Insight into the Self-Assembling Properties of Peptergents: A Molecular Dynamics Simulation Study

 ,
, 
Abstract
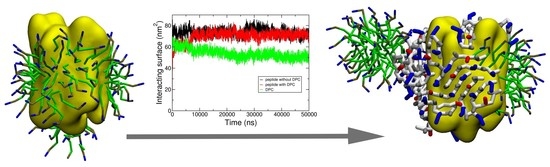
{kind=link}
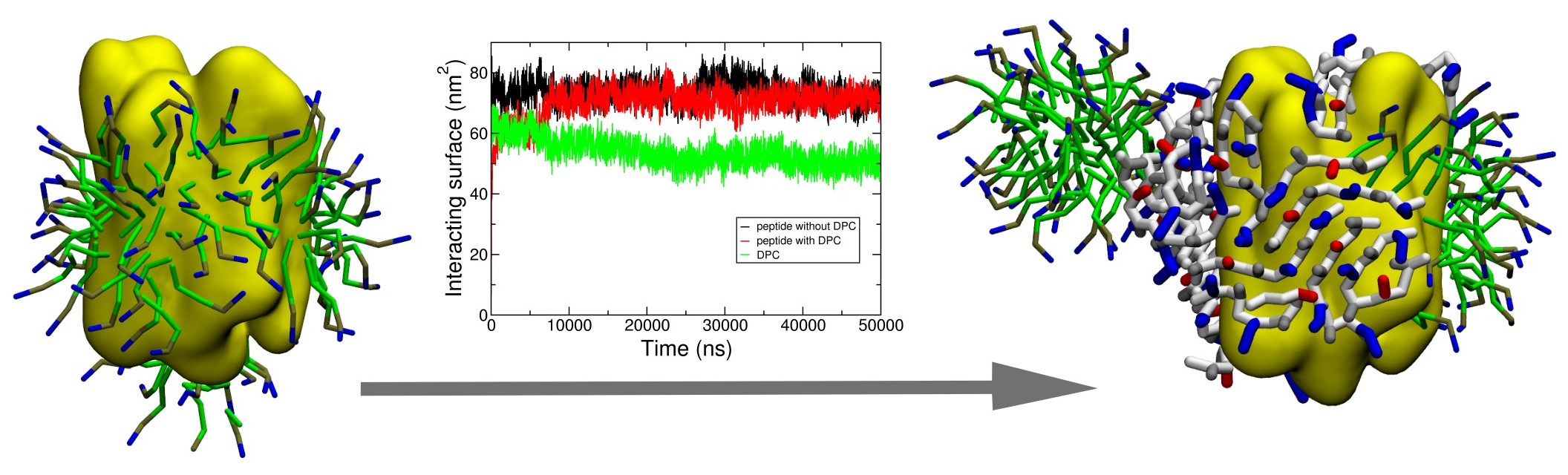
{kind=link}
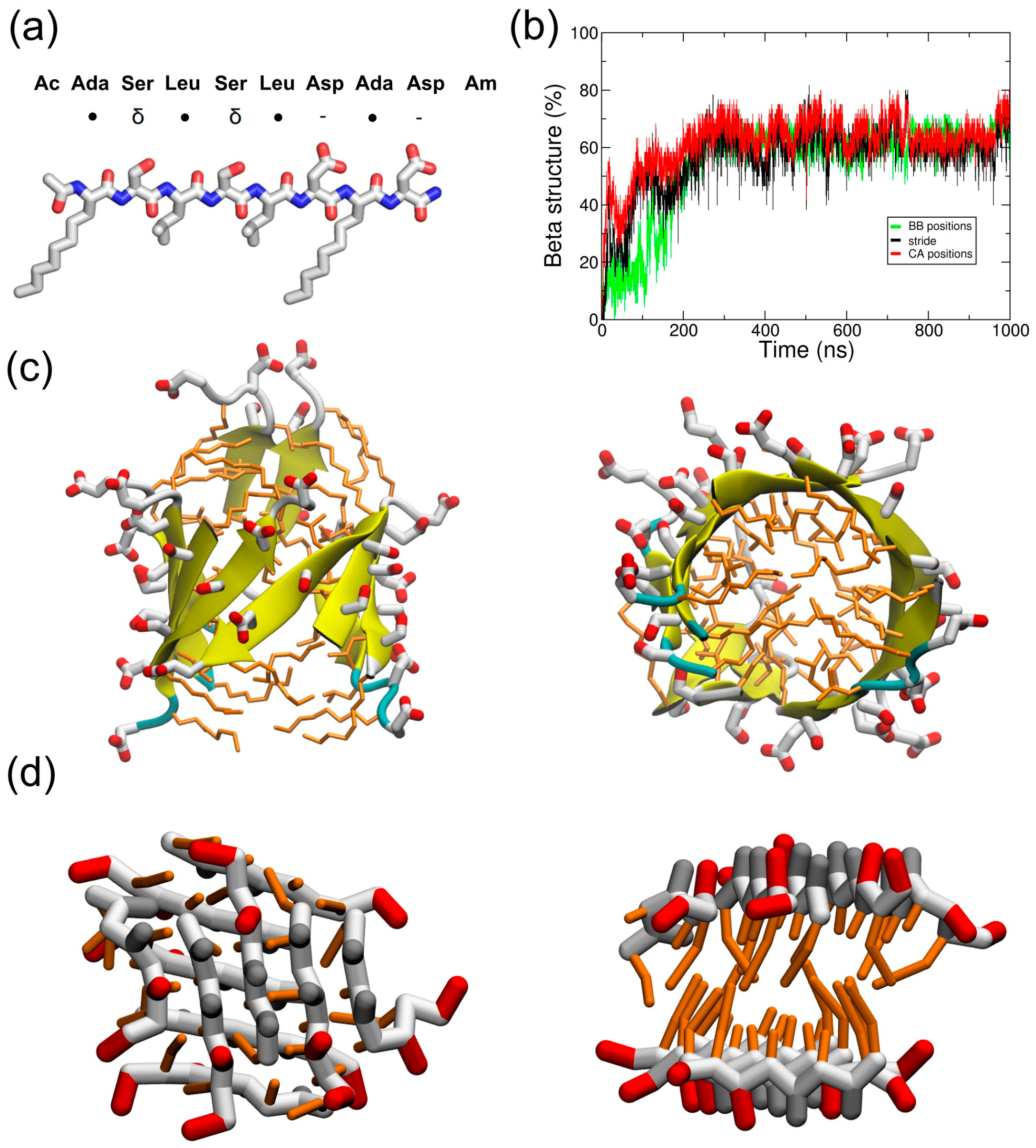
{kind=link}
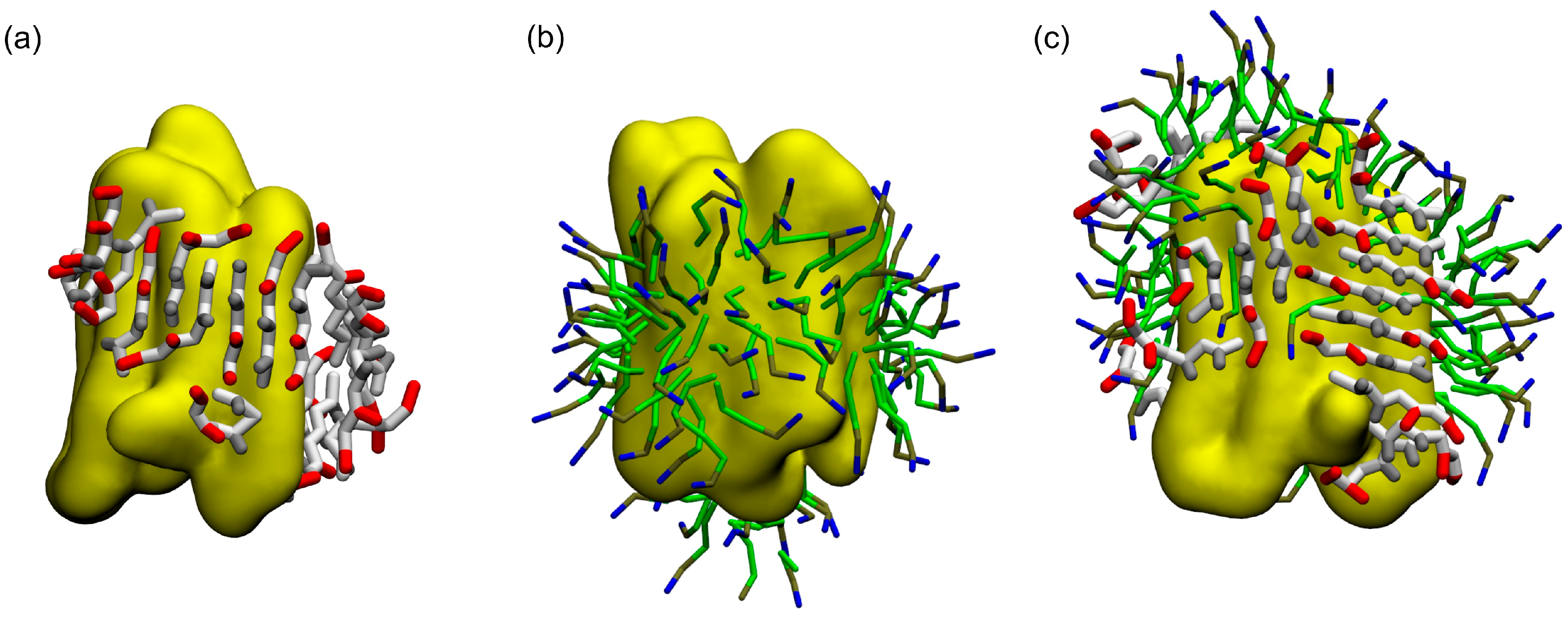
{kind=link}
{kind=link}
1. Introduction
2. Results
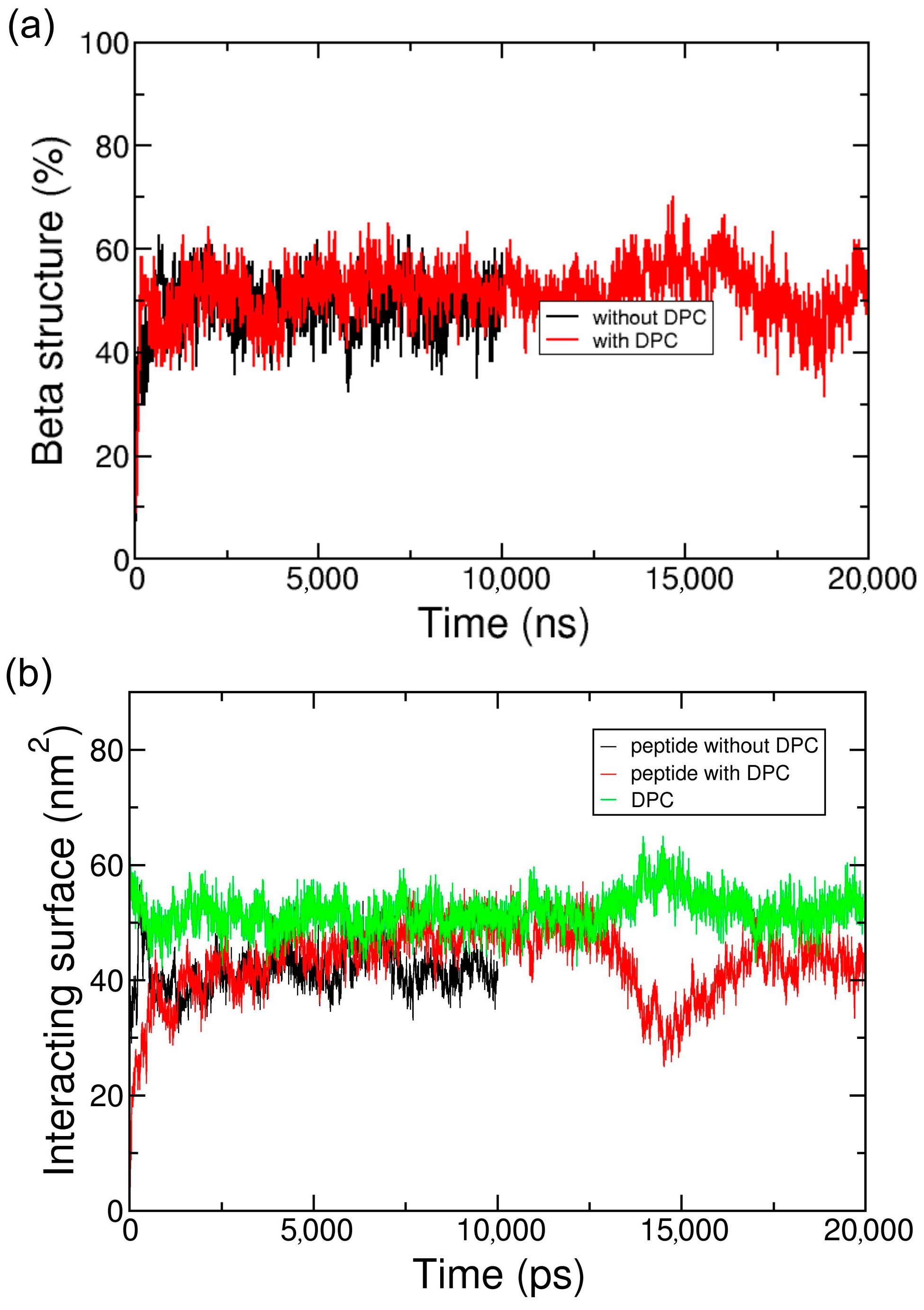
2.1. Simulations in Water
2.2. Coarse-Grained Simulation of the ADA8 Peptide in the Presence of a Membrane Protein
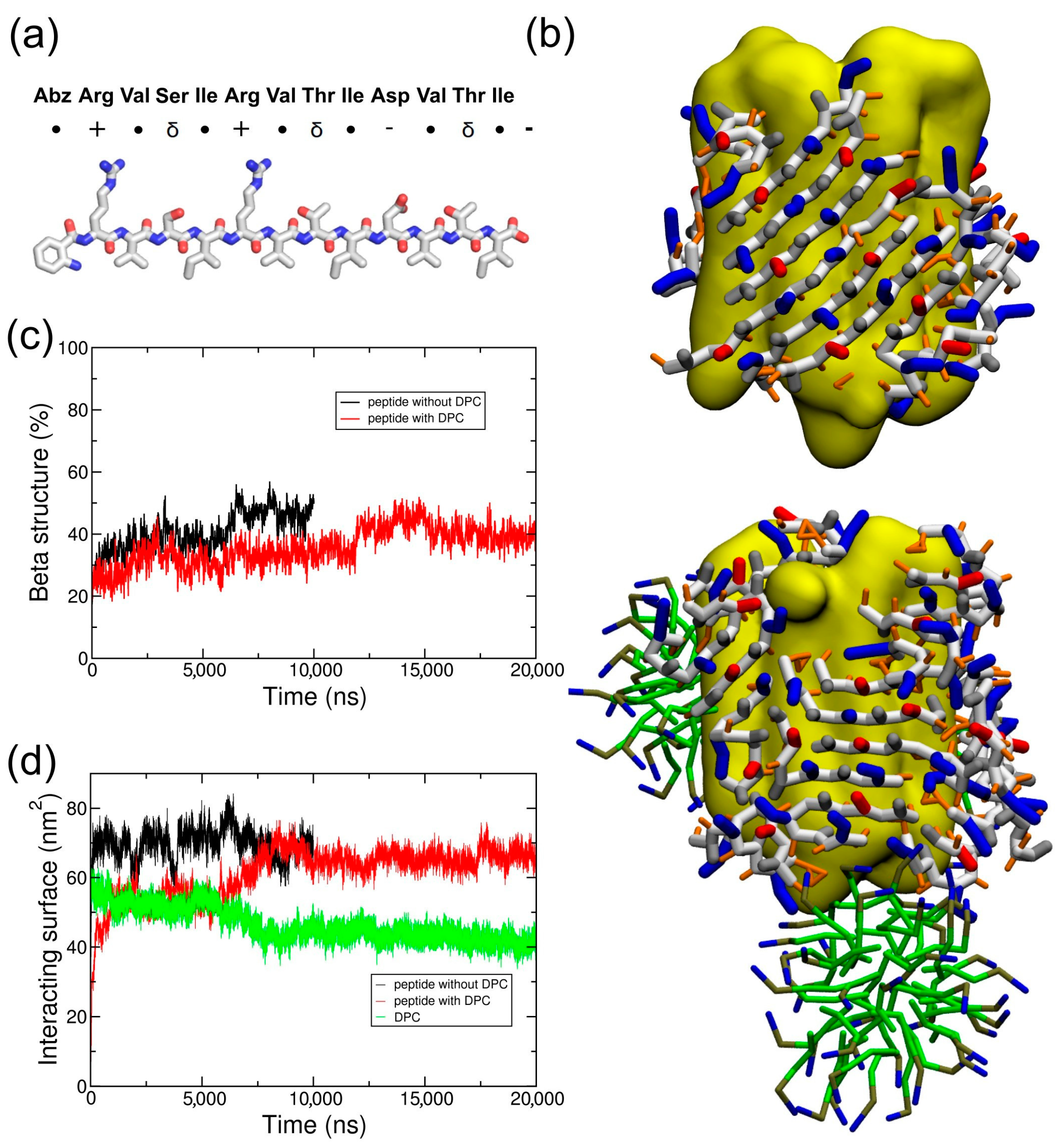
2.3. Coarse-Grained Simulation of the Designed ABZ12 Peptide in the Presence of a Membrane Protein
3. Discussion
4. Materials and Methods
4.1. Peptide Synthesis
4.2. Fourier Transform Infrared (FTIR) Experiments
4.3. Systems Studied
4.4. Atomistic Molecular Dynamic Simulations
4.5. Coarse Grained Molecular Dynamic Simulations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Lakshmanan, A.; Zhang, S.; Hauser, C.A.E. Short self-assembling peptides as building blocks for modern nanodevices. Trends Biotechnol. 2012, 30, 155–165. [Google Scholar] [CrossRef] [PubMed]
- Mandal, D.; Nasrolahi Shirazi, A.; Parang, K. Self-assembly of peptides to nanostructures. Org. Biomol. Chem. 2014, 12, 3544–3561. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Marini, D.M.; Hwang, W.; Santoso, S. Design of nanostructured biological materials through self-assembly of peptides and proteins. Curr. Opin. Chem. Biol. 2002, 6, 865–871. [Google Scholar] [CrossRef]
- Xiong, H.; Buckwalter, B.L.; Shieh, H.M.; Hecht, M.H. Periodicity of polar and nonpolar amino acids is the major determinant of secondary structure in self-assembling oligomeric peptides. Proc. Natl. Acad. Sci. USA 1995, 92, 6349–6353. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Tao, H.; Hong, W.-X. New amphiphiles for membrane protein structural biology. Methods 2011, 55, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Schafmeister, C.E.; Miercke, L.J.; Stroud, R.M. Structure at 2.5 A of a designed peptide that maintains solubility of membrane proteins. Science 1993, 262, 734–738. [Google Scholar] [CrossRef] [PubMed]
- McGregor, C.-L.; Chen, L.; Pomroy, N.C.; Hwang, P.; Go, S.; Chakrabartty, A.; Privé, G.G. Lipopeptide detergents designed for the structural study of membrane proteins. Nat. Biotechnol. 2003, 21, 171–176. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Huang, G.; Yu, D.; Ge, B.; Wang, J.; Xu, F.; Huang, F.; Xu, H.; Lu, J.R. Solubilization and stabilization of isolated photosystem I complex with lipopeptide detergents. PLoS ONE 2013, 8, e76256. [Google Scholar] [CrossRef] [PubMed]
- Ho, D.N.; Pomroy, N.C.; Cuesta-Seijo, J.A.; Privé, G.G. Crystal structure of a self-assembling lipopeptide detergent at 1.20 A. Proc. Natl. Acad. Sci. USA 2008, 105, 12861–12866. [Google Scholar] [CrossRef] [PubMed]
- Kiley, P.; Zhao, X.; Vaughn, M.; Baldo, M.A.; Bruce, B.D.; Zhang, S. Self-assembling peptide detergents stabilize isolated photosystem I on a dry surface for an extended time. PLoS Biol. 2005, 3, e230. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Nagai, Y.; Reeves, P.J.; Kiley, P.; Khorana, H.G.; Zhang, S. Designer short peptide surfactants stabilize G protein-coupled receptor bovine rhodopsin. Proc. Natl. Acad. Sci. USA 2006, 103, 17707–17712. [Google Scholar] [CrossRef] [PubMed]
- Corin, K.; Baaske, P.; Ravel, D.B.; Song, J.; Brown, E.; Wang, X.; Wienken, C.J.; Jerabek-Willemsen, M.; Duhr, S.; Luo, Y.; et al. Designer lipid-like peptides: A class of detergents for studying functional olfactory receptors using commercial cell-free systems. PLoS ONE 2011, 6, e25067. [Google Scholar] [CrossRef] [PubMed]
- Tao, H.; Lee, S.C.; Moeller, A.; Roy, R.S.; Siu, F.Y.; Zimmermann, J.; Stevens, R.C.; Potter, C.S.; Carragher, B.; Zhang, Q. Engineered nanostructured β-sheet peptides protect membrane proteins. Nat. Methods 2013, 10, 759–761. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, E.P.; Beis, K.; Cameron, A.D.; Iwata, S. Overcoming the challenges of membrane protein crystallography. Curr. Opin. Struct. Biol. 2008, 18, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Ge, B.; Yang, F.; Yu, D.; Liu, S.; Xu, H. Designer amphiphilic short peptides enhance thermal stability of isolated photosystem-I. PLoS ONE 2010, 5, e10233. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Lee, S.C.; Tao, H.; Speir, J.A.; Chang, G.; Urbatsch, I.L.; Potter, C.S.; Carragher, B.; Zhang, Q. Distinct conformational spectrum of homologous multidrug ABC transporters. Structure 2015, 23, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.; Kandasamy, S.K.; Periole, X.; Larson, R.G.; Tieleman, D.P.; Marrink, S. The MARTINI coarse-grained force field: Extension to proteins. J. Chem. Theory Comput. 2008, 4, 819–834. [Google Scholar] [CrossRef] [PubMed]
- Hung, A.; Yarovsky, I. Inhibition of peptide aggregation by lipids: Insights from coarse-grained molecular simulations. J. Mol. Graph. Model. 2011, 29, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Sørensen, J.; Periole, X.; Skeby, K.K.; Marrink, S.-J.; Schiøtt, B. Protofibrillar assembly toward the formation of amyloid fibrils. J. Phys. Chem. Lett. 2011, 2, 2385–2390. [Google Scholar] [CrossRef]
- Seo, M.; Rauscher, S.; Pomès, R.; Tieleman, D.P. Improving internal peptide dynamics in the coarse-grained MARTINI model: Toward large-scale simulations of amyloid- and elastin-like peptides. J. Chem. Theory Comput. 2012, 8, 1774–1785. [Google Scholar] [CrossRef] [PubMed]
- Marrink, S.J.; Tieleman, D.P. Perspective on the Martini model. Chem. Soc. Rev. 2013, 42, 6801–6822. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Biswas, P. Position-specific propensities of amino acids in the β-strand. BMC Struct. Biol. 2010, 10, 29. [Google Scholar] [CrossRef] [PubMed]
- Malkov, S.N.; Zivković, M.V.; Beljanski, M.V.; Hall, M.B.; Zarić, S.D. A reexamination of the propensities of amino acids towards a particular secondary structure: Classification of amino acids based on their chemical structure. J. Mol. Model. 2008, 14, 769–775. [Google Scholar] [CrossRef] [PubMed]
- Wimley, W.C. The versatile beta-barrel membrane protein. Curr. Opin. Struct. Biol. 2003, 13, 404–411. [Google Scholar] [CrossRef]
- Jackups, R.; Liang, J. Interstrand pairing patterns in beta-barrel membrane proteins: The positive-outside rule, aromatic rescue, and strand registration prediction. J. Mol. Biol. 2005, 354, 979–993. [Google Scholar] [CrossRef] [PubMed]
- Schmid, N.; Eichenberger, A.P.; Choutko, A.; Riniker, S.; Winger, M.; Mark, A.E.; van Gunsteren, W.F. Definition and testing of the GROMOS force-field versions 54A7 and 54B7. Eur. Biophys. J. 2011, 40, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Song, B.; Kibler, P.; Malde, A.; Kodukula, K.; Galande, A.K. Design of short linear peptides that show hydrogen bonding constraints in water. J. Am. Chem. Soc. 2010, 132, 4508–4509. [Google Scholar] [CrossRef] [PubMed]
- Hermans, J.; Berendsen, H.J.C.; van Gunsteren, W.F.; Postma, J.P.M. A consistent empirical potential for water-protein interactions. Biopolymers 1984, 23, 1513–1518. [Google Scholar] [CrossRef]
- Marrink, S.J.; Risselada, H.J.; Yefimov, S.; Tieleman, D.P.; de Vries, A.H. The MARTINI force field: Coarse grained model for biomolecular simulations. J. Phys. Chem. B 2007, 111, 7812–7824. [Google Scholar] [CrossRef] [PubMed]
- Dony, N.; Crowet, J.M.; Joris, B.; Brasseur, R.; Lins, L. SAHBNET, an Accessible Surface-Based Elastic Network: An Application to Membrane Protein. Int. J. Mol. Sci. 2013, 14, 11510–11526. [Google Scholar] [CrossRef] [PubMed]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Parrinello, M. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182. [Google Scholar] [CrossRef]
- Heinz, T.N.; van Gunsteren, W.F.; Hunenberger, P.H. Comparison of four methods to compute the dielectric permittivity of liquids from molecular dynamics simulations. J. Chem. Phys. 2001, 115, 1125–1136. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A toolkit for the analysis of molecular dynamics simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger, L. The PyMOL Molecular Graphics System. version 1.3. 2010. Available online: https://pymol.org/ (accessed on 1 September 2018).
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Frishman, D.; Argos, P. Incorporation of non-local interactions in protein secondary structure prediction from the amino acid sequence. Protein Eng. 1996, 9, 133–142. [Google Scholar] [CrossRef] [PubMed]
- De Jong, D.H.; Singh, G.; Bennett, W.F.D.; Arnarez, C.; Wassenaar, T.A.; Schäfer, L.V.; Periole, X.; Tieleman, D.P.; Marrink, S.J. Improved Parameters for the Martini Coarse-Grained Protein Force Field. J. Chem. Theory Comput. 2013, 9, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.R.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, T.A.; Pluhackova, K.; Böckmann, R.A.; Marrink, S.J.; Tieleman, D.P. Going backward: A flexible geometric approach to reverse transformation from coarse grained to atomistic models. J. Chem. Theory Comput. 2013, 10, 676–690. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crowet, J.M.; Nasir, M.N.; Dony, N.; Deschamps, A.; Stroobant, V.; Morsomme, P.; Deleu, M.; Soumillion, P.; Lins, L. Insight into the Self-Assembling Properties of Peptergents: A Molecular Dynamics Simulation Study. Int. J. Mol. Sci. 2018, 19, 2772. https://doi.org/10.3390/ijms19092772
Crowet JM, Nasir MN, Dony N, Deschamps A, Stroobant V, Morsomme P, Deleu M, Soumillion P, Lins L. Insight into the Self-Assembling Properties of Peptergents: A Molecular Dynamics Simulation Study. International Journal of Molecular Sciences. 2018; 19(9):2772. https://doi.org/10.3390/ijms19092772
Chicago/Turabian StyleCrowet, Jean Marc, Mehmet Nail Nasir, Nicolas Dony, Antoine Deschamps, Vincent Stroobant, Pierre Morsomme, Magali Deleu, Patrice Soumillion, and Laurence Lins. 2018. "Insight into the Self-Assembling Properties of Peptergents: A Molecular Dynamics Simulation Study" International Journal of Molecular Sciences 19, no. 9: 2772. https://doi.org/10.3390/ijms19092772
APA StyleCrowet, J. M., Nasir, M. N., Dony, N., Deschamps, A., Stroobant, V., Morsomme, P., Deleu, M., Soumillion, P., & Lins, L. (2018). Insight into the Self-Assembling Properties of Peptergents: A Molecular Dynamics Simulation Study. International Journal of Molecular Sciences, 19(9), 2772. https://doi.org/10.3390/ijms19092772