Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers


Abstract
1. Introduction
2. EBV miRNAs in EBV-Associated Cancers
2.1. Nasopharyngeal Carcinoma (NPC)
2.2. Diffuse Large B-Cell Lymphoma (DLBCL) and Natural Killer/T-Cell Lymphoma (NKTL)
2.3. BL and Lymphoblastoid Cells (LCL)
2.4. Gastric Carcinoma (GC)
2.5. Comprehensive Studies
3. EBV miRNAs and EV
4. EV Isolation Methods
4.1. Ultracentrifugation (UC)
4.2. Ultrafiltration (UF)
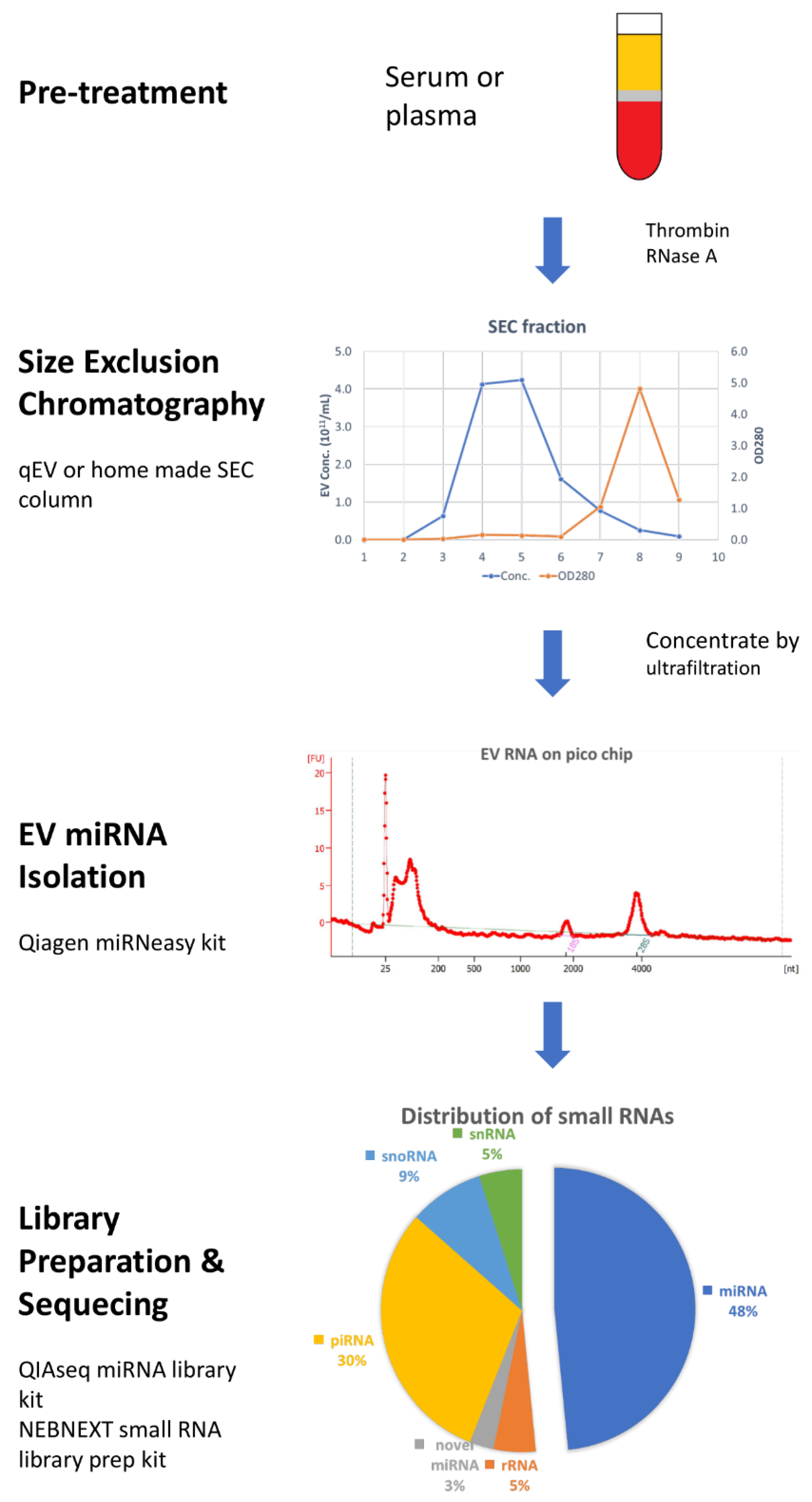
4.3. Size-Exclusion Chromatography (SEC)
4.4. Anion Exchange Chromatography (AIEX)
4.5. Precipitation Methods
4.6. Immunoaffinity Isolation
4.7. Microfluidic Devices
4.8. Combination Approaches
4.9. Comparison of EV Isolation Methods for miRNA Detection
5. Methods for miRNA Detection
5.1. RT-qPCR
5.2. DNA Probes
5.3. NGS
5.4. Surface Plasmon Resonance (SPR)
5.5. Localized Surface Plasmon Resonance (LSPR)
5.6. Surface-Enhanced Raman Scattering (SERS)
6. Conclusions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIEX | Anion exchange chromatography |
| BART | BamHI A rightward transcripts |
| BHRF | BamHI fragment H rightward open reading frame |
| BL | Burkitt′s lymphoma |
| DC | Differential ultracentrifugation |
| DLBCL | Diffuse large B-cell lymphoma |
| EBV | Epstein–Barr-virus |
| EV | Extracellular vesicle |
| GC | Gastric carcinoma |
| HL | Hodgkin’s lymphoma |
| LCL | Lymphoblastoid cell line |
| LSPR | Localized surface plasmon resonance |
| MB | Molecular beacon |
| NGS | Next-generation sequencing |
| NKTL | Nasal NK/T-cell lymphoma |
| NPC | Nasopharyngeal carcinoma |
| NTA | Nanoparticle tracking analysis |
| PAP | Poly(A) polymerase |
| PTLD | Post-transplant lymphoproliferative disease |
| RT-qPCR | Reverse transcription quantitative polymerase chain reaction |
| SEC | Size-exclusion chromatography |
| SERS | Surface-enhanced Raman scattering |
| SPR | Surface plasmon resonance |
| TFF | Tangential flow filtration |
| UC | Ultracentrifugation |
| UF | Ultrafiltration |
| UMI | Unique Molecular Identifier |
References
- Lee, Y.; El Andaloussi, S.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Gen. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed]
- Raposo, G.; Stoorvogel, W. Extracellular vesicles: Exosomes, microvesicles, and friends. J. Cell Biol. 2013, 200, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Akers, J.C.; Gonda, D.; Kim, R.; Carter, B.S.; Chen, C.C. Biogenesis of extracellular vesicles (EV): Exosomes, microvesicles, retrovirus-like vesicles, and apoptotic bodies. J. Neurooncol. 2013, 113, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Szatanek, R.; Baran, J.; Siedlar, M.; Baj-Krzyworzeka, M. Isolation of extracellular vesicles: Determining the correct approach (Review). Int. J. Mol. Med. 2015, 36, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, J.; Wysoczynski, M.; Hayek, F.; Janowska-Wieczorek, A.; Ratajczak, M.Z. Membrane-derived microvesicles: Important and underappreciated mediators of cell-to-cell communication. Leukemia 2006, 20, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Camussi, G.; Deregibus, M.C.; Bruno, S.; Cantaluppi, V.; Biancone, L. Exosomes/microvesicles as a mechanism of cell-to-cell communication. Kidney Int. 2010, 78, 838–848. [Google Scholar] [CrossRef] [PubMed]
- Nishida-Aoki, N.; Ochiya, T. Interactions between cancer cells and normal cells via miRNAs in extracellular vesicles. Cell. Mol. Life Sci. 2015, 72, 1849–1861. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yang, P.; Wang, X.F. Microenvironmental regulation of cancer metastasis by miRNAs. Trends Cell Biol. 2014, 24, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Mei, Q.; Li, X.; Guo, M.; Fu, X.; Han, W. The miRNA network: Micro-regulator of cell signaling in cancer. Expert Rev. Anticancer Ther. 2014, 14, 1515–1527. [Google Scholar] [CrossRef] [PubMed]
- Si, M.; Zhu, S.; Wu, H.; Lu, Z.; Wu, F.; Mo, Y. miR-21-mediated tumor growth. Oncogene 2007, 26, 2799. [Google Scholar] [CrossRef] [PubMed]
- Baranwal, S.; Alahari, S.K. miRNA control of tumor cell invasion and metastasis. Int. J. Cancer 2010, 126, 1283–1290. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W. Circulating microRNA biomarker studies: Pitfalls and potential solutions. Clin. Chem. 2015, 61, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Shin, V.Y.; Chu, K.M. MiRNA as potential biomarkers and therapeutic targets for gastric cancer. World J. Gastroenterol. 2014, 20, 10432–10439. [Google Scholar] [CrossRef] [PubMed]
- Javidi, M.A.; Ahmadi, A.H.; Bakhshinejad, B.; Nouraee, N.; Babashah, S.; Sadeghizadeh, M. Cell-free microRNAs as cancer biomarkers: The odyssey of miRNAs through body fluids. Med. Oncol. 2014, 31, 295. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.; Peruzzi, P.P.; Lawler, S. MicroRNAs in cancer: Biomarkers, functions and therapy. Trends Mol. Med. 2014, 20, 460–469. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Jacob, L.S.; Slack, F.J. Non-coding RNA networks in cancer. Nat. Rev. Cancer 2018, 18, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.D.; Anastasiadou, E.; Esteller, M.; He, L.; Slack, F.J. The inescapable influence of noncoding RNAs in cancer. Cancer Res. 2015, 75, 24. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.R.; Yang, C.H.; Pfeffer, L.M. The Role of miR-21 in Cancer. Drug Dev. Res. 2015, 76, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Stott, S.L.; Hsu, C.-H.; Tsukrov, D.I.; Yu, M.; Miyamoto, D.T.; Waltman, B.A.; Rothenberg, S.M.; Shah, A.M.; Smas, M.E.; Korir, G.K. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proc. Natl. Acad. Sci. USA 2010, 107, 18392–18397. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.W.; Nakanishi, H.; Kumar, V.S.; Bhadkamkar, V.A.; McCormack, R.; Fritsche, H.A.; Handy, B.; Gornet, T.; Babaian, R.J. Circulating tumor cells in peripheral blood samples from patients with increased serum prostate specific antigen: Initial results in early prostate cancer. J. Urol. 2008, 179, 2187–2191. [Google Scholar] [CrossRef] [PubMed]
- Lobb, R.J.; Becker, M.; Wen, S.W.; Wong, C.S.; Wiegmans, A.P.; Leimgruber, A.; Moller, A. Optimized exosome isolation protocol for cell culture supernatant and human plasma. J. Extracell. Vesicles 2015, 4, 27031. [Google Scholar] [CrossRef] [PubMed]
- Malloci, M.; Perdomo, L.; Veerasamy, M.; Andriantsitohaina, R.; Simard, G.; Martinez, M.C. Extracellular Vesicles: Mechanisms in Human Health and Disease. Antioxid Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Maia, J.; Caja, S.; Strano Moraes, M.C.; Couto, N.; Costa-Silva, B. Exosome-Based Cell-Cell Communication in the Tumor Microenvironment. Front. Cell Dev. Biol 2018, 6, 18. [Google Scholar] [CrossRef] [PubMed]
- Katsuda, T.; Kosaka, N.; Ochiya, T. The roles of extracellular vesicles in cancer biology: Toward the development of novel cancer biomarkers. Proteomics 2014, 14, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Sadovska, L.; Eglitis, J.; Line, A. Extracellular Vesicles as Biomarkers and Therapeutic Targets in Breast Cancer. Anticancer Res. 2015, 35, 6379–6390. [Google Scholar] [PubMed]
- Boukouris, S.; Mathivanan, S. Exosomes in bodily fluids are a highly stable resource of disease biomarkers. Proteom. Clin. Appl. 2015, 9, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Shen, Y.; Chen, Z.; Li, R.; Ge, Q. No Significant Difference between Plasma miRNAs and Plasma-Derived Exosomal miRNAs from Healthy People. Biomed. Res. Int. 2017, 2017, 1304816. [Google Scholar] [CrossRef] [PubMed]
- Ge, Q.; Zhou, Y.; Lu, J.; Bai, Y.; Xie, X.; Lu, Z.J.M. miRNA in plasma exosome is stable under different storage conditions. Molecules 2014, 19, 1568–1575. [Google Scholar] [CrossRef] [PubMed]
- Epstein, M.A.; Achong, B.G.; Barr, Y.M. Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma. Lancet 1964, 1, 702–703. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV the prototypical human tumor virus–just how bad is it? J. Allergy Clin. Immunol. 2005, 116, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Masucci, M.G.; Ernberg, I. Epstein-Barr virus: Adaptation to a life within the immune system. Trends Microbiol. 1994, 2, 125–130. [Google Scholar] [CrossRef]
- Chen, S.J.; Chen, G.H.; Chen, Y.H.; Liu, C.Y.; Chang, K.P.; Chang, Y.S.; Chen, H.C. Characterization of Epstein-Barr virus miRNAome in nasopharyngeal carcinoma by deep sequencing. PLoS ONE 2010, 5, e12745. [Google Scholar] [CrossRef] [PubMed]
- Edwards, R.H.; Marquitz, A.R.; Raab-Traub, N.J.J.O.V. Epstein-Barr virus BART microRNAs are produced from a large intron prior to splicing. J. Virol. 2008, 82, 9094–9106. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Zavolan, M.; Grasser, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of virus-encoded microRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.Y.; Siu, K.L.; Kok, K.H.; Lung, R.W.; Tsang, C.M.; To, K.F.; Kwong, D.L.; Tsao, S.W.; Jin, D.Y. An Epstein-Barr virus-encoded microRNA targets PUMA to promote host cell survival. J. Exp. Med. 2008, 205, 2551–2560. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Meister, G.; Grasser, F.A. EBV-encoded miRNAs. Biochim. Biophys. Acta 2011, 1809, 631–640. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.M.; Kong, K.L.; Tsang, J.W.; Kwong, D.L.; Guan, X.Y. Profiling of Epstein-Barr virus-encoded microRNAs in nasopharyngeal carcinoma reveals potential biomarkers and oncomirs. Cancer 2012, 118, 698–710. [Google Scholar] [CrossRef] [PubMed]
- Cullen, B.R. Viral and cellular messenger RNA targets of viral microRNAs. Nature 2009, 457, 421–425. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.K.; To, K.F.; Lo, K.W.; Lung, R.W.; Hui, J.W.; Liao, G.; Hayward, S.D. Modulation of LMP1 protein expression by EBV-encoded microRNAs. Proc. Natl. Acad. Sci. USA 2007, 104, 16164–16169. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Pfuhl, T.; Mamiani, A.; Ehses, C.; Roemer, K.; Kremmer, E.; Jaker, C.; Hock, J.; Meister, G.; Grasser, F.A. Epstein-Barr virus-encoded microRNA miR-BART2 down-regulates the viral DNA polymerase BALF5. Nucleic Acids Res. 2008, 36, 666–675. [Google Scholar] [CrossRef] [PubMed]
- Lung, R.W.; Tong, J.H.; Sung, Y.M.; Leung, P.S.; Ng, D.C.; Chau, S.L.; Chan, A.W.; Ng, E.K.; Lo, K.W.; To, K.F. Modulation of LMP2A expression by a newly identified Epstein-Barr virus-encoded microRNA miR-BART22. Neoplasia 2009, 11, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Nachmani, D.; Stern-Ginossar, N.; Sarid, R.; Mandelboim, O. Diverse herpesvirus microRNAs target the stress-induced immune ligand MICB to escape recognition by natural killer cells. Cell Host Microbe 2009, 5, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.N.; Chae, H.S.; Oh, S.T.; Kang, J.H.; Park, C.H.; Park, W.S.; Takada, K.; Lee, J.M.; Lee, W.K.; Lee, S.K. Expression of viral microRNAs in Epstein-Barr virus-associated gastric carcinoma. J. Virol. 2007, 81, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Iizasa, H.; Wulff, B.E.; Alla, N.R.; Maragkakis, M.; Megraw, M.; Hatzigeorgiou, A.; Iwakiri, D.; Takada, K.; Wiedmer, A.; Showe, L.; et al. Editing of Epstein-Barr virus-encoded BART6 microRNAs controls their dicer targeting and consequently affects viral latency. J. Biol. Chem. 2010, 285, 33358–33370. [Google Scholar] [CrossRef] [PubMed]
- Anastasiadou, E.; Garg, N.; Bigi, R.; Yadav, S.; Campese, A.F.; Lapenta, C.; Spada, M.; Cuomo, L.; Botta, A.; Belardelli, F.; et al. Epstein-Barr virus infection induces miR-21 in terminally differentiated malignant B cells. Int J. Cancer 2015, 137, 1491–1497. [Google Scholar] [CrossRef] [PubMed]
- Tagawa, T.; Albanese, M.; Bouvet, M.; Moosmann, A.; Mautner, J.; Heissmeyer, V.; Zielinski, C.; Lutter, D.; Hoser, J.; Hastreiter, M.; et al. Epstein-Barr viral miRNAs inhibit antiviral CD4+ T cell responses targeting IL-12 and peptide processing. J. Exp. Med. 2016, 213, 2065–2080. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Tagawa, T.; Bouvet, M.; Maliqi, L.; Lutter, D.; Hoser, J.; Hastreiter, M.; Hayes, M.; Sugden, B.; Martin, L.; et al. Epstein-Barr virus microRNAs reduce immune surveillance by virus-specific CD8+ T cells. Proc. Natl. Acad. Sci. USA 2016, 113, E6467–E6475. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Moosmann, A.; Gromminger, S.; Walz, N.; Grundhoff, A.; Hammerschmidt, W. Micro RNAs of Epstein-Barr virus promote cell cycle progression and prevent apoptosis of primary human B cells. PLoS Pathog. 2010, 6, e1001063. [Google Scholar] [CrossRef] [PubMed]
- Albanese, M.; Tagawa, T.; Buschle, A.; Hammerschmidt, W. MicroRNAs of Epstein-Barr Virus Control Innate and Adaptive Antiviral Immunity. J. Virol. 2017, 91, 01667. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Choi, H.; Lee, S.K. Epstein-Barr virus miR-BART20-5p regulates cell proliferation and apoptosis by targeting BAD. Cancer Lett. 2015, 356, 733–742. [Google Scholar] [CrossRef] [PubMed]
- Cosmopoulos, K.; Pegtel, M.; Hawkins, J.; Moffett, H.; Novina, C.; Middeldorp, J.; Thorley-Lawson, D.A. Comprehensive profiling of Epstein-Barr virus microRNAs in nasopharyngeal carcinoma. J. Virol. 2009, 83, 2357–2367. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.Y.; Pfuhl, T.; Motsch, N.; Barth, S.; Nicholls, J.; Grasser, F.; Meister, G. Identification of novel Epstein-Barr virus microRNA genes from nasopharyngeal carcinomas. J. Virol. 2009, 83, 3333–3341. [Google Scholar] [CrossRef] [PubMed]
- Pratt, Z.L.; Kuzembayeva, M.; Sengupta, S.; Sugden, B. The microRNAs of Epstein-Barr Virus are expressed at dramatically differing levels among cell lines. Virology 2009, 386, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Wei, W.I.; Sham, J.S.T. Nasopharyngeal carcinoma. Lancet 2005, 365, 2041–2054. [Google Scholar] [CrossRef]
- Sheng, L.; Shui, Y.; Shen, L.; Wei, Q. Effect of patient-related delay in diagnosis on the extent of disease and prognosis in nasopharyngeal carcinoma. Am. J. Rhinol. 2008, 22, 317–320. [Google Scholar] [CrossRef] [PubMed]
- Arango, B.A.; Castrellon, A.B.; Perez, C.A.; Raez, L.E.; Santos, E.S. Nasopharyngeal carcinoma: Alternative treatment options after disease progression. Expert Rev. Anticancer Ther. 2010, 10, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Kefas, B.; Comeau, L.; Floyd, D.H.; Seleverstov, O.; Godlewski, J.; Schmittgen, T.; Jiang, J.; Li, Y.; Chiocca, E.A.; Lee, J. The neuronal microRNA miR-326 acts in a feedback loop with notch and has therapeutic potential against brain tumors. J. Neurosci. 2009, 29, 15161–15168. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hu, C.; Arnovitz, S.; Bugno, J.; Yu, M.; Zuo, Z.; Chen, P.; Huang, H.; Ulrich, B.; Gurbuxani, S.; et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat. Commun. 2016, 7, 11452. [Google Scholar] [CrossRef] [PubMed]
- Fesler, A.; Liu, H.; Ju, J. Modified miR-15a has therapeutic potential for improving treatment of advanced stage colorectal cancer through inhibition of BCL2, BMI1, YAP1 and DCLK1. Oncotarget 2018, 9, 2367–2383. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zong, J.; Lin, S.; Verhoeven, R.J.; Tong, S.; Chen, Y.; Ji, M.; Cheng, W.; Tsao, S.W.; Lung, M.; et al. Circulating Epstein-Barr virus microRNAs miR-BART7 and miR-BART13 as biomarkers for nasopharyngeal carcinoma diagnosis and treatment. Int. J. Cancer 2015, 136, E301–E312. [Google Scholar] [CrossRef] [PubMed]
- He, M.L.; Luo, M.X.; Lin, M.C.; Kung, H.F. MicroRNAs: Potential diagnostic markers and therapeutic targets for EBV-associated nasopharyngeal carcinoma. Biochim. Biophys. Acta 2012, 1825, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Luo, H.N.; Tian, W.D.; Lu, J.; Li, G.; Wang, L.; Zhang, B.; Liang, B.J.; Peng, X.H.; Lin, S.X.; et al. Diagnostic and prognostic value of plasma microRNA deregulation in nasopharyngeal carcinoma. Cancer Biol. Ther. 2013, 14, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Imig, J.; Motsch, N.; Zhu, J.Y.; Barth, S.; Okoniewski, M.; Reineke, T.; Tinguely, M.; Faggioni, A.; Trivedi, P.; Meister, G.; et al. microRNA profiling in Epstein-Barr virus-associated B-cell lymphoma. Nucleic Acids Res. 2011, 39, 1880–1893. [Google Scholar] [CrossRef] [PubMed]
- Motsch, N.; Alles, J.; Imig, J.; Zhu, J.; Barth, S.; Reineke, T.; Tinguely, M.; Cogliatti, S.; Dueck, A.; Meister, G.; et al. MicroRNA profiling of Epstein-Barr virus-associated NK/T-cell lymphomas by deep sequencing. PLoS ONE 2012, 7, e42193. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Schafer, A.; Lu, S.; Bilello, J.P.; Desrosiers, R.C.; Edwards, R.; Raab-Traub, N.; Cullen, B.R. Epstein-Barr virus microRNAs are evolutionarily conserved and differentially expressed. PLoS Pathog. 2006, 2, e23. [Google Scholar] [CrossRef] [PubMed]
- Grundhoff, A.; Sullivan, C.S.; Ganem, D. A combined computational and microarray-based approach identifies novel microRNAs encoded by human gamma-herpesviruses. RNA 2006, 12, 733–750. [Google Scholar] [CrossRef] [PubMed]
- Marquitz, A.R.; Mathur, A.; Shair, K.H.; Raab-Traub, N. Infection of Epstein-Barr virus in a gastric carcinoma cell line induces anchorage independence and global changes in gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 9593–9598. [Google Scholar] [CrossRef] [PubMed]
- Shinozaki-Ushiku, A.; Kunita, A.; Isogai, M.; Hibiya, T.; Ushiku, T.; Takada, K.; Fukayama, M. Profiling of Virus-Encoded MicroRNAs in Epstein-Barr Virus-Associated Gastric Carcinoma and Their Roles in Gastric Carcinogenesis. J. Virol. 2015, 89, 5581–5591. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.Y.; Liu, Y.Y.; Liu, K.H.; Hsu, J.T.; Chen, T.C.; Chiu, C.T.; Yeh, T.S. Comprehensive profiling of virus microRNAs of Epstein-Barr virus-associated gastric carcinoma: Highlighting the interactions of ebv-Bart9 and host tumor cells. J. Gastroenterol. Hepatol. 2017, 32, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.N.; Seo, M.K.; Choi, H.; Kim, S.Y.; Shin, H.J.; Yoon, A.R.; Tao, Q.; Rha, S.Y.; Lee, S.K. Characterization of naturally Epstein-Barr virus-infected gastric carcinoma cell line YCCEL1. J. Gen. Virol. 2013, 94, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Cosmopoulos, K.; Pegtel, M.; Hopmans, E.; Murray, P.; Middeldorp, J.; Shapiro, M.; Thorley-Lawson, D.A. A novel persistence associated EBV miRNA expression profile is disrupted in neoplasia. PLoS Pathog. 2011, 7, e1002193. [Google Scholar] [CrossRef] [PubMed]
- Amoroso, R.; Fitzsimmons, L.; Thomas, W.A.; Kelly, G.L.; Rowe, M.; Bell, A.I. Quantitative studies of Epstein-Barr virus-encoded microRNAs provide novel insights into their regulation. J. Virol. 2011, 85, 996–1010. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr. Exosomal communication goes viral. J. Virol. 2015, 89, 5200–5203. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Gunawardena, H.P.; Dekroon, R.M.; Heaton, P.R.; Edwards, R.H.; Ozgur, S.; Griffith, J.D.; Damania, B.; Raab-Traub, N. Modulation of B-cell exosome proteins by gamma herpesvirus infection. Proc. Natl. Acad. Sci. USA 2013, 110, E2925–E2933. [Google Scholar] [CrossRef] [PubMed]
- Pegtel, D.M.; Cosmopoulos, K.; Thorley-Lawson, D.A.; van Eijndhoven, M.A.; Hopmans, E.S.; Lindenberg, J.L.; de Gruijl, T.D.; Wurdinger, T.; Middeldorp, J.M. Functional delivery of viral miRNAs via exosomes. Proc. Natl. Acad. Sci. USA 2010, 107, 6328–6333. [Google Scholar] [CrossRef] [PubMed]
- Canitano, A.; Venturi, G.; Borghi, M.; Ammendolia, M.G.; Fais, S. Exosomes released in vitro from Epstein-Barr virus (EBV)-infected cells contain EBV-encoded latent phase mRNAs. Cancer Lett. 2013, 337, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Gourzones, C.; Gelin, A.; Bombik, I.; Klibi, J.; Vérillaud, B.; Guigay, J.; Lang, P.; Témam, S.; Schneider, V.; Amiel, C.; et al. Extra-cellular release and blood diffusion of BART viral micro-RNAs produced by EBV-infected nasopharyngeal carcinoma cells. Virol. J. 2010, 7, 271. [Google Scholar] [CrossRef] [PubMed]
- Meckes, D.G., Jr.; Shair, K.H.; Marquitz, A.R.; Kung, C.P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed]
- Aga, M.; Bentz, G.L.; Raffa, S.; Torrisi, M.R.; Kondo, S.; Wakisaka, N.; Yoshizaki, T.; Pagano, J.S.; Shackelford, J. Exosomal HIF1alpha supports invasive potential of nasopharyngeal carcinoma-associated LMP1-positive exosomes. Oncogene 2014, 33, 4613–4622. [Google Scholar] [CrossRef] [PubMed]
- Nanbo, A.; Kawanishi, E.; Yoshida, R.; Yoshiyama, H. Exosomes derived from Epstein-Barr virus-infected cells are internalized via caveola-dependent endocytosis and promote phenotypic modulation in target cells. J. Virol. 2013, 87, 10334–10347. [Google Scholar] [CrossRef] [PubMed]
- Gutzeit, C.; Nagy, N.; Gentile, M.; Lyberg, K.; Gumz, J.; Vallhov, H.; Puga, I.; Klein, E.; Gabrielsson, S.; Cerutti, A.; et al. Exosomes derived from Burkitt’s lymphoma cell lines induce proliferation, differentiation, and class-switch recombination in B cells. J. Immunol. 2014, 192, 5852–5862. [Google Scholar] [CrossRef] [PubMed]
- Baglio, S.R.; van Eijndhoven, M.A.; Koppers-Lalic, D.; Berenguer, J.; Lougheed, S.M.; Gibbs, S.; Leveille, N.; Rinkel, R.N.; Hopmans, E.S.; Swaminathan, S.; et al. Sensing of latent EBV infection through exosomal transfer of 5′pppRNA. Proc. Natl. Acad. Sci. USA 2016, 113, E587–E596. [Google Scholar] [CrossRef] [PubMed]
- Rechavi, O.; Erlich, Y.; Amram, H.; Flomenblit, L.; Karginov, F.V.; Goldstein, I.; Hannon, G.J.; Kloog, Y. Cell contact-dependent acquisition of cellular and viral nonautonomously encoded small RNAs. Genes Dev. 2009, 23, 1971–1979. [Google Scholar] [CrossRef] [PubMed]
- Haneklaus, M.; Gerlic, M.; Kurowska-Stolarska, M.; Rainey, A.A.; Pich, D.; McInnes, I.B.; Hammerschmidt, W.; O’Neill, L.A.; Masters, S.L. Cutting edge: MiR-223 and EBV miR-BART15 regulate the NLRP3 inflammasome and IL-1beta production. J. Immunol. 2012, 189, 3795–3799. [Google Scholar] [CrossRef] [PubMed]
- Sadeghipour, S.; Mathias, R.A. Herpesviruses hijack host exosomes for viral pathogenesis. Semin. Cell Dev. Biol. 2017, 67, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Kirschner, M.B.; Edelman, J.J.; Kao, S.C.; Vallely, M.P.; van Zandwijk, N.; Reid, G. The Impact of Hemolysis on Cell-Free microRNA Biomarkers. Front. Genet. 2013, 4, 94. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzas, E.I.; Bemis, L.T.; Bora, A.; Lasser, C.; Lotvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Redgrave, T.G.; Roberts, D.C.; West, C.E. Separation of plasma lipoproteins by density-gradient ultracentrifugation. Anal. Biochem. 1975, 65, 42–49. [Google Scholar] [CrossRef]
- Cheruvanky, A.; Zhou, H.; Pisitkun, T.; Kopp, J.B.; Knepper, M.A.; Yuen, P.S.; Star, R.A. Rapid isolation of urinary exosomal biomarkers using a nanomembrane ultrafiltration concentrator. Am. J. Physiol. Renal. Physiol. 2007, 292, F1657–F1661. [Google Scholar] [CrossRef] [PubMed]
- Nordin, J.Z.; Lee, Y.; Vader, P.; Mäger, I.; Johansson, H.J.; Heusermann, W.; Wiklander, O.P.B.; Hällbrink, M.; Seow, Y.; Bultema, J.J.; et al. Ultrafiltration with size-exclusion liquid chromatography for high yield isolation of extracellular vesicles preserving intact biophysical and functional properties. Nanomedicine 2015, 11, 879–883. [Google Scholar] [CrossRef] [PubMed]
- Corso, G.; Mäger, I.; Lee, Y.; Görgens, A.; Bultema, J.; Giebel, B.; Wood, M.J.A.; Nordin, J.Z.; Andaloussi, S.E.L. Reproducible and scalable purification of extracellular vesicles using combined bind-elute and size exclusion chromatography. Sci. Rep. 2017, 7, 11561. [Google Scholar] [CrossRef] [PubMed]
- Heinemann, M.L.; Ilmer, M.; Silva, L.P.; Hawke, D.H.; Recio, A.; Vorontsova, M.A.; Alt, E.; Vykoukal, J. Benchtop isolation and characterization of functional exosomes by sequential filtration. J. Chromatogr. A 2014, 1371, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Pertoft, H. Fractionation of cells and subcellular particles with Percoll. J. Biochem. Biophys. Methods 2000, 44, 1–30. [Google Scholar] [CrossRef]
- Boing, A.N.; van der Pol, E.; Grootemaat, A.E.; Coumans, F.A.; Sturk, A.; Nieuwland, R. Single-step isolation of extracellular vesicles by size-exclusion chromatography. J. Extracell. Vesicles 2014, 3, 23430. [Google Scholar] [CrossRef] [PubMed]
- Welton, J.L.; Webber, J.P.; Botos, L.A.; Jones, M.; Clayton, A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J. Extracell. Vesicles 2015, 4, 27269. [Google Scholar] [CrossRef] [PubMed]
- Koh, Y.Q.; Almughlliq, F.B.; Vaswani, K.; Peiris, H.N.; Mitchell, M.D. Exosome enrichment by ultracentrifugation and size exclusion chromatography. Front Biosci. 2018, 23, 865–874. [Google Scholar]
- Heath, N.; Grant, L.; De Oliveira, T.M.; Rowlinson, R.; Osteikoetxea, X.; Dekker, N.; Overman, R. Rapid isolation and enrichment of extracellular vesicle preparations using anion exchange chromatography. Sci. Rep. 2018, 8, 5730. [Google Scholar] [CrossRef] [PubMed]
- Pastorek, J.; Marcinka, K. Effects of pH and ionic strength on precipitation of phytopathogenic viruses by polyethylene glycol. Acta Virol. 1989, 33, 68–74. [Google Scholar] [PubMed]
- Green, M.R.; Sambrook, J. Preparation of Single-Stranded Bacteriophage M13 DNA by Precipitation with Polyethylene Glycol. Cold Spring Harb. Protoc. 2017, 2017, 093419. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.R.; Alberts, B.M.; Benzinger, R.; Lawhorne, L.; Treiber, G. Rapid bacteriophage sedimentation in the presence of polyethylene glycol and its application to large-scale virus purification. Virology 1970, 40, 734–744. [Google Scholar] [CrossRef]
- Wang, K.; Zhang, S.; Weber, J.; Baxter, D.; Galas, D.J. Export of microRNAs and microRNA-protective protein by mammalian cells. Nucleic Acids Res. 2010, 38, 7248–7259. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Meckes, D.G., Jr. An Adaptable Polyethylene Glycol-Based Workflow for Proteomic Analysis of Extracellar Vesicles. Methods Mol. Biol. 2017, 1660, 303–317. [Google Scholar] [PubMed]
- Tang, Y.T.; Huang, Y.Y.; Zheng, L.; Qin, S.H.; Xu, X.P.; An, T.X.; Xu, Y.; Wu, Y.S.; Hu, X.M.; Ping, B.H.; et al. Comparison of isolation methods of exosomes and exosomal RNA from cell culture medium and serum. Int. J. Mol. Med. 2017, 40, 834–844. [Google Scholar] [CrossRef] [PubMed]
- Van Deun, J.; Mestdagh, P.; Sormunen, R.; Cocquyt, V.; Vermaelen, K.; Vandesompele, J.; Bracke, M.; De Wever, O.; Hendrix, A. The impact of disparate isolation methods for extracellular vesicles on downstream RNA profiling. J. Extracell. Vesicles 2014, 3, 24858. [Google Scholar] [CrossRef] [PubMed]
- Rider, M.A.; Hurwitz, S.N.; Meckes, D.G., Jr. ExtraPEG: A Polyethylene Glycol-Based Method for Enrichment of Extracellular Vesicles. Sci. Rep. 2016, 6, 23978. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Skog, J.; Hsu, C.H.; Lessard, R.T.; Balaj, L.; Wurdinger, T.; Carter, B.S.; Breakefield, X.O.; Toner, M.; Irimia, D. Microfluidic isolation and transcriptome analysis of serum microvesicles. Lab. Chip 2010, 10, 505–511. [Google Scholar] [CrossRef] [PubMed]
- Wubbolts, R.; Leckie, R.S.; Veenhuizen, P.T.; Schwarzmann, G.; Mobius, W.; Hoernschemeyer, J.; Slot, J.W.; Geuze, H.J.; Stoorvogel, W. Proteomic and biochemical analyses of human B cell-derived exosomes. Potential implications for their function and multivesicular body formation. J. Biol. Chem. 2003, 278, 10963–10972. [Google Scholar] [CrossRef] [PubMed]
- Caby, M.P.; Lankar, D.; Vincendeau-Scherrer, C.; Raposo, G.; Bonnerot, C. Exosomal-like vesicles are present in human blood plasma. Int. Immunol. 2005, 17, 879–887. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Blomberg, K.E.M.; Sadik, M.; Alaarg, A.; Smith, C.I.E.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef] [PubMed]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Thery, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef] [PubMed]
- Hurwitz, S.N.; Rider, M.A.; Bundy, J.L.; Liu, X.; Singh, R.K.; Meckes, D.G., Jr. Proteomic profiling of NCI-60 extracellular vesicles uncovers common protein cargo and cancer type-specific biomarkers. Oncotarget 2016, 7, 86999–87015. [Google Scholar] [CrossRef] [PubMed]
- Zarovni, N.; Corrado, A.; Guazzi, P.; Zocco, D.; Lari, E.; Radano, G.; Muhhina, J.; Fondelli, C.; Gavrilova, J.; Chiesi, A. Integrated isolation and quantitative analysis of exosome shuttled proteins and nucleic acids using immunocapture approaches. Methods 2015, 87, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Enderle, D.; Spiel, A.; Coticchia, C.M.; Berghoff, E.; Mueller, R.; Schlumpberger, M.; Sprenger-Haussels, M.; Shaffer, J.M.; Lader, E.; Skog, J.; et al. Characterization of RNA from Exosomes and Other Extracellular Vesicles Isolated by a Novel Spin Column-Based Method. PLoS ONE 2015, 10, e0136133. [Google Scholar] [CrossRef] [PubMed]
- Nakai, W.; Yoshida, T.; Diez, D.; Miyatake, Y.; Nishibu, T.; Imawaka, N.; Naruse, K.; Sadamura, Y.; Hanayama, R. A novel affinity-based method for the isolation of highly purified extracellular vesicles. Sci. Rep. 2016, 6, 33935. [Google Scholar] [CrossRef] [PubMed]
- Balaj, L.; Atai, N.A.; Chen, W.; Mu, D.; Tannous, B.A.; Breakefield, X.O.; Skog, J.; Maguire, C.A. Heparin affinity purification of extracellular vesicles. Sci. Rep. 2015, 5, 10266. [Google Scholar] [CrossRef] [PubMed]
- Bjork, I.; Lindahl, U. Mechanism of the anticoagulant action of heparin. Mol. Cell Biochem. 1982, 48, 161–182. [Google Scholar] [CrossRef] [PubMed]
- Edward Conrad, H. (Ed.) Heparin-Binding Proteins in Lipoprotein Metabolism. In Heparin-Binding Proteins; Academic Press: San Diego, CA, USA, 1998; Chapter 11; p. 367-IV. [Google Scholar]
- Edward Conrad, H. (Ed.) Fibroblast Growth Factors. In Heparin-Binding Proteins; Academic Press: San Diego, CA, USA, 1998; Chapter 9; p. 301-III. [Google Scholar]
- Edward Conrad, H. (Ed.) Heparin-Binding Proteins in Hemostasis. In Heparin-Binding Proteins; Academic Press: San Diego, CA, USA, 1998; Chapter 8; pp. 239–300. [Google Scholar]
- Edward Conrad, H. (Ed.) Antithrombin, the Prototypic Heparin-Binding Protein. In Heparin-Binding Proteins; Academic Press: San Diego, CA, USA, 1998; Chapter 7; pp. 203–238. [Google Scholar]
- Lee, K.; Shao, H.; Weissleder, R.; Lee, H. Acoustic purification of extracellular microvesicles. ACS Nano 2015, 9, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Guo, J.; Tian, F.; Yang, N.; Yan, F.; Ding, Y.; Wei, J.; Hu, G.; Nie, G.; Sun, J. Field-Free Isolation of Exosomes from Extracellular Vesicles by Microfluidic Viscoelastic Flows. ACS Nano 2017, 11, 6968–6976. [Google Scholar] [CrossRef] [PubMed]
- Rekker, K.; Saare, M.; Roost, A.M.; Kubo, A.L.; Zarovni, N.; Chiesi, A.; Salumets, A.; Peters, M. Comparison of serum exosome isolation methods for microRNA profiling. Clin. Biochem. 2014, 47, 135–138. [Google Scholar] [CrossRef] [PubMed]
- Eldh, M.; Lotvall, J.; Malmhall, C.; Ekstrom, K. Importance of RNA isolation methods for analysis of exosomal RNA: Evaluation of different methods. Mol. Immunol. 2012, 50, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, D.; Kirchner, B.; Hermann, S.; Märte, M.; Wurmser, C.; Brandes, F.; Kotschote, S.; Bonin, M.; Steinlein, O.K.; Pfaffl, M.W.; et al. Evaluation of serum extracellular vesicle isolation methods for profiling miRNAs by next-generation sequencing. J. Extracell. Vesicles 2018, 7, 1481321. [Google Scholar] [CrossRef] [PubMed]
- Schageman, J.; Zeringer, E.; Li, M.; Barta, T.; Lea, K.; Gu, J.; Magdaleno, S.; Setterquist, R.; Vlassov, A.V. The complete exosome workflow solution: From isolation to characterization of RNA cargo. Biomed. Res. Int. 2013, 2013, 253957. [Google Scholar] [CrossRef] [PubMed]
- Stranska, R.; Gysbrechts, L.; Wouters, J.; Vermeersch, P.; Bloch, K.; Dierickx, D.; Andrei, G.; Snoeck, R. Comparison of membrane affinity-based method with size-exclusion chromatography for isolation of exosome-like vesicles from human plasma. J. Transl. Med. 2018, 16, 29316942. [Google Scholar] [CrossRef] [PubMed]
- Andreu, Z.; Rivas, E.; Sanguino-Pascual, A.; Lamana, A.; Marazuela, M.; Gonzalez-Alvaro, I.; Sanchez-Madrid, F.; de la Fuente, H.; Yanez-Mo, M. Comparative analysis of EV isolation procedures for miRNAs detection in serum samples. J. Extracell. Vesicles 2016, 5, 31655. [Google Scholar] [CrossRef] [PubMed]
- Wachalska, M.; Koppers-Lalic, D.; van Eijndhoven, M.; Pegtel, M.; Geldof, A.A.; Lipinska, A.D.; van Moorselaar, R.J.; Bijnsdorp, I.V. Protein Complexes in Urine Interfere with Extracellular Vesicle Biomarker Studies. J. Circ. Biomark. 2016, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Royo, F.; Diwan, I.; Tackett, M.R.; Zuniga, P.; Sanchez-Mosquera, P.; Loizaga-Iriarte, A.; Ugalde-Olano, A.; Lacasa, I.; Perez, A.; Unda, M.; et al. Comparative miRNA Analysis of Urine Extracellular Vesicles Isolated through Five Different Methods. Cancers 2016, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Channavajjhala Sarath, K.; Rossato, M.; Morandini, F.; Castagna, A.; Pizzolo, F.; Bazzoni, F.; Olivieri, O. Optimizing the purification and analysis of miRNAs from urinary exosomes. Clin. Chem. Lab. Med. 2014, 52, 345. [Google Scholar] [CrossRef] [PubMed]
- Hunt, E.A.; Broyles, D.; Head, T.; Deo, S.K. MicroRNA Detection: Current Technology and Research Strategies. Annu. Rev. Anal. Chem. 2015, 8, 217–237. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Schmittgen, T.D.; Lee, E.J.; Jiang, J.; Sarkar, A.; Yang, L.; Elton, T.S.; Chen, C. Real-time PCR quantification of precursor and mature microRNA. Methods 2008, 44, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Kramer, M.F. Stem-loop RT-qPCR for miRNAs. Curr. Protoc. Mol. Biol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Shi, R.; Chiang, V.L. Facile means for quantifying microRNA expression by real-time PCR. Biotechniques 2005, 39, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Benes, V.; Collier, P.; Kordes, C.; Stolte, J.; Rausch, T.; Muckentaler, M.U.; Häussinger, D.; Castoldi, M. Identification of cytokine-induced modulation of microRNA expression and secretion as measured by a novel microRNA specific qPCR assay. Sci. Rep. 2015, 5, 11590. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Yao, B.; Huang, H.; Wang, Z.; Sun, C.; Fan, Y.; Chang, Q.; Li, S.; Wang, X.; Xi, J. Real-Time Polymerase Chain Reaction MicroRNA Detection Based on Enzymatic Stem-Loop Probes Ligation. Anal. Chem. 2009, 81, 5446–5451. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Z.; Wang, H.; Wang, Y.; Jia, H.; Yan, J. Ultrasensitive quantification of mature microRNAs by real-time PCR based on ligation of a ribonucleotide-modified DNA probe. Chem. Commun. 2011, 47, 9465–9467. [Google Scholar] [CrossRef] [PubMed]
- Androvic, P.; Valihrach, L.; Elling, J.; Sjoback, R.; Kubista, M. Two-tailed RT-qPCR: A novel method for highly accurate miRNA quantification. Nucleic Acids Res. 2017, 45, e144. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Hammerle-Fickinger, A.; Riedmaier, I.; Pfaffl, M.W. mRNA and microRNA quality control for RT-qPCR analysis. Methods 2010, 50, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Mestdagh, P.; Van Vlierberghe, P.; De Weer, A.; Muth, D.; Westermann, F.; Speleman, F.; Vandesompele, J. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009, 10, R64. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.B.; Bao, G.; Searles, C.D. In vitro quantification of specific microRNA using molecular beacons. Nucleic Acids Res. 2011, 40, e13. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.; Rhee, W.J.; Tsourkas, A. Fluorescent probes for live-cell RNA detection. Annu. Rev. Biomed. Eng. 2009, 11, 25–47. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, J.A.; Kwon, M.H.; Kang, J.Y.; Rhee, W.J. In situ single step detection of exosome microRNA using molecular beacon. Biomaterials 2015, 54, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Kim, J.A.; Jeong, S.; Rhee, W.J. Simultaneous and multiplexed detection of exosome microRNAs using molecular beacons. Biosens. Bioelectron. 2016, 86, 202–210. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Kwak, K.J.; Agarwal, K.; Marras, A.; Wang, C.; Mao, Y.; Huang, X.; Ma, J.; Yu, B.; Lee, R.; et al. Detection of extracellular RNAs in cancer and viral infection via tethered cationic lipoplex nanoparticles containing molecular beacons. Anal. Chem. 2013, 85, 11265–11274. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, S.; Deighan, C.; Bandeira, E.; Kwak, K.J.; Rahman, M.; Nana-Sinkam, P.; Lee, L.J.; Paulaitis, M.E. Analyzing the miRNA content of extracellular vesicles by fluorescence nanoparticle tracking. Nanomedicine 2017, 13, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.G.; Calin, G.A.; Volinia, S.; Croce, C.M. MicroRNA expression profiling using microarrays. Nat. Protoc. 2008, 3, 563–578. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Gelfond, J.A.; McManus, L.M.; Shireman, P.K. Reproducibility of quantitative RT-PCR array in miRNA expression profiling and comparison with microarray analysis. BMC Genom. 2009, 10, 407. [Google Scholar] [CrossRef] [PubMed]
- Hesse, J.; Jacak, J.; Kasper, M.; Regl, G.; Eichberger, T.; Winklmayr, M.; Aberger, F.; Sonnleitner, M.; Schlapak, R.; Howorka, S. RNA expression profiling at the single molecule level. Genome Res. 2006, 16, 1041–1045. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.D.; Jatkoe, T.A.; Stumpf, C.R.; Lu, J.; Thomas, J.D.; Madore, S.J. Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Res. 2000, 28, 4552–4557. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, F.; Fuchs, R.T.; Sun, Z.; Zheng, Y.; Robb, G.B. Structural bias in T4 RNA ligase-mediated 3′-adapter ligation. Nucleic Acids Res. 2012, 40, e54. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Zheng, K.-X.; Duan, D.; Jiang, L.; Li, J. Label-Free MicroRNA Profiling Not Biased by 3′ End 2′-O-Methylation. Anal. Chem. 2012, 84, 6361–6365. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Cho, H.; Jung, Y. Fabrication of a structure-specific RNA binder for array detection of label-free microRNA. Angew. Chem. Int. Ed. Engl. 2010, 49, 8662–8665. [Google Scholar] [CrossRef] [PubMed]
- Nelson, P.T.; Baldwin, D.A.; Scearce, L.M.; Oberholtzer, J.C.; Tobias, J.W.; Mourelatos, Z. Microarray-based, high-throughput gene expression profiling of microRNAs. Nat. Methods 2004, 1, 155. [Google Scholar] [CrossRef] [PubMed]
- Iizuka, R.; Ueno, T.; Funatsu, T. Detection and Quantification of MicroRNAs by Ligase-Assisted Sandwich Hybridization on a Microarray. Methods Mol. Biol. 2016, 1368, 53–65. [Google Scholar] [PubMed]
- Ueno, T.; Funatsu, T. Label-free quantification of microRNAs using ligase-assisted sandwich hybridization on a DNA microarray. PLoS ONE 2014, 9, e90920. [Google Scholar] [CrossRef] [PubMed]
- van Balkom, B.W.; Eisele, A.S.; Pegtel, D.M.; Bervoets, S.; Verhaar, M.C. Quantitative and qualitative analysis of small RNAs in human endothelial cells and exosomes provides insights into localized RNA processing, degradation and sorting. J. Extracell. Vesicles 2015, 4, 26760. [Google Scholar] [CrossRef] [PubMed]
- Lahens, N.F.; Ricciotti, E.; Smirnova, O.; Toorens, E.; Kim, E.J.; Baruzzo, G.; Hayer, K.E.; Ganguly, T.; Schug, J.; Grant, G.R. A comparison of Illumina and Ion Torrent sequencing platforms in the context of differential gene expression. BMC Genom. 2017, 18, 602. [Google Scholar] [CrossRef] [PubMed]
- Leshkowitz, D.; Horn-Saban, S.; Parmet, Y.; Feldmesser, E. Differences in microRNA detection levels are technology and sequence dependent. RNA 2013, 19, 527–538. [Google Scholar] [CrossRef] [PubMed]
- Islam, S.; Zeisel, A.; Joost, S.; La Manno, G.; Zajac, P.; Kasper, M.; Lönnerberg, P.; Linnarsson, S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat. Methods 2013, 11, 163–166. [Google Scholar] [CrossRef] [PubMed]
- van Dijk, E.L.; Jaszczyszyn, Y.; Thermes, C. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res. 2014, 322, 12–20. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Yuan, T.; Tschannen, M.; Sun, Z.; Jacob, H.; Du, M.; Liang, M.; Dittmar, R.L.; Liu, Y.; Liang, M.; et al. Characterization of human plasma-derived exosomal RNAs by deep sequencing. BMC Genom. 2013, 14, 319. [Google Scholar] [CrossRef] [PubMed]
- Im, H.; Shao, H.; Park, Y.I.; Peterson, V.M.; Castro, C.M.; Weissleder, R.; Lee, H. Label-free detection and molecular profiling of exosomes with a nano-plasmonic sensor. Nat. Biotechnol. 2014, 32, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V.; Bartel, B.; Bartel, D.P.; Burge, C.B.; Carrington, J.C.; Chen, X.; Dreyfuss, G.; Eddy, S.R.; Griffiths-Jones, S.; Marshall, M.; et al. A uniform system for microRNA annotation. RNA 2003, 9, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Wahid, F.; Shehzad, A.; Khan, T.; Kim, Y.Y. MicroRNAs: Synthesis, mechanism, function, and recent clinical trials. Biochim. Biophys. Acta 2010, 1803, 1231–1243. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Lee, H.J.; Wark, A.W.; Corn, R.M. Attomole microarray detection of microRNAs by nanoparticle-amplified SPR imaging measurements of surface polyadenylation reactions. J. Am. Chem. Soc. 2006, 128, 14044–14046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Yan, Y.; Cheng, W.; Zhang, W.; Li, Y.; Ju, H.; Ding, S. Streptavidin-enhanced surface plasmon resonance biosensor for highly sensitive and specific detection of microRNA. Microchim. Acta 2013, 180, 397–403. [Google Scholar] [CrossRef]
- Ding, X.; Yan, Y.; Li, S.; Zhang, Y.; Cheng, W.; Cheng, Q.; Ding, S. Surface plasmon resonance biosensor for highly sensitive detection of microRNA based on DNA super-sandwich assemblies and streptavidin signal amplification. Anal. Chim. Acta 2015, 874, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Hao, K.; He, Y.; Lu, H.; Pu, S.; Zhang, Y.; Dong, H.; Zhang, X. High-sensitive surface plasmon resonance microRNA biosensor based on streptavidin functionalized gold nanorods-assisted signal amplification. Anal. Chim. Acta 2017, 954, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Rycenga, M.; Cobley, C.M.; Zeng, J.; Li, W.; Moran, C.H.; Zhang, Q.; Qin, D.; Xia, Y. Controlling the Synthesis and Assembly of Silver Nanostructures for Plasmonic Applications. Chem. Rev. 2011, 111, 3669–3712. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.K.; Deitz-McElyea, S.; Johnson, M.; Mali, S.; Korc, M.; Sardar, R. Highly specific plasmonic biosensors for ultrasensitive microRNA detection in plasma from pancreatic cancer patients. Nano Lett. 2014, 14, 6955–6963. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Park, J.; Lee, J.Y.; Yeo, J.S. Contact Transfer Printing of Side Edge Prefunctionalized Nanoplasmonic Arrays for Flexible microRNA Biosensor. Adv. Sci. 2015, 2, 1500121. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Lin, Y.; Chen, C.; Qiu, B.; Lin, Z.; Chen, G. Direct visualization of sub-femtomolar circulating microRNAs in serum based on the duplex-specific nuclease-amplified oriented assembly of gold nanoparticle dimers. Chem. Commun. 2016, 52, 11347–11350. [Google Scholar] [CrossRef] [PubMed]
- Blackie, E.J.; Le Ru, E.C.; Etchegoin, P.G. Single-molecule surface-enhanced Raman spectroscopy of nonresonant molecules. J. Am. Chem. Soc. 2009, 131, 14466–14472. [Google Scholar] [CrossRef] [PubMed]
- Driskell, J.D.; Seto, A.G.; Jones, L.P.; Jokela, S.; Dluhy, R.A.; Zhao, Y.P.; Tripp, R.A. Rapid microRNA (miRNA) detection and classification via surface-enhanced Raman spectroscopy (SERS). Biosens. Bioelectron. 2008, 24, 923–928. [Google Scholar] [CrossRef] [PubMed]
- Guven, B.; Dudak, F.C.; Boyaci, I.H.; Tamer, U.; Ozsoz, M. SERS-based direct and sandwich assay methods for mir-21 detection. Analyst 2014, 139, 1141–1147. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Tumor Type | Sample | Latency Type | BHRF1 Cluster | BART Cluster 1 | BART Cluster 2 | BART-2 | Method | Ref. |
|---|---|---|---|---|---|---|---|---|
| BL | MUTUI/ OUS/ BL41/95, Marm. B95.8 | I/ II/ III | −/ +/ + | 1 | + | Northern blot | [35] | |
| BC-1/ Jijoye/ Raji/ BL41/95/ MUTU III, Namalwa | I/ III/ III/ III/ III | −/ 1-2/ 1-1, 1-2/ 1-1, 1-2/ 1-1, 1-2/ | 1, 3, 5/ 1, 3, 5/ −/ 1, 3/ 1, 3, 5/ | 7, 10, 12/ 7, 10, 12/ −/ −/ 7, 10, 12/ | Northern blot | [66] | ||
| Jijoye | III | 3, 4, 5, 6, 15, 17 | 7, 8, 9, 10, 11, 12, 13, 14, 19, 20 | Northern blot, microarray | [67] | |||
| BL-5, Savl, KemI, Akata, Dante, Daudi/ OkuI, GG68, Raji, MutuIII | I/ III | −/ + | 3, 4, 1-5p, 15 | 7, 10, 12, 20-5p | qPCR | [54] | ||
| 2A8.1, RaeI | + | qPCR | [52] | |||||
| B95.8-transformed cells/ Daudi/ Namalwa B | I/ I/ III | 15/ 15, 16/ 16 | −/ 22/ 22 | qPCR | [33] | |||
| HL | RPMI6666 | II | + | 1 | + | Northern blot | [35] | |
| NPC | C666-1 | II | − | 3, 4, 1-5p, 15 | 7, 10, 12, 20-5p | qPCR | [54] | |
| C666 | II | − | 1, 3, 5 | 7, 10, 12 | Northern blot | [66] | ||
| C666-1 | II | 1-1, 1-2, 1-3 | + | + | + | qPCR | [52] | |
| C666-1 | II | + | + | + | qPCR | [70] | ||
| C-15 | − | 1, 3, 5 | 7, 10, 12 | Northern blot | [66] | |||
| clinical tissue; C666; NP460hTERT + EBV | 1-3p, 5, 6-5p, 6-3p,17-5p | 7, 8, 9, 14,18-5p, 19-3p | + | Microarray, qPCR | [38] | |||
| clinical tissue/ HK1-EBV, C666-1 | − | +/ 15, 16 | +/ 22 | + | qPCR, Deep sequencing | [33] | ||
| clinical tissue | − | + | + | + | qPCR, Deep sequencing | [52] | ||
| clinical tissue | − | + | + | Northern blot, Deep sequencing | [53] | |||
| GC | SNU-719 | I | − | 3, 4, 1-5p, 15 | 7, 10, 12, 20-5p | qPCR | [54] | |
| SNU-719 | I | − | + | + | + | qPCR | [69] | |
| AGS-EBV | I | 3, 1-3p, 5, 17-5p | 7, 9 | qPCR | [68] | |||
| clinical tissue, SNU-719 | + | + | + | qPCR | [70] | |||
| clinical tissue | − | + | + | + | qPCR | [69] | ||
| YCCEL1 | I | − | 1-3p, 15-3p | 9-3p, 5-5p, 7-3p, 22-3p, 19-3p | Northern blot, qPCR | [71] | ||
| SNU-719, AGS-EBV/ Animal model/ clinical tissue | I/ / / | −/ −/ −/ | 1, 3, 5/ 1, 5/ 1 | 7, 10, 12/ 7, 10, 12/ / | +/ / + | Northern blot | [44] | |
| NKTL | Lymphoma clinical tissue | − | + | + | + | Deep sequencing | [65] | |
| LCL | 721, B958IID6 | III | + | 3, 4, 1-5p, 15 | − | qPCR | [54] | |
| IM-9 | III | 1-1, 1-2 | 1, 3, 5/ | 7, 10, 12/ | Northern blot | [66] | ||
| IM-9 | III | + | + | + | qPCR | [70] | ||
| AT | + | qPCR | [52] | |||||
| DLBCL | clinical tissue | − | + | + | + | Deep sequencing | [64] | |
| PTLD | PTLD1 | III | + | 3, 4, 1-5p, 15 | 7, 10, 12, 20-5p | qPCR | [54] |
| Method | Sample Volume | Yield | Purity | Cost | Advantage | Disadvantage |
|---|---|---|---|---|---|---|
| UC | 1–100 mL | ++ | ++ | + | No chemical additives | Time and labor intensive; Low throughput; EVs/protein aggregates |
| UF | Variety | ++ | + | ++ | Flexible volume; No chemical additives | Low purity; Low throughput; Efficiency dependent on the type of ultra-membrane |
| Precipitation | Variety | +++ | + | + | Flexible volume; Time and labor saving; No expensive equipment needed | Low purity; Sample contamination by polymer particles; Co-isolation of nonspecific proteins; EV/protein aggregates |
| SEC | <10 mL | ++ | +++ | + | High purity; Time saving; No chemical additives; Physiological buffer | Sample volume limited; Low throughput; Sample diluted |
| Immunoaffinity | <5 mL | + | +++ | +++ | High purity; Physiological buffer; Integration with downstream biological analysis | Sample volume limited; Low throughput; Very selective; Dependent on antibody/protein; Contaminated with antibody/protein; Pre-enrichment needed |
| AIEX | Variety | ++ | ++ | ++ | Label free; Flexible volume | Low throughput; Sample diluted |
| Microfluidic | <1 mL | ++ | N.A. | +++ | Label free; Integration with downstream biological analysis | Sample volume extremely limited; Low throughput |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, L.; Meckes, D.G., Jr. Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers. Int. J. Mol. Sci. 2018, 19, 2810. https://doi.org/10.3390/ijms19092810
Sun L, Meckes DG Jr. Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers. International Journal of Molecular Sciences. 2018; 19(9):2810. https://doi.org/10.3390/ijms19092810
Chicago/Turabian StyleSun, Li, and David G. Meckes, Jr. 2018. "Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers" International Journal of Molecular Sciences 19, no. 9: 2810. https://doi.org/10.3390/ijms19092810
APA StyleSun, L., & Meckes, D. G., Jr. (2018). Methodological Approaches to Study Extracellular Vesicle miRNAs in Epstein–Barr Virus-Associated Cancers. International Journal of Molecular Sciences, 19(9), 2810. https://doi.org/10.3390/ijms19092810