GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill)

 ,
,
Abstract
1. Introduction
2. Results
2.1. Variation and Heritability in Branch Number of the Soybean Core Collection
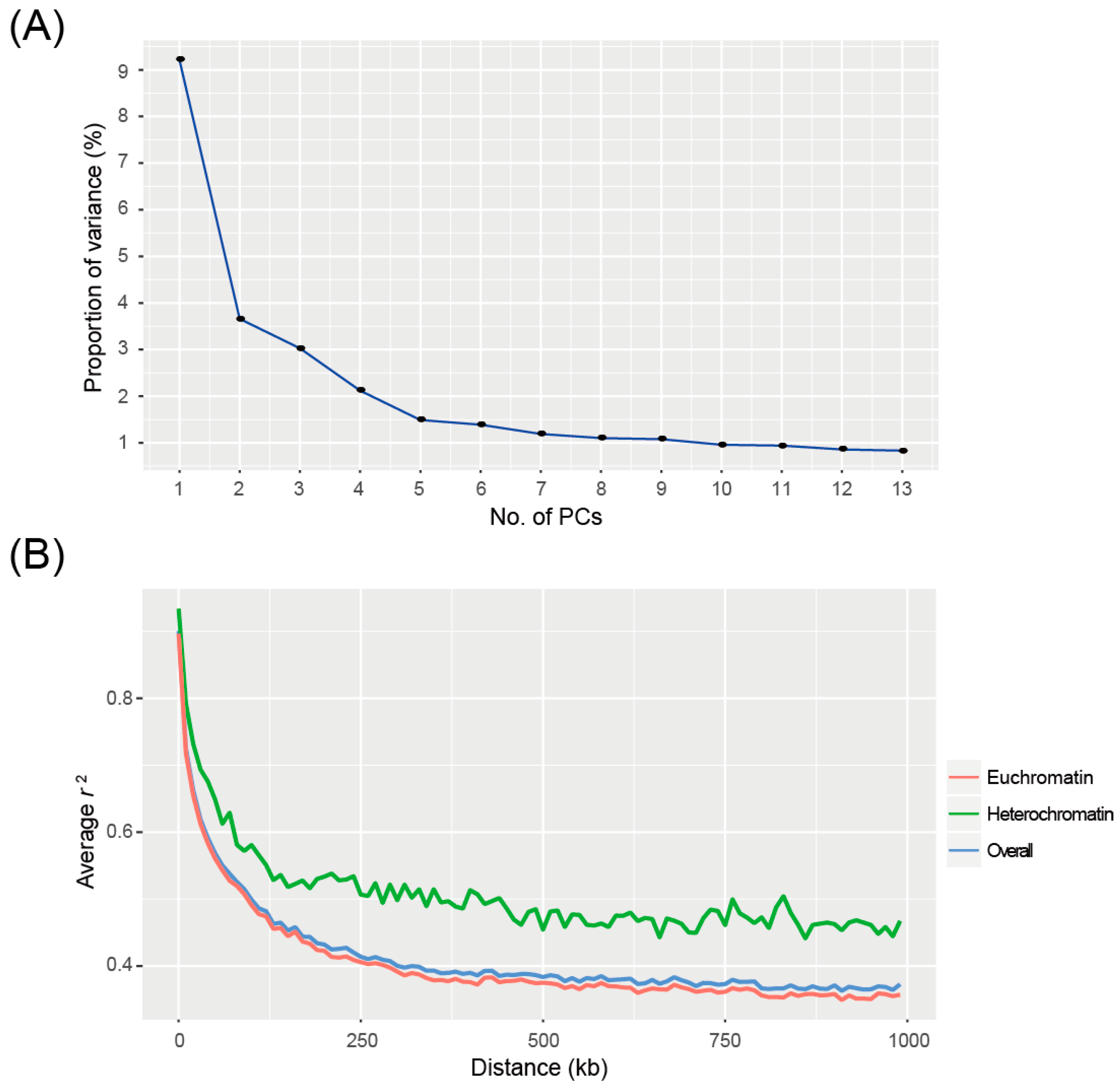
2.2. Population Structure and LD
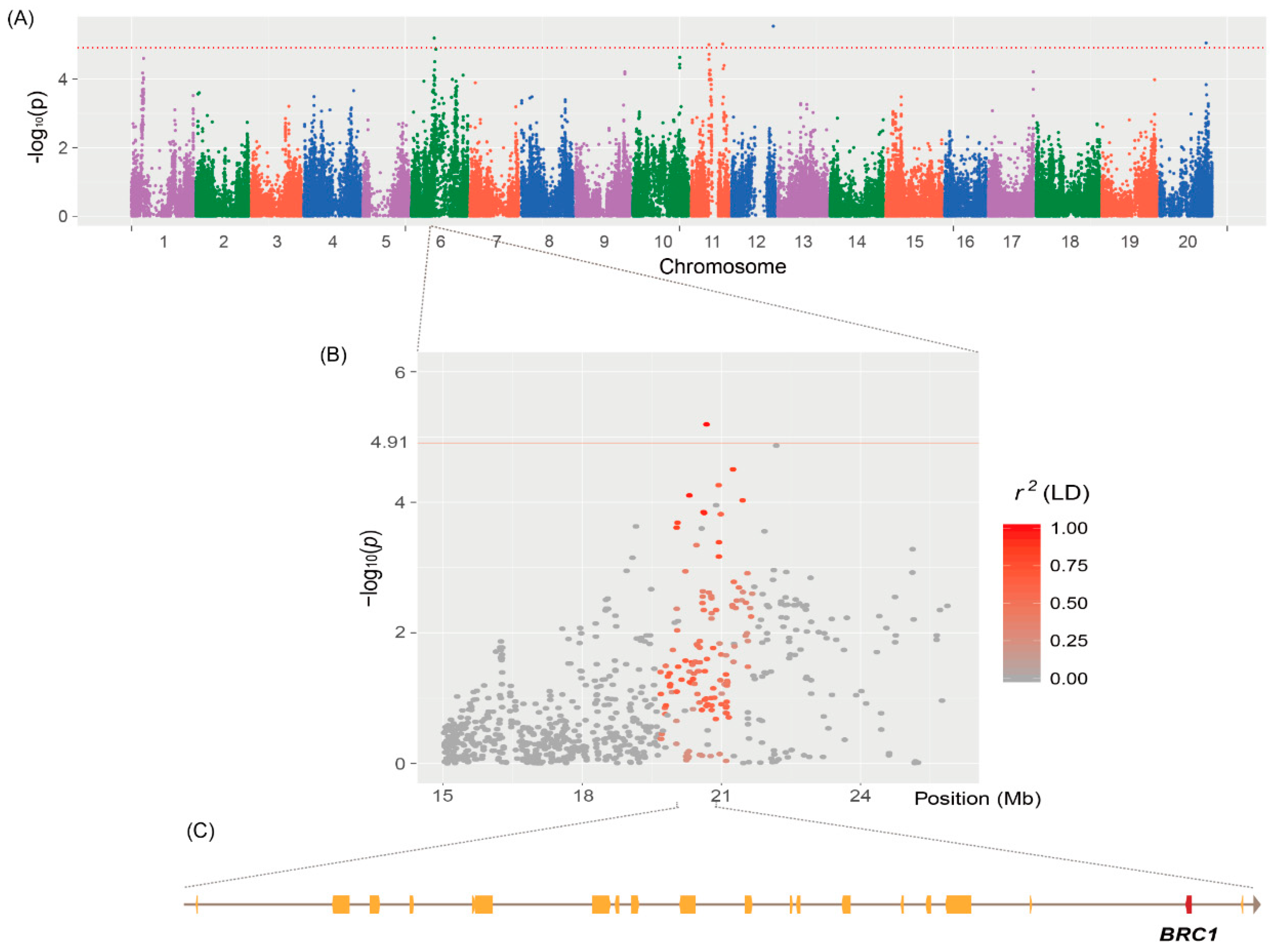
2.3. Determination of Genetic Association with Branch Numbers Using GWAS
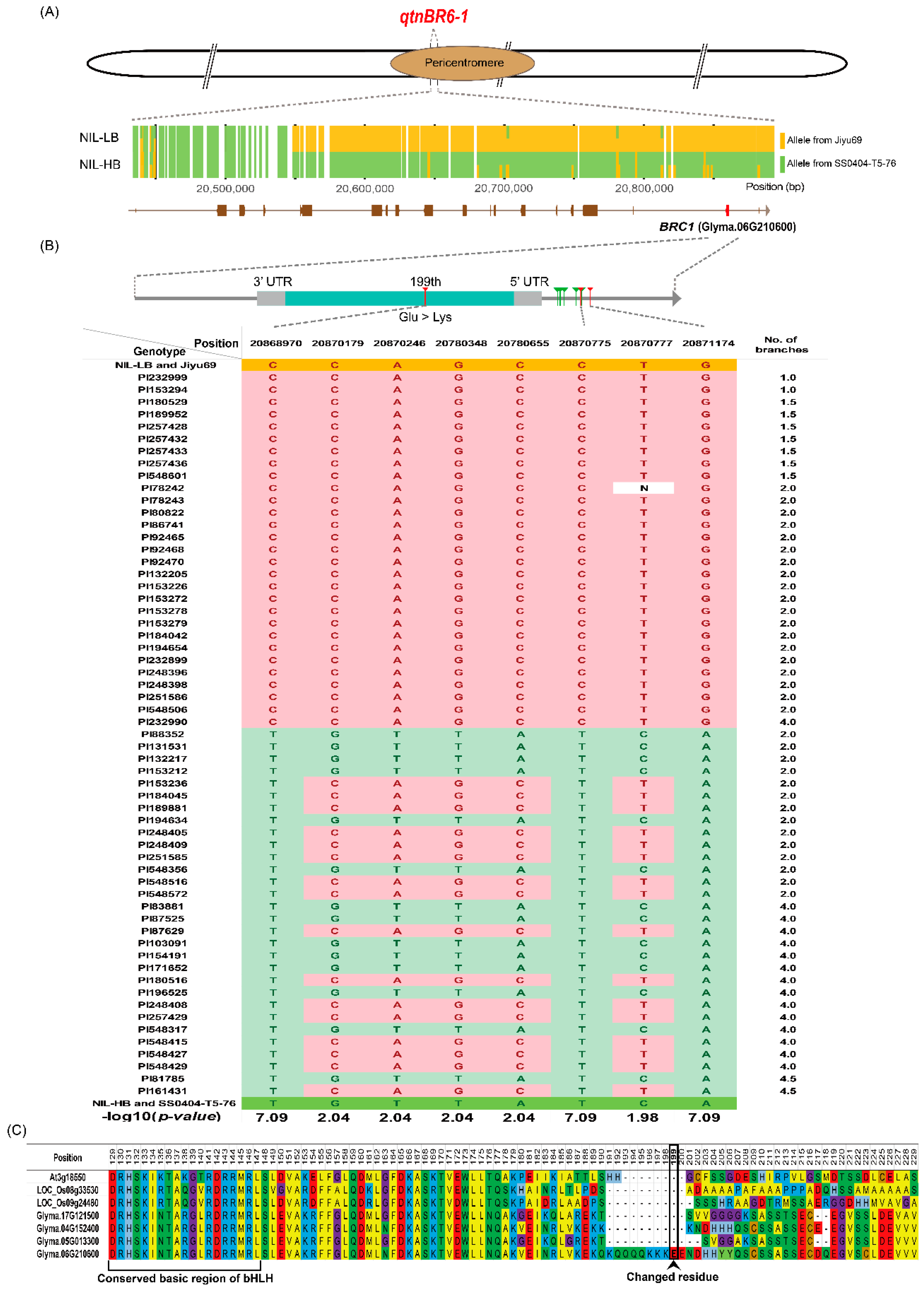
2.4. Isogenicity and Phenotypic Differences Between NIL-HB and NIL-LB Associated with qBR6-1
2.5. Candidate Gene Identification and Allelic Association Analysis
3. Discussion
4. Materials and Methods
4.1. Plant Materials
4.2. Phenotyping of Branch Number and Statistical Analysis
4.3. Population Structure and LD Analysis
4.4. GWAS
4.5. Expression Patterns of Genes in LD Block of qtnBR6-1
4.6. Resequencing of NIL-HB, NIL-LB, and Parental Genotypes
4.7. qRT-PCR Analysis of Candidate Genes in NILs
4.8. Analysis of BRC1 SNPs and Amino Acid Sequence
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| BLUP | Best linear unbiased predictor/prediction |
| BRC1 | BRANCHED1 |
| GLM | Generalized linear model |
| GWAS | Genome-wide association study |
| LD | Linkage disequilibrium |
| MLM | Mixed linear model |
| NIL | Near-isogenic line |
| QTL | Quantitative trait locus |
| QTN | Quantitative trait nucleotide |
| RHL | Residual heterozygous line |
| RIL | Recombinant inbred line |
| SAM | Shoot apical meristem |
| SNP | Single nucleotide polymorphism |
| TCP | TEOSINTE BRANCHED1/CYCLODEA/PROLIFERATING CELL FACTOR 1 and 2 |
References
- Heatherly, L.G.; Elmore, R.W. Managing Inputs for Peak Production. In Soybeans: Improvement, Production, and Uses; Agronomy Monograph 16; American Society of Agronomy, Inc.; Crop Science Society of America, Inc.; Soil Science Society of America, Inc.: Madison, WI, USA, 2004; pp. 451–536. [Google Scholar] [CrossRef]
- Cho, Y.; Kim, S. Growth parameters and seed yield compenets by seeding time and seed density of non-/few branching soybean cultivars in drained paddy field. Asian J. Plant Sci. 2010, 9, 140–145. [Google Scholar]
- Agudamu; Yoshihira, T.; Shiraiwa, T. Branch development responses to planting density and yield stability in soybean cultivars. Plant Prod. Sci. 2016, 19, 331–339. [Google Scholar] [CrossRef]
- Board, J. Light Interception Efficiency and Light Quality Affect Yield Compensation of Soybean at Low Plant Populations. Crop Sci. 2000, 40, 1285–1294. [Google Scholar] [CrossRef]
- Cox, W.J.; Cherney, J.H.; Shields, E. Soybeans Compensate at Low Seeding Rates but not at High Thinning Rates. Agron. J. 2010, 102, 1238–1243. [Google Scholar] [CrossRef]
- Board, J.E.; Kahlon, C.S. Morphological Responses to Low Plant Population Differ Between Soybean Genotypes. Crop Sci. 2013, 53, 1109–1119. [Google Scholar] [CrossRef]
- Sayama, T.; Hwang, T.-Y.; Yamazaki, H.; Yamaguchi, N.; Komatsu, K.; Takahashi, M.; Suzuki, C.; Miyoshi, T.; Tanaka, Y.; Xia, Z.; et al. Mapping and comparison of quantitative trait loci for soybean branching phenotype in two locations. Breeding Sci. 2010, 60, 380–389. [Google Scholar] [CrossRef]
- Shim, S.; Kim, M.Y.; Ha, J.; Lee, Y.-H.; Lee, S.-H. Identification of QTLs for branching in soybean (Glycine max (L.) Merrill). Euphytica 2017, 213, 225. [Google Scholar] [CrossRef]
- Yao, D.; Liu, Z.Z.; Zhang, J.; Liu, S.Y.; Qu, J.; Guan, S.Y.; Pan, L.D.; Wang, D.; Liu, J.W.; Wang, P.W. Analysis of quantitative trait loci for main plant traits in soybean. Genet. Mol. Res. 2015, 14, 6101–6109. [Google Scholar] [CrossRef]
- Chen, Q.; Zhang, Z.; Liu, C.; Xin, D.; Qiu, H.; Shan, D.; Shan, C.; Hu, G. QTL Analysis of Major Agronomic Traits in Soybean. Agr. Sci. China 2007, 6, 399–405. [Google Scholar] [CrossRef]
- Li, W.; Zheng, D.; Van, K.; Lee, S. QTL Mapping for Major Agronomic Traits across Two Years in Soybean (Glycine max L. Merr.). J. Crop Sci. Biotech. 2008, 11, 171. [Google Scholar]
- Rafalski, J.A. Association genetics in crop improvement. Curr. Opin. Plant Biol. 2010, 13, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Tung, C.-W.; Eizenga, G.C.; Wright, M.H.; Ali, M.L.; Price, A.H.; Norton, G.J.; Islam, M.R.; Reynolds, A.; Mezey, J.; et al. Genome-wide association mapping reveals a rich genetic architecture of complex traits in Oryza sativa. Nat. Commun. 2011, 2, 467. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Bradbury, P.J.; Brown, P.J.; Hung, H.; Sun, Q.; Flint-Garcia, S.; Rocheford, T.R.; McMullen, M.D.; Holland, J.B.; Buckler, E.S. Genome-wide association study of leaf architecture in the maize nested association mapping population. Nat. Genet. 2011, 43, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Aranzana, M.J.; Kim, S.; Lister, C.; Shindo, C.; Tang, C.; Toomajian, C.; Zheng, H.; Dean, C.; Marjoram, P.; et al. An Arabidopsis Example of Association Mapping in Structured Samples. PLoS Genet. 2007, 3, e4. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Li, S.; Wang, Z.; Chang, F.; Kong, J.; Gai, J.; Zhao, T. Identification of Major Quantitative Trait Loci for Seed Oil Content in Soybeans by Combining Linkage and Genome-Wide Association Mapping. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Zhang, J.; Song, Q.; Cregan, P.B.; Nelson, R.L.; Wang, X.; Wu, J.; Jiang, G.-L. Genome-wide association study for flowering time, maturity dates and plant height in early maturing soybean (Glycine max) germplasm. BMC Genomics 2015, 16, 217. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- Schmutz, J.; Cannon, S.B.; Schlueter, J.; Ma, J.; Mitros, T.; Nelson, W.; Hyten, D.L.; Song, Q.; Thelen, J.J.; Cheng, J.; et al. Genome sequence of the palaeopolyploid soybean. Nature 2010, 463, 178–183. [Google Scholar] [CrossRef]
- Aguilar-Martínez, J.A.; Poza-Carrión, C.; Cubas, P. Arabidopsis BRANCHED1 Acts as an Integrator of Branching Signals within Axillary Buds. Plant Cell 2007, 19, 458–472. [Google Scholar] [CrossRef]
- Poza-Carrión, C.; Aguilar-Martínez, J.A.; Cubas, P. Role of TCP Gene BRANCHED1 in the Control of Shoot Branching in Arabidopsis. Plant Signal Behav. 2007, 2, 551–552. [Google Scholar] [CrossRef] [PubMed]
- Ghodrati, G. Study of genetic variation and broad sense heritability for some qualitative and quantitative traits in soybean (Glycine max L.) genotypes. Curr. Opin. Agr. 2013, 2. [Google Scholar]
- Rameau, C.; Bertheloot, J.; Leduc, N.; Andrieu, B.; Foucher, F.; Sakr, S. Multiple pathways regulate shoot branching. Front Plant Sci. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Board, J.E.; Settimi, J.R. Photoperiod Effect before and after Flowering on Branch Development in Determinate Soybean. Agron. J. 1986, 78, 995–1002. [Google Scholar] [CrossRef]
- Frederick, J.R.; Camp, C.R.; Bauer, P.J. Drought-Stress Effects on Branch and Mainstem Seed Yield and Yield Components of Determinate Soybean. Crop Sci. 2001, 41, 759–763. [Google Scholar] [CrossRef]
- Linkemer, G.; Board, J.E.; Musgrave, M.E. Waterlogging effects on growth and yield components in late-planted soybean. Crop Sci. 1998, 38, 1576–1584. [Google Scholar] [CrossRef] [PubMed]
- Thimann, K.V.; Skoog, F. Studies on the growth hormone of plants the inhibiting action of the growth substance on bud development. Proc. Natl. Acad. Sci. USA 1933, 19, 714–716. [Google Scholar] [CrossRef] [PubMed]
- Niwa, M.; Daimon, Y.; Kurotani, K.; Higo, A.; Pruneda-Paz, J.L.; Breton, G.; Mitsuda, N.; Kay, S.A.; Ohme-Takagi, M.; Endo, M.; et al. BRANCHED1 Interacts with FLOWERING LOCUS T to Repress the Floral Transition of the Axillary Meristems in Arabidopsis[C][W][OA]. Plant Cell 2013, 25, 1228–1242. [Google Scholar] [CrossRef]
- Braun, N.; Germain, A. de S.; Pillot, J.-P.; Boutet-Mercey, S.; Dalmais, M.; Antoniadi, I.; Li, X.; Maia-Grondard, A.; Signor, C.L.; Bouteiller, N.; et al. The Pea TCP Transcription Factor PsBRC1 Acts Downstream of Strigolactones to Control Shoot Branching. Plant Physiol. 2012, 158, 225–238. [Google Scholar] [CrossRef]
- Doebley, J.; Stec, A.; Hubbard, L. The evolution of apical dominance in maize. Nature 1997, 386, 485–488. [Google Scholar] [CrossRef]
- Takeda, T.; Suwa, Y.; Suzuki, M.; Kitano, H.; Ueguchi-Tanaka, M.; Ashikari, M.; Matsuoka, M.; Ueguchi, C. The OsTB1 gene negatively regulates lateral branching in rice. Plant J. 2003, 33, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Finlayson, S.A. Arabidopsis TEOSINTE BRANCHED1-LIKE 1 Regulates Axillary Bud Outgrowth and is Homologous to Monocot TEOSINTE BRANCHED1. Plant Cell Physiol. 2007, 48, 667–677. [Google Scholar] [CrossRef] [PubMed]
- Kebrom, T.H.; Brutnell, T.P.; Finlayson, S.A. Suppression of sorghum axillary bud outgrowth by shade, phyB and defoliation signalling pathways. Plant Cell Environ. 2010, 33, 48–58. [Google Scholar] [CrossRef]
- Minakuchi, K.; Kameoka, H.; Yasuno, N.; Umehara, M.; Luo, L.; Kobayashi, K.; Hanada, A.; Ueno, K.; Asami, T.; Yamaguchi, S.; et al. FINE CULM1 (FC1) Works Downstream of Strigolactones to Inhibit the Outgrowth of Axillary Buds in Rice. Plant Cell Physiol. 2010, 51, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Cubas, P.; Lauter, N.; Doebley, J.; Coen, E. The TCP domain: a motif found in proteins regulating plant growth and development. Plant J. 1999, 18, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Sorefan, K.; Booker, J.; Haurogné, K.; Goussot, M.; Bainbridge, K.; Foo, E.; Chatfield, S.; Ward, S.; Beveridge, C.; Rameau, C.; et al. MAX4 and RMS1 are orthologous dioxygenase-like genes that regulate shoot branching in Arabidopsis and pea. Genes Dev. 2003, 17, 1469–1474. [Google Scholar] [CrossRef]
- Clark, R.M.; Linton, E.; Messing, J.; Doebley, J.F. Pattern of diversity in the genomic region near the maize domestication gene tb1. Proc. Natl. Acad. Sci. USA 2004, 101, 700–707. [Google Scholar] [CrossRef]
- Clark, R.M.; Wagler, T.N.; Quijada, P.; Doebley, J. A distant upstream enhancer at the maize domestication gene tb1 has pleiotropic effects on plant and inflorescent architecture. Nat. Genet. 2006, 38, 594–597. [Google Scholar] [CrossRef]
- Bates, D.; Sarkar, D. lme4: Linear mixed-effects models using S4 classes. 2007. [Google Scholar]
- Lee, Y.-G.; Jeong, N.; Kim, J.H.; Lee, K.; Kim, K.H.; Pirani, A.; Ha, B.-K.; Kang, S.-T.; Park, B.-S.; Moon, J.-K.; et al. Development, validation and genetic analysis of a large soybean SNP genotyping array. Plant J. 2015, 81, 625–636. [Google Scholar] [CrossRef]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Bradbury, P.J.; Zhang, Z.; Kroon, D.E.; Casstevens, T.M.; Ramdoss, Y.; Buckler, E.S. TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 2007, 23, 2633–2635. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. The variant call format and VCFtools. Bioinformatics 2011, 27, 2156–2158. [Google Scholar] [CrossRef]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Nijveen, H.; Rao, X.; Bisseling, T.; Geurts, R.; Leunissen, J.A.M. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 2007, 35, W71–W74. [Google Scholar] [CrossRef] [PubMed]
- Rotmistrovsky, K.; Jang, W.; Schuler, G.D. A web server for performing electronic PCR. Nucleic Acids Res. 2004, 32, W108–W112. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| QTN ID | Marker ID | Chr a | Marker Position (bp) | p-Value | Phenotypic R2 (%) | Chromosomal Location | Linkage Disequilibrium Block | No. of Genes | Known QTL | Reference b | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| qtnBR6-1 | AX-90305605 | 6 | 20,663,101 | 6.43 × 10−6 | 5.8 | Heterochromatin | 20,433,101 | 20,893,101 | 20 | qBR6-1 | [8] |
| qtnBR11-1 | AX-90512426 | 11 | 16,074,992 | 9.98 × 10−6 | 5.0 | Heterochromatin | 15,844,992 | 16,304,992 | 13 | qBR11-1 | [8] |
| qtnBR11-2 | AX-90472718 | 11 | 28,613,118 | 9.51 × 10−6 | 5.6 | Heterochromatin | 28,383,118 | 28,843,118 | 23 | qBR11-1 | [8] |
| qtnBR12-1 | AX-90419363 | 12 | 38,057,780 | 2.89 × 10−6 | 6.4 | Euchromatin | 37,987,780 | 38,127,780 | 14 | ||
| qtnBR20-1 | AX-90519199 | 20 | 42,471,316 | 8.88 × 10−6 | 4.9 | Euchromatin | 42,401,316 | 42,541,316 | 13 | ||
| Year | Planting Density | Growth Condition | NIL-HB | NIL-LB | p-Value |
|---|---|---|---|---|---|
| 2017 | Low | Greenhouse | 5.8 ± 1.7 | 3.4 ± 0.7 | 0.0001 |
| Field | 14.5 ± 1.1 | 12.2 ± 2.1 | 0.019 | ||
| 2018 | Low | Field | 7.1 ± 1.4 | 5.1 ± 1.6 | 0.0003 |
| High | 3.3 ± 1.9 | 2.6 ± 0.8 | 0.057 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shim, S.; Ha, J.; Kim, M.Y.; Choi, M.S.; Kang, S.-T.; Jeong, S.-C.; Moon, J.-K.; Lee, S.-H. GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill). Int. J. Mol. Sci. 2019, 20, 135. https://doi.org/10.3390/ijms20010135
Shim S, Ha J, Kim MY, Choi MS, Kang S-T, Jeong S-C, Moon J-K, Lee S-H. GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill). International Journal of Molecular Sciences. 2019; 20(1):135. https://doi.org/10.3390/ijms20010135
Chicago/Turabian StyleShim, Sangrea, Jungmin Ha, Moon Young Kim, Man Soo Choi, Sung-Taeg Kang, Soon-Chun Jeong, Jung-Kyung Moon, and Suk-Ha Lee. 2019. "GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill)" International Journal of Molecular Sciences 20, no. 1: 135. https://doi.org/10.3390/ijms20010135
APA StyleShim, S., Ha, J., Kim, M. Y., Choi, M. S., Kang, S.-T., Jeong, S.-C., Moon, J.-K., & Lee, S.-H. (2019). GmBRC1 is a Candidate Gene for Branching in Soybean (Glycine max (L.) Merrill). International Journal of Molecular Sciences, 20(1), 135. https://doi.org/10.3390/ijms20010135