A.; Abraham, T.P.; O’Rourke, B. Inhibiting mitochondrial Na+/Ca2+ exchange prevents sudden death in a Guinea pig model of heart failure. Circ. Res. 2014, 115, 44–54. [Google Scholar] [CrossRef]
- Sloan, R.C.; Moukdar, F.; Frasier, C.R.; Patel, H.D.; Bostian, P.A.; Lust, R.M.; Brown, D.A. Mitochondrial permeability transition in the diabetic heart: Contributions of thiol redox state and mitochondrial calcium to augmented reperfusion injury. J. Mol. Cell. Cardiol. 2012, 52, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Luongo, T.S.; Lambert, J.P.; Gross, P.; Nwokedi, M.; Lombardi, A.A.; Shanmughapriya, S.; Carpenter, A.C.; Kolmetzky, D.; Gao, E.; van Berlo, J.H.; et al. The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 2017, 545, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Paillard, M.; Tubbs, E.; Thiebaut, P.-A.; Gomez, L.; Fauconnier, J.; Da Silva, C.C.; Teixeira, G.; Mewton, N.; Belaidi, E.; Durand, A.; et al. Depressing Mitochondria-Reticulum Interactions Protects Cardiomyocytes from Lethal Hypoxia-Reoxygenation Injury. Circulation 2013, 128, 1555–1565. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, E.J.; Balaska, D.; Cheng, W.H. The ups and downs of mitochondrial calcium signalling in the heart. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 856–864. [Google Scholar] [CrossRef]
- Uchi, J.; Jhun, B.S.; Xu, S.; Hurst, S.; Raffaello, A.; Liu, X.; Yi, B.; Zhang, H.; Gross, P.; Mishra, J.; et al. Adrenergic signaling regulates mitochondrial Ca2+ uptake through Pyk2-dependent tyrosine phosphorylation of the mitochondrial Ca2+ uniporter. Antioxid. Redox Signal. 2014, 21, 863–879. [Google Scholar] [CrossRef]
- Suárez, J.; Cividini, F.; Scott, B.T.; Lehmann, K.; Díaz-Juárez, J.; Diemer, T.; Dai, A.; Suarez, J.A.; Jain, M.; Dillmann, W.H. Restoring mitochondrial calcium uniporter expression in diabetic mouse heart improves mitochondrial calcium handling and cardiac function. J. Biol. Chem. 2018, 293, 8182–8195. [Google Scholar] [CrossRef]
- Kettlewell, S.; Cabrero, P.; Nicklin, S.A.; Dow, J.A.; Davies, S.; Smith, G.L. Changes of intra-mitochondrial Ca2+ in adult ventricular cardiomyocytes examined using a novel fluorescent Ca2+ indicator targeted to mitochondria. J. Mol. Cell. Cardiol. 2009, 46, 891–901. [Google Scholar] [CrossRef]
- Kaestner, L.; Scholz, A.; Tian, Q.; Ruppenthal, S.; Tabellion, W.; Wiesen, K.; Katus, H.A.; Müller, O.J.; Kotlikoff, M.I.; Lipp, P. Genetically encoded Ca2+ indicators in cardiac myocytes. Circ. Res. 2014, 114, 1623–1639. [Google Scholar] [CrossRef]
- Jones, J.S.; Small, D.M.; Nishimura, N. In Vivo Calcium Imaging of Cardiomyocytes in the Beating Mouse Heart with Multiphoton Microscopy. Front. Physiol. 2018, 9, 969. [Google Scholar] [CrossRef]
- Bovo, E.; Nikolaieko, R.; Bhayani, S.; Kahn, D.; Cao, Q.; Martin, J.L.; Kuo, I.Y.; Robia, S.; Zima, A.V. Novel approach for quantification of endoplasmic reticulum Ca2+ transport. Am. J. Physiol. Heart Circ. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- No, M.-H.; Heo, J.-W.; Yoo, S.-Z.; Jo, H.-S.; Park, D.-H.; Kang, J.-H.; Seo, D.-Y.; Han, J.; Kwak, H.-B. Effects of aging on mitochondrial hydrogen peroxide emission and calcium retention capacity in rat heart. J. Exerc. Rehabil. 2018, 14, 920–926. [Google Scholar] [CrossRef]
- Harman, D. Aging: A Theory Based on Free Radical and Radiation Chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Adlam, V.J.; Harrison, J.C.; Porteous, C.M.; James, A.M.; Smith, R.A.J.; Murphy, M.P.; Sammut, I. Targeting an antioxidant to mitochondria decreases cardiac ischemia-reperfusion injury. FASEB J. 2005, 19, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Graham, D.; Huynh, N.N.; Hamilton, C.A.; Beattie, E.; Smith, R.A.; Cochemé, H.M.; Murphy, M.; Dominiczak, A.F. Mitochondria-Targeted Antioxidant MitoQ10 Improves Endothelial Function and Attenuates Cardiac Hypertrophy. Hypertension 2009, 54, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Gioscia-Ryan, R.A.; Battson, M.L.; Cuevas, L.M.; Eng, J.S.; Murphy, M.P.; Seals, D.R. Mitochondria-targeted antioxidant therapy with MitoQ ameliorates aortic stiffening in old mice. J. Appl. Physiol. (1985) 2018, 124, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Rossman, M.J.; Santos-Parker, J.R.; Steward, C.A.; Bispham, N.Z.; Cuevas, L.M.; Rosenberg, H.L.; Woodward, K.A.; Chonchol, M.; Gioscia-Ryan, R.A.; Murphy, M.P.; et al. Chronic Supplementation with a Mitochondrial Antioxidant (MitoQ) Improves Vascular Function in Healthy Older Adults. Hypertension 2018, 71, 1056–1063. [Google Scholar] [CrossRef] [PubMed]
- Nickel, A.G.; Von Hardenberg, A.; Hohl, M.; Löffler, J.R.; Kohlhaas, M.; Becker, J.; Reil, J.-C.; Kazakov, A.; Bonnekoh, J.; Stadelmaier, M.; et al. Reversal of Mitochondrial Transhydrogenase Causes Oxidative Stress in Heart Failure. Cell Metab. 2015, 22, 472–484. [Google Scholar] [CrossRef]
- Dai, D.-F.; Hsieh, E.J.; Chen, T.; Menendez, L.G.; Basisty, N.B.; Tsai, L.; Beyer, R.P.; Crispin, D.A.; Shulman, N.J.; Szeto, H.H.; et al. Global Proteomics and Pathway Analysis of Pressure-overload Induced Heart Failure and Its Attenuation by Mitochondrial Targeted Peptides. Circ. Heart Fail. 2013, 6, 1067–1076. [Google Scholar] [CrossRef]
- Dai, D.-F.; Chiao, Y.A.; Marcinek, D.J.; Szeto, H.H.; Rabinovitch, P.S. Mitochondrial oxidative stress in aging and healthspan. Longev. Healthspan 2014, 3, 6. [Google Scholar] [CrossRef]
- Aon, M.; Cortassa, S.; O’Rourke, B. Redox-optimized ROS balance: A unifying hypothesis. Biochim. Biophys. Acta (BBA)-Bioenerg. 2010, 1797, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Aldosari, S.; Awad, M.; Harrington, E.O.; Sellke, F.W.; Abid, M.R. Subcellular Reactive Oxygen Species (ROS) in Cardiovascular Pathophysiology. Antioxidants 2018, 7, 14. [Google Scholar] [CrossRef]
- García-Rivas, G.D.J.; Carvajal, K.; Correa, F.; Zazueta, C.; García-Rivas, G.J. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post-ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Schweitzer, M.K.; Wilting, F.; Sedej, S.; Dreizehnter, L.; Dupper, N.J.; Tian, Q.; Moretti, A.; My, I.; Kwon, O.; Priori, S.G.; et al. Suppression of Arrhythmia by Enhancing Mitochondrial Ca2+ Uptake in Catecholaminergic Ventricular Tachycardia Models. JACC Basic Transl. Sci. 2017, 2, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Zhou, A.; Liu, H.; Shi, G.; Liu, M.; Boheler, K.R.; Dudley, S.C., Jr. Mitochondrial Ca2+ flux modulates spontaneous electrical activity in ventricular cardiomyocytes. PLoS ONE 2018, 13, e0200448. [Google Scholar] [CrossRef] [PubMed]
- Dedkova, E.N.; Blatter, L.A. Mitochondrial Ca2+ and the heart. Cell Calcium 2008, 44, 77–91. [Google Scholar] [CrossRef] [PubMed]
- O’Rourke, B.; Blatter, L.A. Mitochondrial Ca2+ uptake: Tortoise or hare? J. Mol. Cell. Cardiol. 2009, 46, 767–774. [Google Scholar] [CrossRef]
- Hohendanner, F.; Ljubojević, S.; MacQuaide, N.; Sacherer, M.; Sedej, S.; Biesmans, L.; Wakula, P.; Platzer, D.; Sokolow, S.; Herchuelz, A.; et al. Intracellular dyssynchrony of diastolic cytosolic [Ca2+] decay in ventricular cardiomyocytes in cardiac remodeling and human heart failure. Circ. Res. 2013, 113, 527–538. [Google Scholar] [CrossRef]
- Williams, G.S.; Boyman, L.; Chikando, A.C.; Khairallah, R.J.; Lederer, W.J. Mitochondrial calcium uptake. Proc. Natl. Acad. Sci. USA 2013, 110, 10479–10486. [Google Scholar] [CrossRef]
- Lu, X.; Ginsburg, K.S.; Kettlewell, S.; Bossuyt, J.; Smith, G.L.; Bers, D.M. Measuring local gradients of intramitochondrial [Ca(2+)] in cardiac myocytes during sarcoplasmic reticulum Ca(2+) release. Circ. Res. 2013, 112, 424–431. [Google Scholar] [CrossRef]
- De La Fuente, S.; Lambert, J.P.; Nichtova, Z.; Sanz, C.F.; Elrod, J.W.; Sheu, S.-S.; Csordás, G. Spatial Separation of Mitochondrial Calcium Uptake and Extrusion for Energy-Efficient Mitochondrial Calcium Signaling in the Heart. Cell Rep. 2018, 24, 3099–3107.e4. [Google Scholar] [CrossRef]
- Bonilla, I.M.; Belevych, A.E.; Sridhar, A.; Nishijima, Y.; Ho, H.T.; He, Q.; Kukielka, M.; Terentyev, D.; Terentyeva, R.; Liu, B.; et al. Endurance exercise training normalizes repolarization and calcium-handling abnormalities, preventing ventricular fibrillation in a model of sudden cardiac death. J. Appl. Physiol. (1985) 2012, 113, 1772–1783. [Google Scholar] [CrossRef]
- Manotheepan, R.; Danielsen, T.K.; Sadredini, M.; Anderson, M.E.; Carlson, C.R.; Lehnart, S.E.; Sjaastad, I.; Stokke, M.K. Exercise training prevents ventricular tachycardia in CPVT1 due to reduced CaMKII-dependent arrhythmogenic Ca2+ release. Cardiovasc. Res. 2016, 111, 295–306. [Google Scholar] [CrossRef]
- Carneiro-Júnior, M.A.; Quintão-Júnior, J.F.; Drummond, L.R.; Lavorato, V.N.; Drummond, F.R.; Amadeu, M.A.; Oliveira, E.M.; Felix, L.B.; Cruz, J.S.; Mill, J.G.; et al. Effect of exercise training on Ca2+ release units of left ventricular myocytes of spontaneously hypertensive rats. Braz. J. Med. Biol. Res. 2014, 47, 960–965. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Groban, L.; Jobe, H.; Lin, M.; Houle, T.; Kitzman, D.A.; Sonntag, W. Effects of Short-Term Treadmill Exercise Training or Growth Hormone Supplementation on Diastolic Function and Exercise Tolerance in Old Rats. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2008, 63, 911–920. [Google Scholar] [CrossRef][Green Version]
- Thomas, M.M.; Vigna, C.; Betik, A.C.; Tupling, A.R.; Hepple, R.T. Cardiac calcium pump inactivation and nitrosylation in senescent rat myocardium are not attenuated by long-term treadmill training. Exp. Gerontol. 2011, 46, 803–810. [Google Scholar] [CrossRef] [PubMed]
- Walton, R.D.; Jones, S.A.; Rostron, K.A.; Kayani, A.C.; Close, G.L.; McArdle, A.; Lancaster, M.K. Interactions of Short-Term and Chronic Treadmill Training with Aging of the Left Ventricle of the Heart. J. Gerontol. A Biol. Sci. Med. Sci. 2016, 71, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Edelmann, F.; Gelbrich, G.; Düngen, H.D.; Fröhling, S.; Wachter, R.; Stahrenberg, R.; Binder, L.; Töpper, A.; Lashki, D.J.; Schwarz, S.; et al. Exercise training improves exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction: Results of the Ex-DHF (Exercise training in Diastolic Heart Failure) pilot study. J. Am. Coll. Cardiol. 2011, 58, 1780–1791. [Google Scholar] [CrossRef]
- Spina, R.J.; Turner, M.J.; A Ehsani, A. Beta-adrenergic-mediated improvement in left ventricular function by exercise training in older men. Am. J. Physiol. 1998, 274, H397–H404. [Google Scholar]
- Howden, E.J.; Sarma, S.; Lawley, J.S.; Opondo, M.; Cornwell, W.; Stoller, D.; Urey, M.A.; Adams-Huet, B.; Levine, B.D. Reversing the Cardiac Effects of Sedentary Aging in Middle Age-A Randomized Controlled Trial: Implications for Heart Failure Prevention. Circulation 2018, 137, 1549–1560. [Google Scholar] [CrossRef]
- Leosco, D.; Parisi, V.; Femminella, G.D.; Formisano, R.; Petraglia, L.; Allocca, E.; Bonaduce, D.; Femminella, D.G. Effects of exercise training on cardiovascular adrenergic system. Front. Physiol. 2013, 4, 348. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Rhee, J.; Chaudhari, V.; Rosenzweig, A. The Role of Exercise in Cardiac Aging: From Physiology to Molecular Mechanisms. Circ. Res. 2016, 118, 279–295. [Google Scholar] [CrossRef]
- McGuire, D.K.; Levine, B.D.; Williamson, J.W.; Snell, P.G.; Blomqvist, C.G.; Saltin, B.; Mitchell, J.H. A 30-year follow-up of the Dallas Bedrest and Training Study: II. Effect of age on cardiovascular adaptation to exercise training. Circulation 2001, 104, 1358–1366. [Google Scholar] [CrossRef] [PubMed]
- Fujimoto, N.; Prasad, A.; Hastings, J.L.; Arbab-Zadeh, A.; Bhella, P.S.; Shibata, S.; Palmer, D.; Levine, B.D. Cardiovascular Effects of 1 Year of Progressive and Vigorous Exercise Training in Previously Sedentary Individuals Older Than 65 Years of Age. Circulation 2010, 122, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Haykowsky, M.J.; Brubaker, P.H.; Stewart, K.P.; Morgan, T.M.; Eggebeen, J.; Kitzman, D.W. Effect of Endurance Training on the Determinants of Peak Exercise Oxygen Consumption in Elderly Patients with Stable Compensated Heart Failure and Preserved Ejection Fraction. J. Am. Coll. Cardiol. 2012, 60, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Most, J.; Tosti, V.; Redman, L.M.; Fontana, L. Calorie restriction in humans: An update. Ageing Res. Rev. 2017, 39, 36–45. [Google Scholar] [CrossRef]
- Zullo, A.; Simone, E.; Grimaldi, M.; Musto, V.; Mancini, F.P. Sirtuins as Mediator of the Anti-Ageing Effects of Calorie Restriction in Skeletal and Cardiac Muscle. Int. J. Mol. Sci. 2018, 19, 928. [Google Scholar] [CrossRef]
- Tanno, M.; Kuno, A.; Horio, Y.; Miura, T. Emerging beneficial roles of sirtuins in heart failure. Basic Res. Cardiol. 2012, 107, 273. [Google Scholar] [CrossRef] [PubMed]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Parodi-Rullán, R.M.; Chapa-Dubocq, X.R.; Javadov, S. Acetylation of Mitochondrial Proteins in the Heart: The Role of SIRT3. Front. Physiol. 2018, 9, 1094. [Google Scholar] [CrossRef] [PubMed]
- Marcelli, S.; Corbo, M.; Iannuzzi, F.; Negri, L.; Blandini, F.; Nisticò, R.; Feligioni, M. The Involvement of Post-Translational Modifications in Alzheimer’s Disease. Curr. Res. 2018, 15, 313–335. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Feller, C.; Ladurner, A.G.; Imhof, A. The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem. Sci. 2016, 41, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Treviño-Saldaña, N.; García-Rivas, G. Regulation of Sirtuin-Mediated Protein Deacetylation by Cardioprotective Phytochemicals. Oxidative Med. Cell. Longev. 2017, 2017, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Horton, J.L.; Martin, O.J.; Riley, N.M.; Richards, A.L.; Vega, R.B.; Leone, T.C.; Bedi, K.C.; Margulies, K.B.; Lai, L.; Pagliarini, D.J.; et al. Mitochondrial protein hyperacetylation in the failing heart. JCI Insight 2016, 1, e84897. [Google Scholar] [CrossRef] [PubMed]
- Sulaiman, M.; Matta, M.J.; Sunderesan, N.R.; Gupta, M.P.; Periasamy, M.; Gupta, M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H833–H843. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Wu, Z.; Li, X.; Yan, J.; Zhao, L.; Yang, C.; Lu, J.; Deng, J.; Chen, M. Resveratrol ameliorates cardiac dysfunction induced by pressure overload in rats via structural protection and modulation of Ca(2+) cycling proteins. J. Transl. Med. 2014, 12, 323. [Google Scholar] [CrossRef]
- Gorski, P.A.; Jang, S.P.; Jeong, D.; Lee, A.; Lee, P.; Oh, J.G.; Chepurko, V.; Yang, D.K.; Kwak, T.H.; Eom, S.H.; et al. Role of SIRT1 in Modulating Acetylation of the Sarco-Endoplasmic Reticulum Ca2+-ATPase in Heart Failure. Circ. Res. 2019, 124, e63–e80. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Change in Function | Species | Sex | Ages Studied | Myocyte Type | Comments | Reference |
|---|---|---|---|---|---|---|
| Ryanodine Receptor (RyR2) | ||||||
| ↑ | Mouse | Both | 6 vs. 24 mo (young adult vs. old) | Ventricular | Increased spontaneous Ca2+ spark activity | [26] |
| Mouse | Unreported | 6 vs. 24 mo (young adult vs. old) | Ventricular | Increased single channel open probability, increased spontaneous Ca2+ spark activity | [27] | |
| Mouse | Male | 3–4 vs. 24–26 mo (young vs. old) | Ventricular | Increased RyR2-mediated diastolic sparks and waves | [28] | |
| Rabbit | Female | 5–9 mo vs. 4–6 yrs (young adult vs. old) | Ventricular | Increased oxidation and SR Ca2+ leak, unchanged PKA- but increased CaMKII-mediated phosphorylation | [9] | |
| Mouse | Male | 4–6 vs. >20 mo (young adult vs. old) | Atrial | Increased glycation | [58] | |
| Mouse | Male | 2–2.5 vs. 24–32 mo (young vs. old) | Atrial | Increased JNK2/CaMKII activity, enhanced SR Ca2+ leak | [39] | |
| Mouse | Male | 4–5 vs. 24 mo (young adult vs. old) | Atrial | Increased CaMKII-mediated phosphorylation and oxidation | [40] | |
| Human | Both | <75 vs. >75 yrs (adult vs. old) | Atrial | Increased glycation | [58] | |
| Human | Both | <55, 55–74, >75 yrs (young, middle aged, old) | Atrial | Reduced CSQ2 expression and increased spontaneous RyR2 activity | [67] | |
| Sarco-endoplasmic reticulum Ca2+-ATPase (SERCa2a) | ||||||
| ↔ | Rabbit | Female | 5–9 mo vs. 4–6 yrs (young adult vs. old) | Ventricular | Unchanged protein levels | [9] |
| Rat | Unreported | 3 vs. 6 mo (young vs. adult) | Ventricular | Unchanged protein levels | [53] | |
| Rat | Male | 5, 15, 26 mo (young adult, adult, old) | Ventricular | Unchanged mRNA levels | [74] | |
| Rabbit | Male | 6 vs. 26 mo (young vs. adult) | Atrial and ventricular | No change in protein expression | [72] | |
| ↓ | Rat | Male | 6 vs. 26 mo (young vs. adult) | Ventricular | Reduced protein expression and pump activity | [76] |
| Rat | Male | 6–8 vs. 26–28 mo (adult vs. old) | Ventricular | Reduced PKA-dependent PLB phosphorylation | [82] | |
| Mouse | Male | 5, 24, and 34 mo (young adult, old, senescent) | Ventricular | Increased PLB expression | [79] | |
| Rat | Male | 6–8 vs. 26–28 mo (adult vs. old) | Ventricular | Depressed activity associated with reduced CaMKII expression | [61] | |
| Rat | Male | 5 vs. 26 mo (young adult vs. old) | Ventricular | Increased 3-Nitrotyrosine modification | [89] | |
| Rat | Male | 2–26 mo (young-old) | Whole heart | Oxidative damage associated with reduced activity | [90] | |
| Mouse | Unreported | 5 vs. 21 mo (adult vs. old) | Ventricular | Increased SERCa2a oxidation | [91] | |
| Human | Both | <55, 55–74, >75 yrs (young, middle aged, old) | Atrial | Decreased expression levels associated with reduced SR Ca2+ content | [67] | |
| L-Type Ca2+ Channel (LTCC) | ||||||
| ↔ | Mouse | Female | 7 vs. 24 mo (adult vs. old) | Ventricular | No alterations in activation or peak ICa | [96] |
| Rabbit | Female | 5–9 mo vs. 4–6 yrs (young adult vs. old) | Ventricular | No change in peak ICa, reduced responsiveness to β-adrenegic stimulation | [9] | |
| ↓ | Mouse | Male | 3 vs. 24 mo (young vs. old) | Ventricular | Reduced ICa density at T-tubules | [110] |
| Rat | Male | 2–3, 8–9, 25–26 mo (young, middle aged, old) | Ventricular | Delayed activation | [93] | |
| Rat | Male | 6 vs. >27 mo (adult vs. old) | Ventricular | Delayed inactivation and reduced peak ICa density | [94] | |
| Rat | Male | 3, 6–8, 24 mo (young, adult, old) | Ventricular | Delayed activation | [95] | |
| Mouse | Male | 7 vs. 24 mo (adult vs. old) | Ventricular | Slower activation and reduced peak ICa | [96] | |
| Rabbit | Male | 6 vs. 26 mo (young vs. adult) | Ventricular | Reduced ICa and maximal conductance, enhanced late component | [72] | |
| Sheep | Female | 18 mo vs. >8 yrs (young vs. old) | Ventricular | Increased peak/integrated ICa | [101] | |
| Dog | Unreported | 2–5, >8 yrs (adult vs. old) | Atrial | Reduced ICa, increased Ito | [97] | |
| Dog | Both | 2–5, >8 yrs (adult vs. old) | Atria | Decreased mRNA and protein expression levels, reduced ICa | [98] | |
| Dog | Unreported | 1–3, >8 yrs (adult vs old) | Atria | Decreased mRNA and protein expression levels, lower peak ICa density | [99,100] | |
| Sheep | Female | 18 mo vs. >8yrs (young adult vs. old) | Atrial | Decreased peak ICa | [102] | |
| Human | Both | <55, 55–74, >75 yrs (young, middle aged, old) | Atrial | Decreased peak ICa | [67] | |
| Na+/Ca+ exchanger (NCX1) | ||||||
| ↔ | Rats | Male | 6 vs. 26 mo (adult vs. old) | Ventricular | Unchanged expression levels | [62] |
| Rabbits | Male | 6 vs. 26 mo (young vs. adult) | Atrial and ventricular | No change in protein expression | [72] | |
| Rabbits | Female | 5–9 mo vs. 4–6 yrs (young adult vs. old) | Ventricular | No change in protein expression | [9] | |
| Rat | Male | 14–15 vs. 27–31 mo (adult vs. old) | Atrial and ventricular | No change in protein expression | [116] | |
| Mice | Unreported | 3 vs. 26–28 mo (young vs. old) | Ventricular | No change in protein levels | [117] | |
| ↑ | Rat | Male | 14–15 vs. 27–31 mo (middle aged vs. old) | Atrial and ventricular | Increased forward activity | [116] |
| Rat | Female | 3 vs. 24 mo (young vs. old) | Ventricular | Increased integrated current | [118] | |
| ↓ | Rat | Male | 4 vs. 24 mo (young vs. old) | Ventricular | Reduced protein expression levels | [114,115] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamilton, S.; Terentyev, D. Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. Int. J. Mol. Sci. 2019, 20, 2386. https://doi.org/10.3390/ijms20102386
Hamilton S, Terentyev D. Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. International Journal of Molecular Sciences. 2019; 20(10):2386. https://doi.org/10.3390/ijms20102386
Chicago/Turabian StyleHamilton, Shanna, and Dmitry Terentyev. 2019. "Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart" International Journal of Molecular Sciences 20, no. 10: 2386. https://doi.org/10.3390/ijms20102386
APA StyleHamilton, S., & Terentyev, D. (2019). Altered Intracellular Calcium Homeostasis and Arrhythmogenesis in the Aged Heart. International Journal of Molecular Sciences, 20(10), 2386. https://doi.org/10.3390/ijms20102386