NFκB Inhibition Mitigates Serum Amyloid A-Induced Pro-Atherogenic Responses in Endothelial Cells and Leukocyte Adhesion and Adverse Changes to Endothelium Function in Isolated Aorta

 ,
, 


Abstract
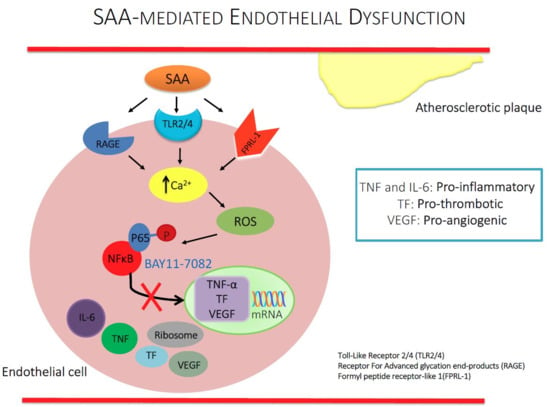
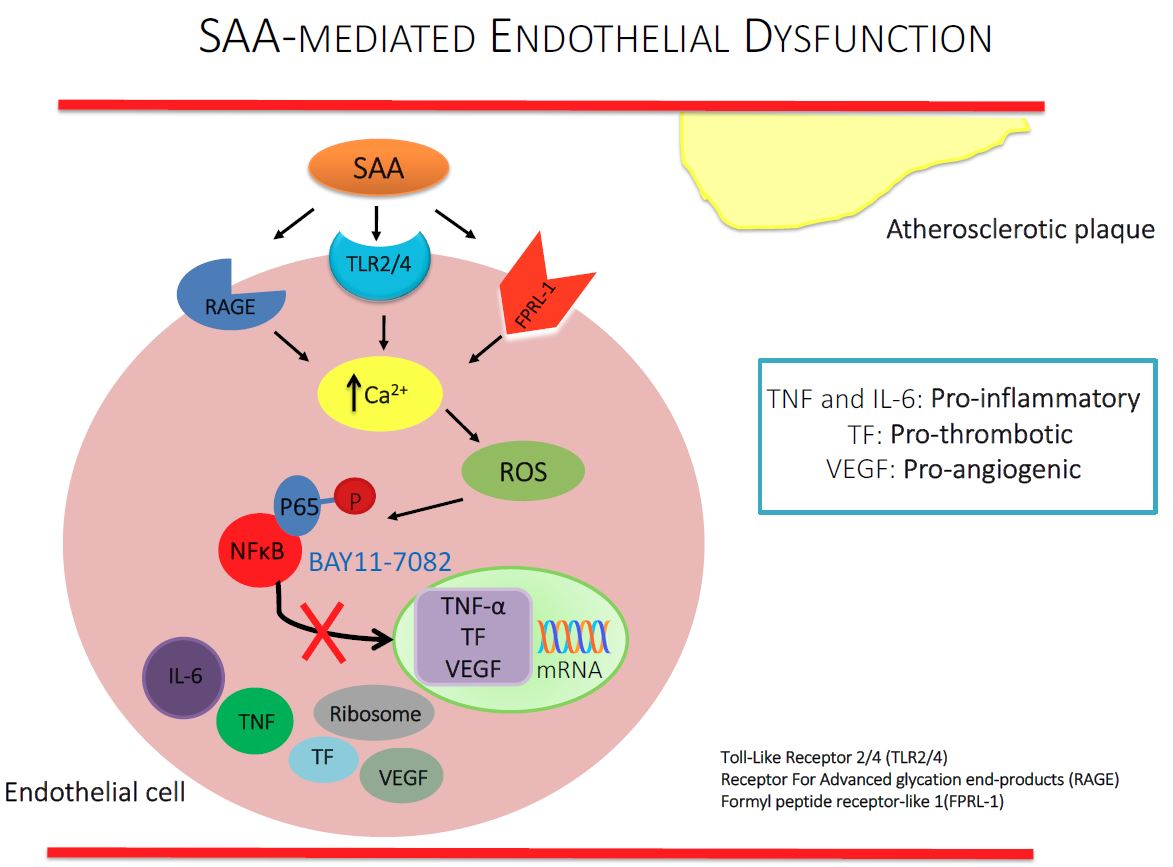
1. Introduction
2. Results
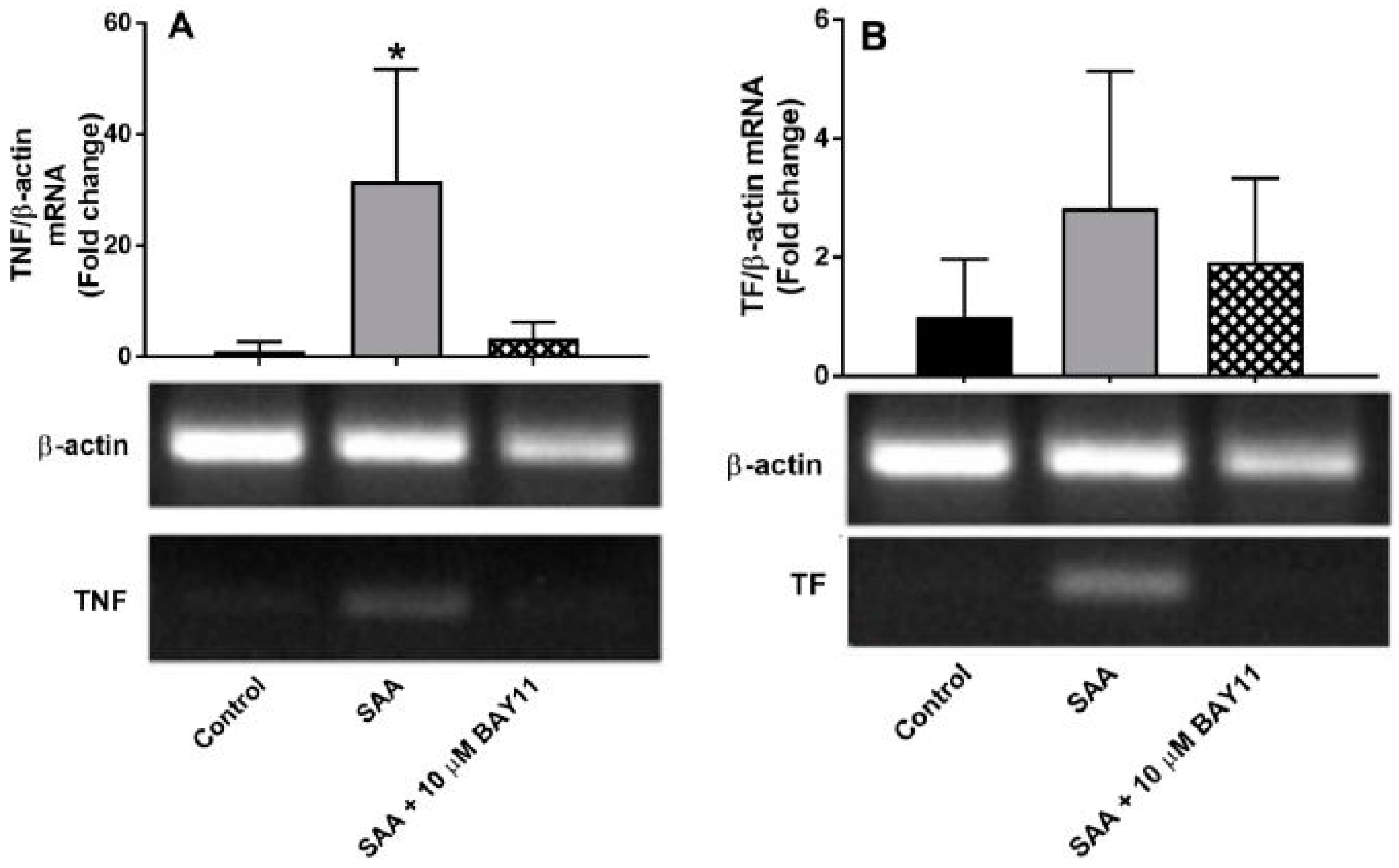
2.1. Gene Expression
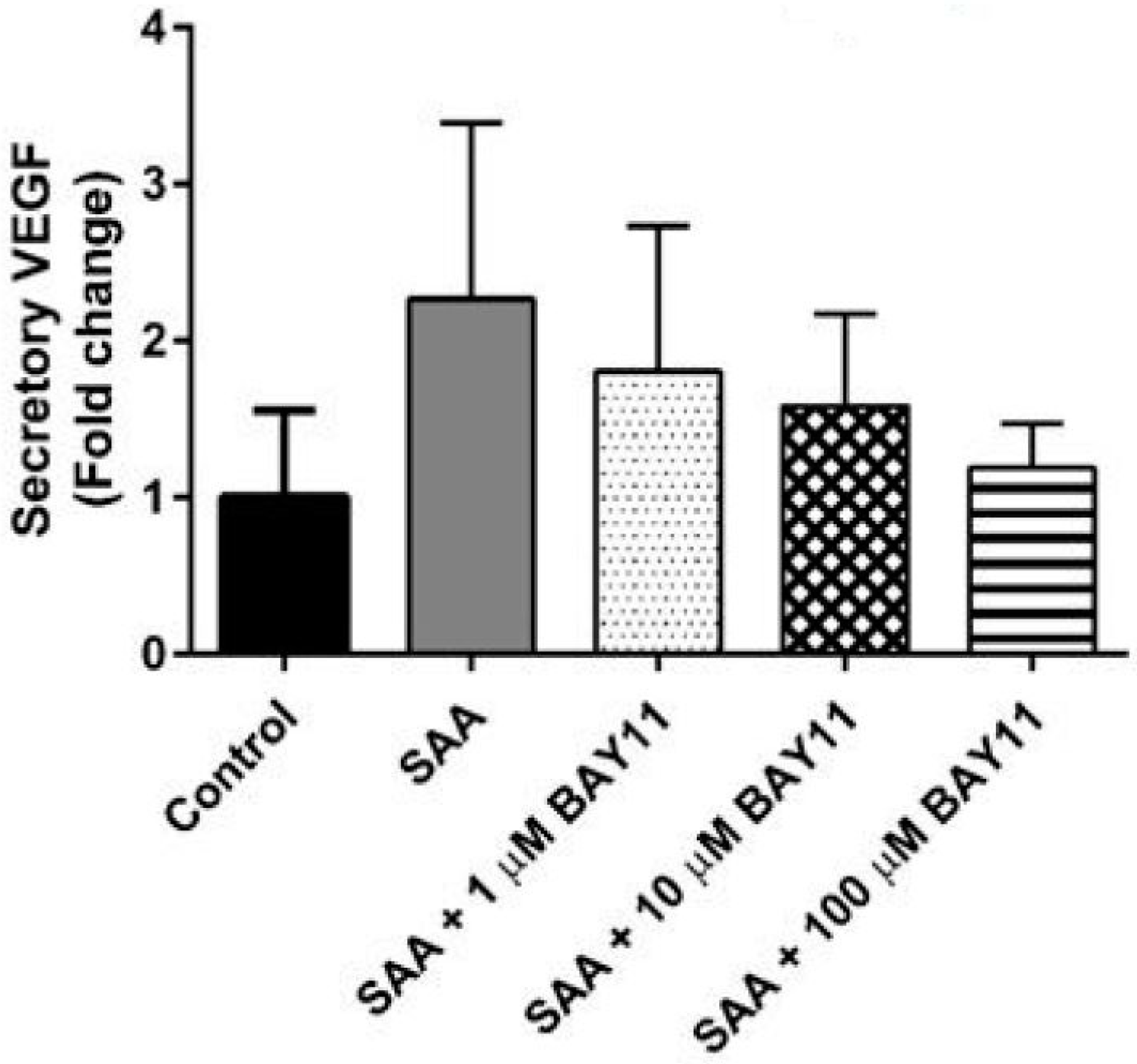
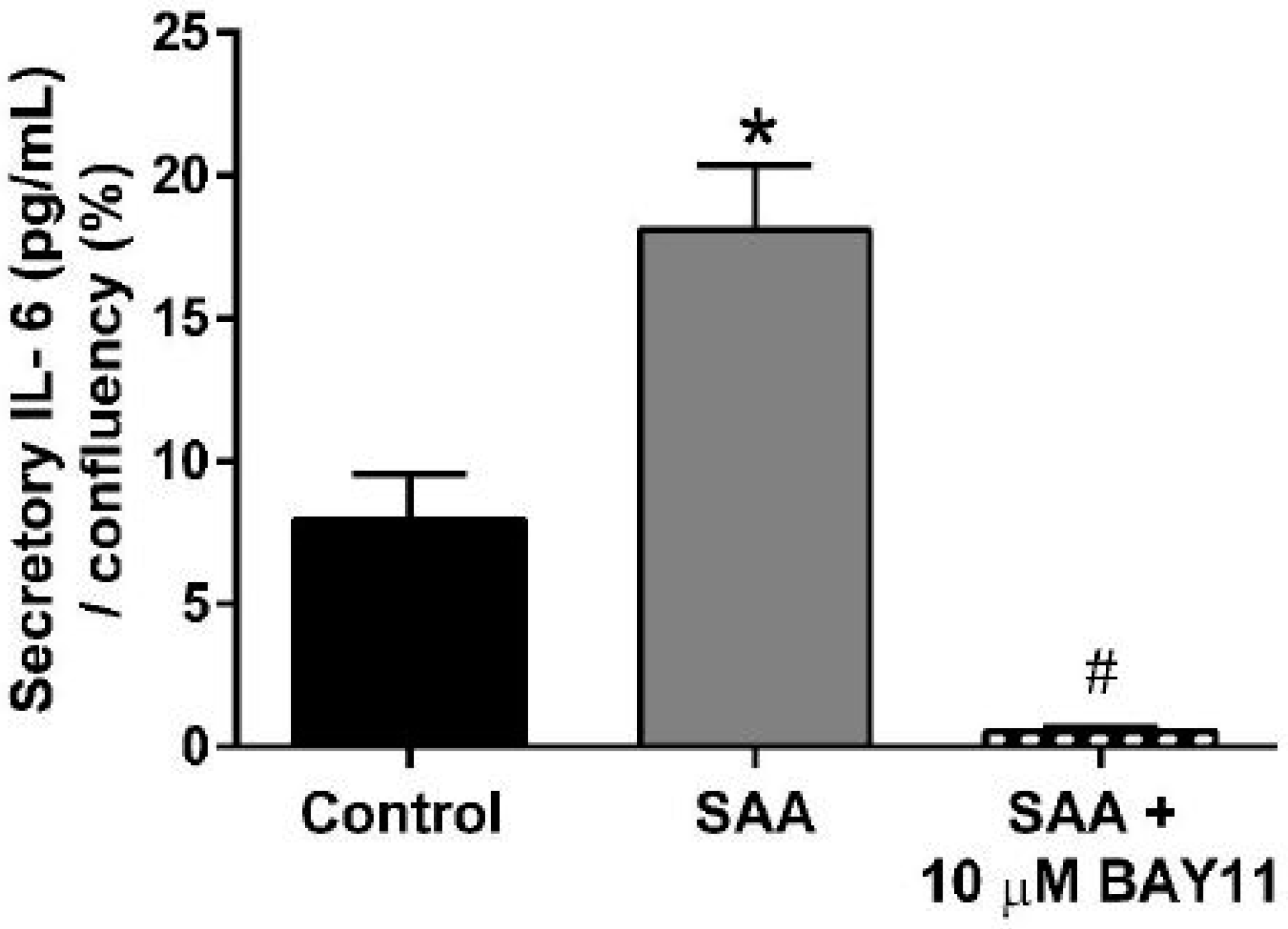
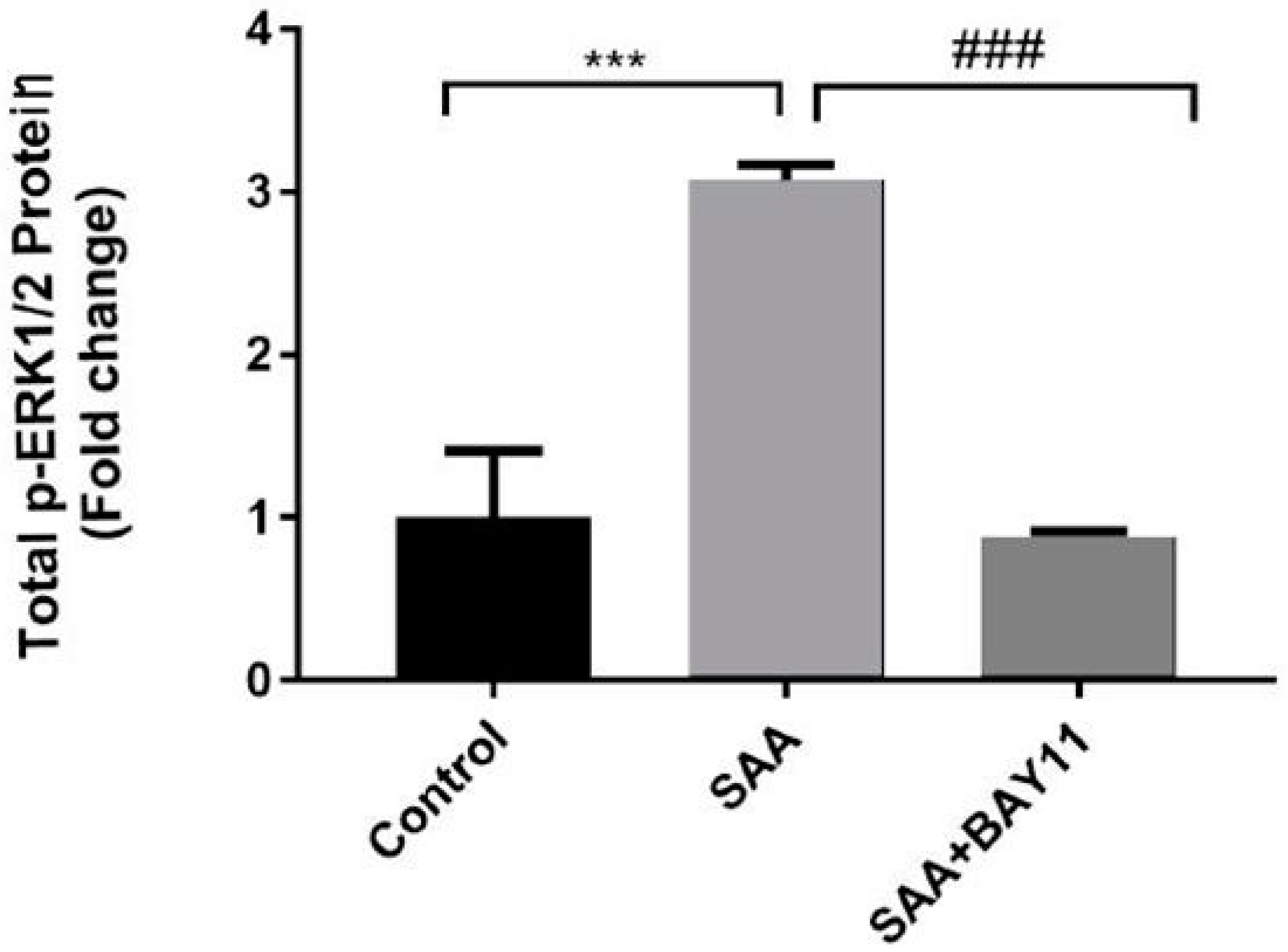
2.2. Inflammatory Protein Expression
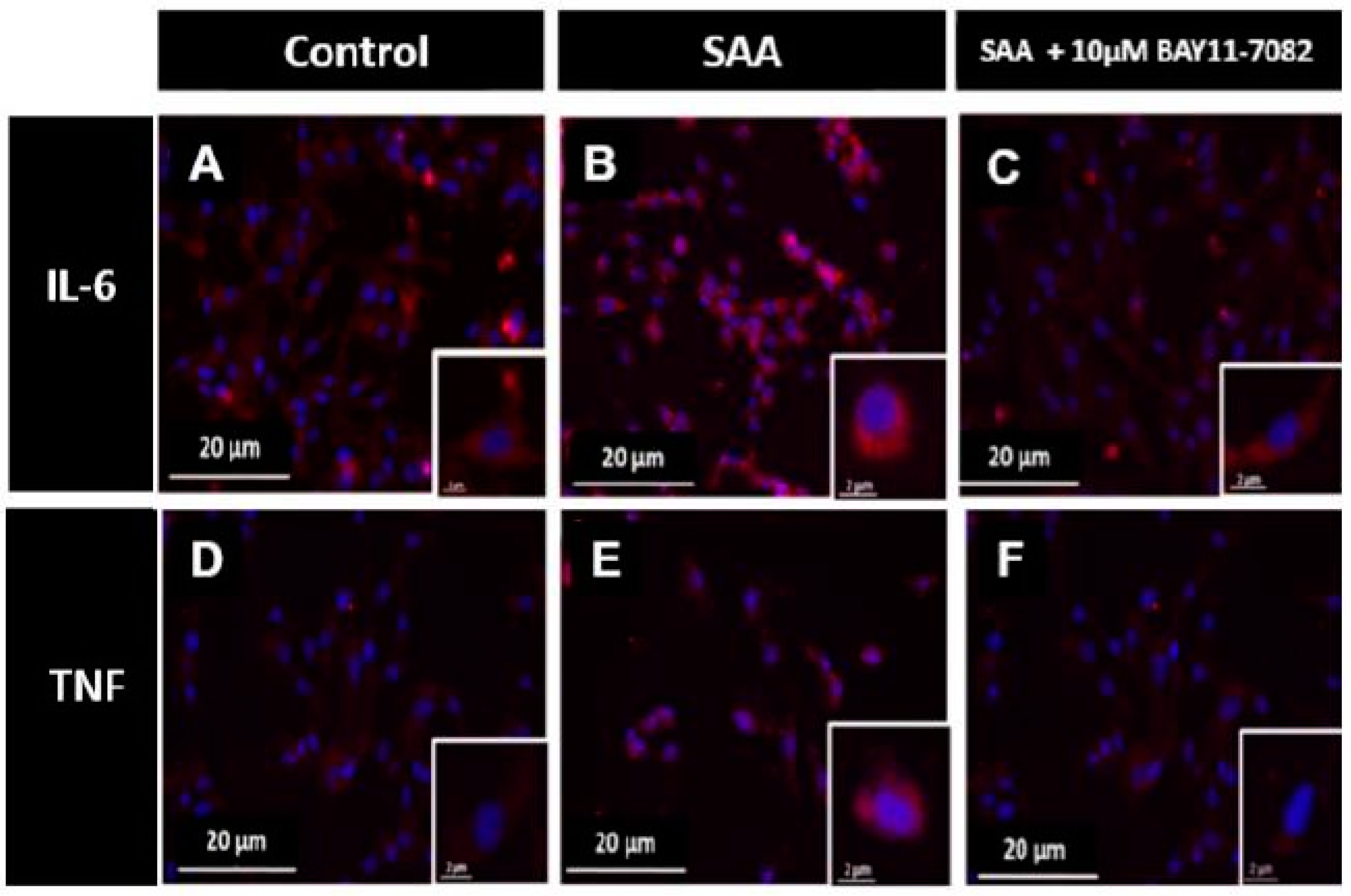
2.3. Immunocytochemistry Analysis
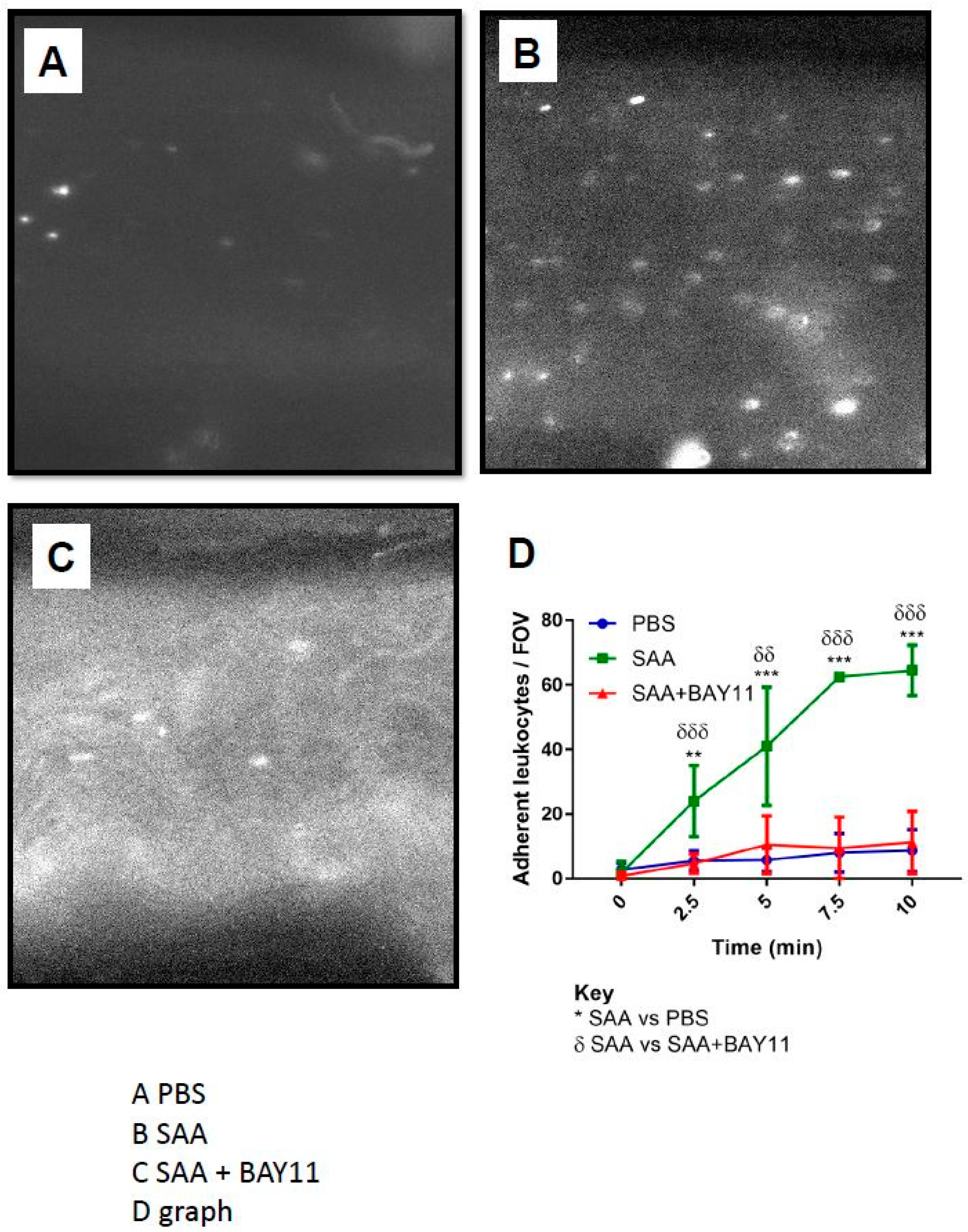
2.4. Leukocyte Endothelial Adhesion
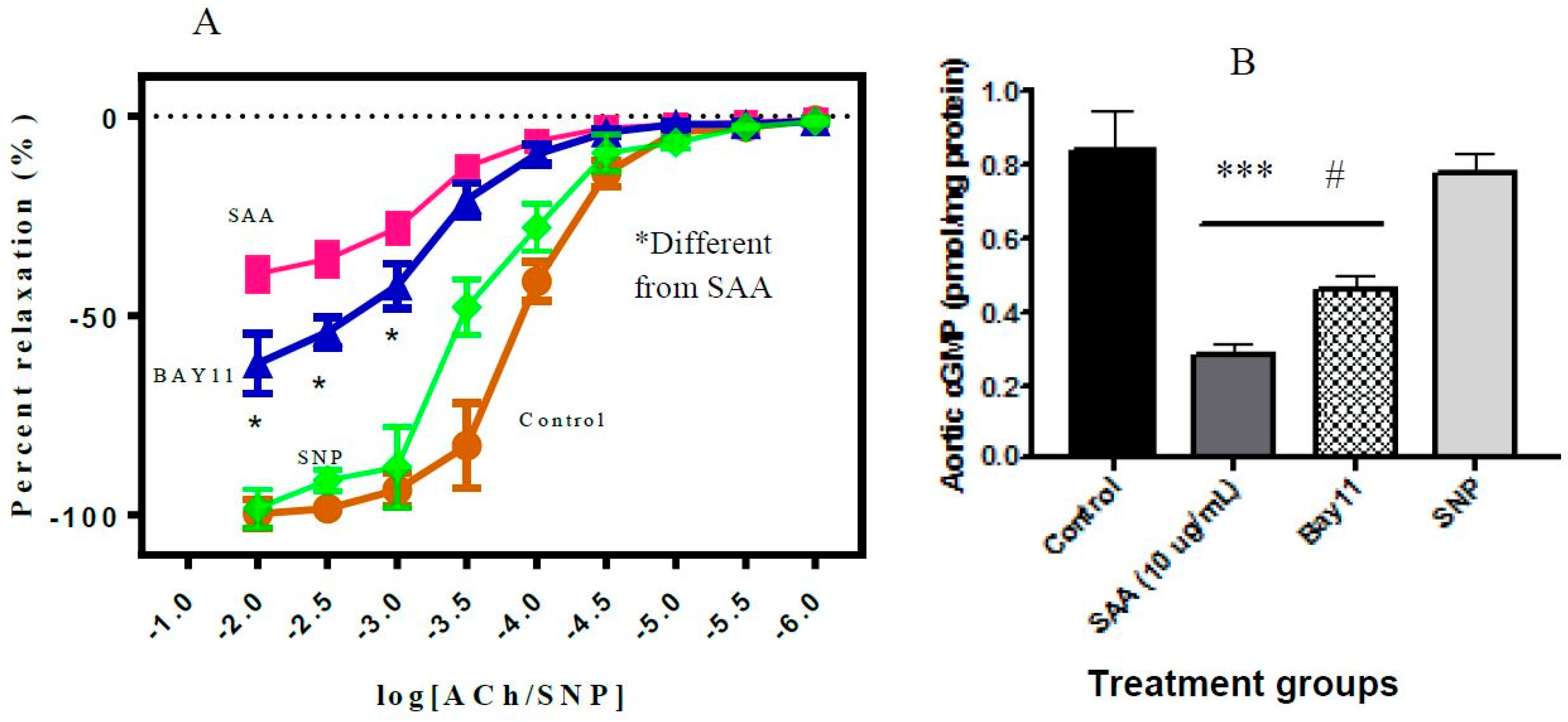
2.5. Vascular Function Test
3. Discussion
4. Limitations for the Study
5. Conclusions
6. Materials and Methods
6.1. Materials
6.2. Cell Culture
6.3. Dose Selection for In Vitro Cell Culture Experiments with BAY11-7082
6.4. Gene Analysis
6.5. Sandwich Enzyme-Linked Immunosorbent Assay (ELISA)
6.6. Immunofluorescence
6.7. Animal Studies
6.7.1. Leukocyte Adhesion
6.7.2. Vascular Function Studies
6.8. Measurement of Aortic cGMP
6.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galley, H.F.; Webster, N.R. Physiology of the endothelium. Br. J. Anaesth. 2004, 93, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.R.; Witting, P.K.; Drummond, G.R. Redox control of endothelial function and dysfunction: Molecular mechanisms and therapeutic opportunities. Antioxid. Redox Signal. 2008, 10, 1713–1765. [Google Scholar] [CrossRef] [PubMed]
- Stocker, R.; Keaney, J.F., Jr. Role of oxidative modifications in atherosclerosis. Physiol. Rev. 2004, 84, 1381–1478. [Google Scholar] [CrossRef] [PubMed]
- Maron, B.A.; Zhang, Y.Y.; White, K.; Chan, S.Y.; Handy, D.E.; Mahoney, C.E.; Loscalzo, J.; Leopold, J.A. Aldosterone inactivates the endothelin-B receptor via a cysteinyl thiol redox switch to decrease pulmonary endothelial nitric oxide levels and modulate pulmonary arterial hypertension. Circulation 2012, 126, 963–974. [Google Scholar] [CrossRef] [PubMed]
- van Bussel, B.C.; Soedamah-Muthu, S.S.; Henry, R.M.; Schalkwijk, C.G.; Ferreira, I.; Chaturvedi, N.; Toeller, M.; Fuller, J.H.; Stehouwer, C.D. Unhealthy dietary patterns associated with inflammation and endothelial dysfunction in type 1 diabetes: The EURODIAB study. Nutr. Metab. Cardiovasc. Dis. 2013, 23, 758–764. [Google Scholar] [CrossRef] [PubMed]
- Murdaca, G.; Colombo, B.M.; Cagnati, P.; Gulli, R.; Spano, F.; Puppo, F. Endothelial dysfunction in rheumatic autoimmune diseases. Atherosclerosis 2012, 224, 309–317. [Google Scholar] [CrossRef]
- Hua, S.; Song, C.; Geczy, C.L.; Freedman, S.B.; Witting, P.K. A role for acute-phase serum amyloid A and high-density lipoprotein in oxidative stress, endothelial dysfunction and atherosclerosis. Redox. Rep. 2009, 14, 187–196. [Google Scholar] [CrossRef]
- Selinger, M.J.; McAdam, K.P.; Kaplan, M.M.; Sipe, J.D.; Vogel, S.N.; Rosenstreich, D.L. Monokine-induced synthesis of serum amyloid A protein by hepatocytes. Nature 1980, 285, 498–500. [Google Scholar] [CrossRef]
- Lakota, K.; Lakota, K.; Resnik, N.; Mrak-Poljsak, K.; Sodin-Semrl, S.; Veranic, P. Colocalization of Serum Amyloid A with Microtubules in Human Coronary Artery Endothelial Cells. J. Biomed. Biotechnol. 2011, 2011, 8. [Google Scholar] [CrossRef]
- Kumon, Y.; Suehiro, T.; Itahara, T.; Ikeda, Y.; Hashimoto, K. Serum amyloid A protein in patients with non-insulin-dependent diabetes mellitus. Clin. Biochem. 1994, 27, 469–473. [Google Scholar] [CrossRef]
- Maier, W.; Altwegg, L.A.; Corti, R.; Gay, S.; Hersberger, M.; Maly, F.E.; Sutsch, G.; Roffi, M.; Neidhart, M.; Eberli, F.R. Inflammatory markers at the site of ruptured plaque in acute myocardial infarction: Locally increased interleukin-6 and serum amyloid A but decreased C-reactive protein. Circulation 2005, 111, 1355–1361. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Miida, T. Serum amyloid A remains at physiological concentrations in coronary atherosclerosis. Clin. Chem. 1997, 43, 193. [Google Scholar] [PubMed]
- Witting, P.K.; Song, C.; Hsu, K.; Hua, S.; Parry, S.N.; Aran, R.; Geczy, C.; Freedman, S.B. The acute-phase protein serum amyloid A induces endothelial dysfunction that is inhibited by high-density lipoprotein. Free Radic. Biol. Med. 2011, 51, 1390–1398. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Witting, P.K.; Stocker, R. Oxidants and antioxidants in atherosclerosis. Curr. Opin. Lipidol. 2001, 12, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Shen, Y.; Yamen, E.; Hsu, K.; Yan, W.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A induction of cytokines in monocytes/macrophages and lymphocytes. Atherosclerosis 2009, 207, 374–383. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.Z.; Ooi, D.S.; Shen, H.M.; Heng, C.K. The atherogenic effects of serum amyloid A are potentially mediated via inflammation and apoptosis. J. Atheroscler. Thromb. 2014, 21, 854–867. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Shen, Y.; Yamen, E.; Hsu, K.; Yan, W.; Witting, P.K.; Geczy, C.L.; Freedman, S.B. Serum amyloid A may potentiate prothrombotic and proinflammatory events in acute coronary syndromes. Atherosclerosis 2009, 202, 596–604. [Google Scholar] [CrossRef]
- Dong, Z.; Wu, T.; Qin, W.; An, C.; Wang, Z.; Zhang, M.; Zhang, Y.; Zhang, C.; An, F. Serum amyloid A directly accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Mol. Med. 2011, 17, 1357–1364. [Google Scholar] [CrossRef]
- Hartman, J.; Frishman, W.H. Inflammation and atherosclerosis: A review of the role of interleukin-6 in the development of atherosclerosis and the potential for targeted drug therapy. Cardiol. Rev. 2014, 22, 147–151. [Google Scholar] [CrossRef]
- Lee, H.Y.; Kim, S.D.; Baek, S.H.; Choi, J.H.; Bae, Y.S. Role of formyl peptide receptor 2 on the serum amyloid A-induced macrophage foam cell formation. Biochem. Biophys. Res. Commun. 2013, 433, 255–259. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J. Free Radicals in Biology and Medicine; Oxford University Press: New York, NY, USA, 2007. [Google Scholar]
- Cai, X.; Freedman, S.B.; Witting, P.K. Serum amyloid A stimulates cultured endothelial cells to migrate and proliferate: Inhibition by the multikinase inhibitor BIBF1120. Clin. Exp. Pharmacol. Physiol. 2013, 40, 662–670. [Google Scholar] [CrossRef] [PubMed]
- Chami, B.; Barrie, N.; Cai, X.; Wang, X.; Paul, M.; Morton-Chandra, R.; Sharland, A.; Dennis, J.M.; Freedman, S.B.; Witting, P.K. Serum Amyloid A Receptor Blockade and Incorporation into High-Density Lipoprotein Modulates Its Pro-Inflammatory and Pro-Thrombotic Activities on Vascular Endothelial Cells. Int. J. Mol. Sci. 2015, 16, 11101–11124. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Song, C.; Endoh, I.; Goyette, J.; Jessup, W.; Freedman, S.B.; McNeil, H.P.; Geczy, C.L. Serum Amyloid A Induces Monocyte Tissue Factor. J. Immunol. 2007, 178, 1852–1860. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhou, S.; Heng, C.K. Impact of serum amyloid A on tissue factor and tissue factor pathway inhibitor expression and activity in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1645–1650. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.G.; Witting, P.K.; Verma, M.; Wu, B.J.; Shanu, A.; Kairaitis, K.; Amis, T.C.; Wheatley, J.R. Tissue vibration induces carotid artery endothelial dysfunction: A mechanism linking snoring and carotid atherosclerosis? Sleep 2011, 34, 751–757. [Google Scholar] [CrossRef] [PubMed]
- Ashander, L.M.; Appukuttan, B.; Ma, Y.; Gardner-Stephen, D.; Smith, J.R. Targeting Endothelial Adhesion Molecule Transcription for Treatment of Inflammatory Disease: A Proof-of-Concept Study. Mediat. Inflamm. 2016, 2016, 7945848. [Google Scholar] [CrossRef] [PubMed]
- Lappas, M.; Yee, K.; Permezel, M.; Rice, G.E. Sulfasalazine and BAY 11-7082 interfere with the nuclear factor-kappa B and I kappa B kinase pathway to regulate the release of proinflammatory cytokines from human adipose tissue and skeletal muscle in vitro. Endocrinology 2005, 146, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Zhang, A.; Wang, K.; Ding, L.; Bao, X.; Wang, X.; Qiu, X.; Liu, J. Bay11-7082 attenuates neuropathic pain via inhibition of nuclear factor-kappa B and nucleotide-binding domain-like receptor protein 3 inflammasome activation in dorsal root ganglions in a rat model of lumbar disc herniation. J. Pain Res. 2017, 10, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Bergh, N.; Ulfhammer, E.; Glise, K.; Jern, S.; Karlsson, L. Influence of TNF-alpha and biomechanical stress on endothelial anti- and prothrombotic genes. Biochem. Biophys. Res. Commun. 2009, 385, 314–318. [Google Scholar] [CrossRef]
- Creager, M.A.; Cooke, J.P.; Mendelsohn, M.E.; Gallagher, S.J.; Coleman, S.M.; Loscalzo, J.; Dzau, V.J. Impaired vasodilation of forearm resistance vessels in hypercholesterolemic humans. J. Clin. Investig. 1990, 86, 228–234. [Google Scholar] [CrossRef]
- Halcox, J.P.; Schenke, W.H.; Zalos, G.; Mincemoyer, R.; Prasad, A.; Waclawiw, M.A.; Nour, K.R.; Quyyumi, A.A. Prognostic value of coronary vascular endothelial dysfunction. Circulation 2002, 106, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Heusch, G.; Schulz, R. TNFalpha in atherosclerosis, myocardial ischemia/reperfusion and heart failure. Pharmacol. Ther. 2010, 127, 295–314. [Google Scholar] [CrossRef] [PubMed]
- McGee, D.W.; Bamberg, T.; Vitkus, S.J.; McGhee, J.R. A synergistic relationship between TNF-alpha, IL-1 beta, and TGF-beta 1 on IL-6 secretion by the IEC-6 intestinal epithelial cell line. Immunology 1995, 86, 6–11. [Google Scholar] [PubMed]
- Zhong, X.; Li, X.; Liu, F.; Tan, H.; Shang, D. Omentin inhibits TNF-alpha-induced expression of adhesion molecules in endothelial cells via ERK/NF-kappaB pathway. Biochem. Biophys. Res. Commun. 2012, 425, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.F.; Fang, R.Y.; Hsieh, H.L.; Chi, P.L.; Lin, C.C.; Hsiao, L.D.; Wu, C.C.; Wang, J.S.; Yang, C.M. Involvement of MAPKs and NF-kappaB in tumor necrosis factor alpha-induced vascular cell adhesion molecule 1 expression in human rheumatoid arthritis synovial fibroblasts. Arthrit. Rheum. 2010, 62, 105–116. [Google Scholar] [CrossRef] [PubMed]
- de Jong, A.L.; Green, D.M.; Trial, J.A.; Birdsall, H.H. Focal effects of mononuclear leukocyte transendothelial migration: TNF-alpha production by migrating monocytes promotes subsequent migration of lymphocytes. J. Leukoc. Biol. 1996, 60, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Friedl, J.; Puhlmann, M.; Bartlett, D.L.; Libutti, S.K.; Turner, E.N.; Gnant, M.F.; Alexander, H.R. Induction of permeability across endothelial cell monolayers by tumor necrosis factor (TNF) occurs via a tissue factor-dependent mechanism: Relationship between the procoagulant and permeability effects of TNF. Blood 2002, 100, 1334–1339. [Google Scholar]
- Ho, A.W.; Wong, C.K.; Lam, C.W. Tumor necrosis factor-alpha up-regulates the expression of CCL2 and adhesion molecules of human proximal tubular epithelial cells through MAPK signaling pathways. Immunobiology 2008, 213, 533–544. [Google Scholar] [CrossRef]
- Polgar, J.; Matuskova, J.; Wagner, D.D. The P-selectin, tissue factor, coagulation triad. J. Thromb. Haemost. 2005, 3, 1590–1596. [Google Scholar] [CrossRef]
- Mach, F.; Schönbeck, U.; Bonnefoy, J.Y.; Pober, J.S.; Libby, P. Activation of Monocyte/Macrophage Functions Related to Acute Atheroma Complication by Ligation of CD40: Induction of Collagenase, Stromelysin, and Tissue Factor. Circulation 1997, 96, 396–399. [Google Scholar] [CrossRef]
- Song, C.J.; Nakagomi, A.; Chandar, S.; Cai, H.; Lim, I.G.; McNeil, H.P.; Freedman, S.B.; Geczy, C.L. C-reactive protein contributes to the hypercoagulable state in coronary artery disease. J. Thromb. Haemost. 2006, 4, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Yudkin, J.S.; Kumari, M.; Humphries, S.E.; Mohamed-Ali, V. Inflammation, obesity, stress and coronary heart disease: Is interleukin-6 the link? Atherosclerosis 2000, 148, 209–214. [Google Scholar] [CrossRef]
- Neumann, F.J.; Ott, I.; Marx, N.; Luther, T.; Kenngott, S.; Gawaz, M.; Kotzsch, M.; Schomig, A. Effect of human recombinant interleukin-6 and interleukin-8 on monocyte procoagulant activity. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 3399–3405. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.; Sironi, M.; Toniatti, C.; Polentarutti, N.; Fruscella, P.; Ghezzi, P.; Faggioni, R.; Luini, W.; van Hinsbergh, V.; Sozzani, S.; et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity 1997, 6, 315–325. [Google Scholar] [CrossRef]
- Huber, S.A.; Sakkinen, P.; Conze, D.; Hardin, N.; Tracy, R. Interleukin-6 exacerbates early atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 2364–2367. [Google Scholar] [CrossRef] [PubMed]
- Maruo, N.; Morita, I.; Shirao, M.; Murota, S. IL-6 increases endothelial permeability in vitro. Endocrinology 1992, 131, 710–714. [Google Scholar] [PubMed]
- Jensen, L.E.; Whitehead, A.S. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem. J. 1998, 334 Pt 3, 489–503. [Google Scholar] [CrossRef]
- Yamada, T.; Wada, A.; Itoh, K.; Igar, J. Serum amyloid A secretion from monocytic leukaemia cell line THP-1 and cultured human peripheral monocytes. Scand. J. Immunol. 2000, 52, 7–12. [Google Scholar] [CrossRef]
- Marin, V.; Montero-Julian, F.A.; Gres, S.; Boulay, V.; Bongrand, P.; Farnarier, C.; Kaplanski, G. The IL-6-soluble IL-6Ralpha autocrine loop of endothelial activation as an intermediate between acute and chronic inflammation: An experimental model involving thrombin. J. Immunol. 2001, 167, 3435–3442. [Google Scholar] [CrossRef]
- Schuerwegh, A.J.; De Clerck, L.S.; Bridts, C.H.; Stevens, W.J. Comparison of intracellular cytokine production with extracellular cytokine levels using two flow cytometric techniques. Cytometry B Clin. Cytom. 2003, 55, 52–58. [Google Scholar] [CrossRef]
- Koga, T.; Torigoshi, T.; Motokawa, S.; Miyashita, T.; Maeda, Y.; Nakamura, M.; Komori, A.; Aiba, Y.; Uemura, T.; Yatsuhashi, H.; et al. Serum amyloid A-induced IL-6 production by rheumatoid synoviocytes. FEBS Lett. 2008, 582, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Nagaya, T.; Imai, T.; Funahashi, H.; Nakao, A.; Seo, H. Inhibition of NF-kappaB activity decreases the VEGF mRNA expression in MDA-MB-231 breast cancer cells. Breast Cancer Res. Treat. 2002, 73, 237–243. [Google Scholar] [CrossRef]
- Ross, R. Atherosclerosis—An inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Ashby, D.; Gamble, J.; Vadas, M.; Fidge, N.; Siggins, S.; Rye, K.; Barter, P.J. Lack of effect of serum amyloid A (SAA) on the ability of high-density lipoproteins to inhibit endothelial cell adhesion molecule expression. Atherosclerosis 2001, 154, 113–121. [Google Scholar] [CrossRef]
- Chiba, T.; Chang, M.Y.; Wang, S.; Wight, T.N.; McMillen, T.S.; Oram, J.F.; Vaisar, T.; Heinecke, J.W.; De Beer, F.C.; De Beer, M.C.; et al. Serum amyloid A facilitates the binding of high-density lipoprotein from mice injected with lipopolysaccharide to vascular proteoglycans. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1326–1332. [Google Scholar] [CrossRef] [PubMed]
- Hwang, Y.G.; Balasubramani, G.K.; Metes, I.D.; Levesque, M.C.; Bridges, S.L., Jr.; Moreland, L.W. Differential response of serum amyloid A to different therapies in early rheumatoid arthritis and its potential value as a disease activity biomarker. Arthrit. Res. Ther. 2016, 18, 108. [Google Scholar] [CrossRef] [PubMed]
- McEneny, J.; Daniels, J.A.; McGowan, A.; Gunness, A.; Moore, K.; Stevenson, M.; Young, I.S.; Gibney, J. A Cross-Sectional Study Demonstrating Increased Serum Amyloid A Related Inflammation in High-Density Lipoproteins from Subjects with Type 1 Diabetes Mellitus and How this Association Was Augmented by Poor Glycaemic Control. J. Diabetes Res. 2015, 2015, 351601. [Google Scholar] [CrossRef] [PubMed]
- Marz, W.; Kleber, M.E.; Scharnagl, H.; Speer, T.; Zewinger, S.; Ritsch, A.; Parhofer, K.G.; von Eckardstein, A.; Landmesser, U.; Laufs, U. HDL cholesterol: Reappraisal of its clinical relevance. Clin. Res. Cardiol. 2017, 106, 663–675. [Google Scholar] [CrossRef] [PubMed]
- Franco, A.G.; Sandri, S.; Campa, A. High-density lipoprotein prevents SAA-induced production of TNF-alpha in THP-1 monocytic cells and peripheral blood mononuclear cells. Mem. Inst. Oswaldo Cruz 2011, 106, 986–992. [Google Scholar] [CrossRef] [PubMed]
- Heinonen, S.E.; Kivela, A.M.; Huusko, J.; Dijkstra, M.H.; Gurzeler, E.; Makinen, P.I.; Leppanen, P.; Olkkonen, V.M.; Eriksson, U.; Jauhiainen, M.; et al. The effects of VEGF-A on atherosclerosis, lipoprotein profile, and lipoprotein lipase in hyperlipidaemic mouse models. Cardiovasc. Res. 2013, 99, 716–723. [Google Scholar] [CrossRef] [PubMed]
- Michell, D.L.; Andrews, K.L.; Woollard, K.J.; Chin-Dusting, J.P. Imaging leukocyte adhesion to the vascular endothelium at high intraluminal pressure. J. Vis. Exp. 2011, 3221. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Yuen, D.; Huet, O.; Pickering, R.; Stefanovic, N.; Bernatchez, P.; de Haan, J.B. Lack of glutathione peroxidase-1 facilitates a pro-inflammatory and activated vascular endothelium. Vascul. Pharmacol. 2016, 79, 32–42. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) | Accession # |
|---|---|---|---|
| β-actin | AGCACTGTGTTGGCGTACAG | GGACTTCGAGCAAGAGATGG | XM_006715764.1 |
| TNF-α | CAGAGGGCCTGTACCTCATC | GGAAGACCCCTCCCAGATAG | NM_000594.3 |
| TF | GTGACCTCACCGACGAGATT | CCGAGGTTTGTCTCCAGGTA | NM_001178096.1 |
| NFκB | TGGAAGCACGAATGACAGA | TGAGGTCCATCTCCTTGGTC | NM_001319226.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vallejo, A.; Chami, B.; Dennis, J.M.; Simone, M.; Ahmad, G.; Abdo, A.I.; Sharma, A.; Shihata, W.A.; Martin, N.; Chin-Dusting, J.P.F.; et al. NFκB Inhibition Mitigates Serum Amyloid A-Induced Pro-Atherogenic Responses in Endothelial Cells and Leukocyte Adhesion and Adverse Changes to Endothelium Function in Isolated Aorta. Int. J. Mol. Sci. 2019, 20, 105. https://doi.org/10.3390/ijms20010105
Vallejo A, Chami B, Dennis JM, Simone M, Ahmad G, Abdo AI, Sharma A, Shihata WA, Martin N, Chin-Dusting JPF, et al. NFκB Inhibition Mitigates Serum Amyloid A-Induced Pro-Atherogenic Responses in Endothelial Cells and Leukocyte Adhesion and Adverse Changes to Endothelium Function in Isolated Aorta. International Journal of Molecular Sciences. 2019; 20(1):105. https://doi.org/10.3390/ijms20010105
Chicago/Turabian StyleVallejo, Abigail, Belal Chami, Joanne M. Dennis, Martin Simone, Gulfam Ahmad, Adrian I. Abdo, Arpeeta Sharma, Waled A. Shihata, Nathan Martin, Jaye P. F. Chin-Dusting, and et al. 2019. "NFκB Inhibition Mitigates Serum Amyloid A-Induced Pro-Atherogenic Responses in Endothelial Cells and Leukocyte Adhesion and Adverse Changes to Endothelium Function in Isolated Aorta" International Journal of Molecular Sciences 20, no. 1: 105. https://doi.org/10.3390/ijms20010105
APA StyleVallejo, A., Chami, B., Dennis, J. M., Simone, M., Ahmad, G., Abdo, A. I., Sharma, A., Shihata, W. A., Martin, N., Chin-Dusting, J. P. F., De Haan, J. B., & Witting, P. K. (2019). NFκB Inhibition Mitigates Serum Amyloid A-Induced Pro-Atherogenic Responses in Endothelial Cells and Leukocyte Adhesion and Adverse Changes to Endothelium Function in Isolated Aorta. International Journal of Molecular Sciences, 20(1), 105. https://doi.org/10.3390/ijms20010105