Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management


Abstract
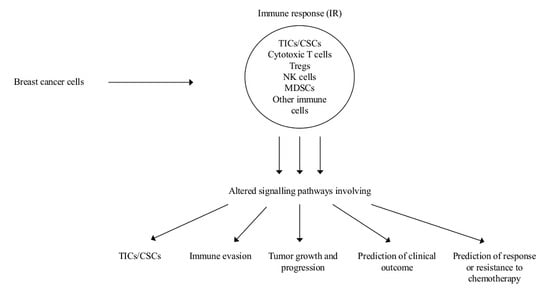
1. Introduction
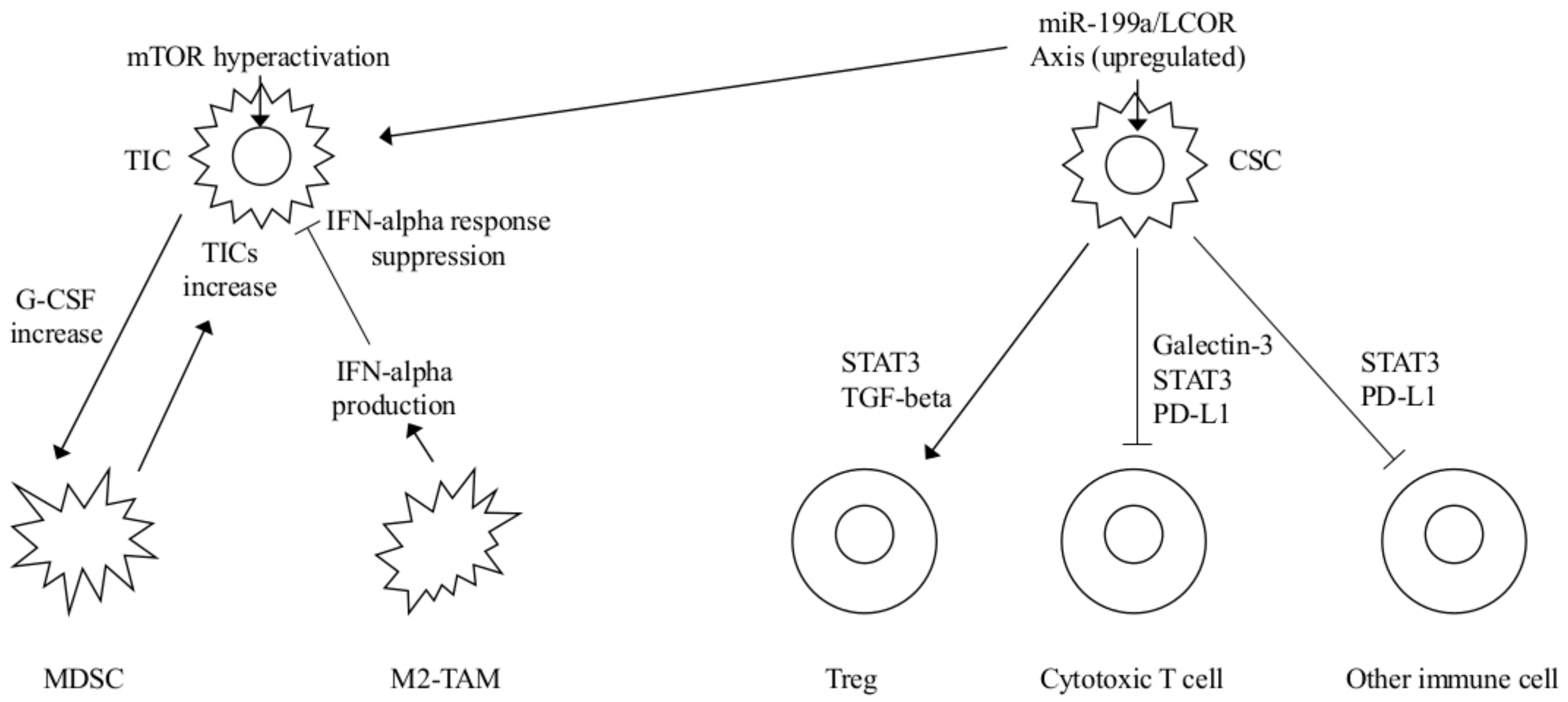
2. Role of Myeloid Derived Suppressor Cells (MDSCs) on Tumor Initiating Cells (TICs) and of INFs on Cancer Stem Cells (CSCs)
2.1. MDSCs and TICs-Notch Signaling
2.2. INF, TIC Activities, and CSCs
2.3. Stabilization of PD-L1, Up-Regulation of CD47 in Cancer Cells, and ShcA Signaling as Mechanisms of Immune Evasion
2.3.1. PD-L1 Stabilization
2.3.2. CD47 Upregulation
2.3.3. Type III Chaperone Protein ShcA (ShcA) Signaling
2.4. Altered Intra-and Inter-Cellular Signaling in the Immune Microenvironment Affects Tumor Growth and Progression
2.4.1. C-C motif Chemokine Receptor (CCR)7 and Its Chemokine Ligands (CCL)19/(CCL)21
2.4.2. Annexin 1 (ANKA1) and Macrophages
2.4.3. Inflammatory Cells, NR4A1 TGF-β/SMAD Signaling
2.5. Prediction of Clinical Outcome
2.5.1. The NF-κB Pathway
2.5.2. Prognostic HTICS Signature Involving an IR
2.5.3. Long-Noncoding (Lnc) RNAs
2.6. Prediction of Response or Resistance to Chemotherapy
2.6.1. Two Immune-Based Gene Modules
2.6.2. Plasma Cells Inhibit Immunogenic Cell Death (ICD)
3. Perspectives and Conclusion
Conflicts of Interest
References
- Nicolini, A.; Carpi, A. Immune manipulation of advanced breast cancer: An interpretative model of the relationship between immune system and tumor cell biology. Med. Res. Rev. 2009, 29, 436–471. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Carpi, A.; Rossi, G. Cytokines in breast cancer. Cytokine Growth Factor Rev. 2006, 17, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Al-Hajj, M.; Clarke, M.F. Self-renewal and solid tumor stem cells. Oncogene 2004, 23, 7274–7282. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Fini, M.; Borsari, V.; Fallahi, P.; Antonelli, A.; Berti, P.; Carpi, A.; Miccoli, P. Stem cells: Their role in breast cancer development and resistance to treatment. Curr. Pharm. Biotechnol. 2011, 12, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Welte, T.; Kim, I.S.; Tian, L.; Gao, X.; Wang, H.; Li, J.; Holdman, X.B.; Herschkowitz, J.I.; Pond, A.; Xie, G. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat. Cell Biol. 2016, 18, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Waight, J.D.; Hu, Q.; Miller, A.; Liu, S.; Abrams, S.I. Tumor-derived G-CSF facilitates neoplastic growth through a granulocytic myeloid-derived suppressor cell-dependent mechanism. PLoS ONE 2011, 6, e27690. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Krelin, Y.; Dvorkin, T.; Bjorkdahl, O.; Segal, S.; Dinarello, C.A.; Voronov, E.; Apte, R.N. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1β-secreting cells. J. Immunol. 2005, 175, 8200–8208. [Google Scholar] [CrossRef] [PubMed]
- Bunt, S.K.; Yang, L.; Sinha, P.; Clements, V.K.; Leips, J.; Ostrand-Rosenberg, S. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res. 2007, 67, 10019–10026. [Google Scholar] [CrossRef] [PubMed]
- Sinha, P.; Clements, V.K.; Fulton, A.M.; Ostrand-Rosenberg, S. Prostaglandin E2 promotes tumor progression by inducing myeloid-derived suppressor cells. Cancer Res. 2007, 67, 4507–4513. [Google Scholar] [CrossRef] [PubMed]
- Gallina, G.; Dolcetti, L.; Serafini, P.; De Santo, C.; Marigo, I.; Colombo, M.P.; Basso, G.; Brombacher, F.; Borrello, I.; Zanovello, P.; et al. Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J. Clin. Investig. 2006, 116, 2777–2790. [Google Scholar] [CrossRef] [PubMed]
- Bronte, V.; Serafini, P.; De Santo, C.; Marigo, I.; Tosello, V.; Mazzoni, A.; Segal, D.M.; Staib, C.; Lowel, M.; Sutter, G. IL-4-induced arginase 1 suppresses alloreactive T cells in tumor-bearing mice. J. Immunol. 2003, 170, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.; Ishida, T.; Oyama, T.; Ran, S.; Kravtsov, V.; Nadaf, S.; Carbone, D.P. Vascular endothelial growth factor inhibits the development of dendritic cells and dramatically affects the differentiation of multiple hematopoietic lineages in vivo. Blood 1998, 92, 4150–4166. [Google Scholar] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Liu, D.D.; Choudhury, A.; Hang, X.; Wei, Y.; Zamalloa, J.; Alfaro-Aco, R.; Chakrabarti, R.; Jiang, Y.Z.; Koh, B.I.; et al. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat. Cell Biol. 2017, 19, 711–723. [Google Scholar] [CrossRef] [PubMed]
- Nicolini, A.; Ferrari, P.; Rossi, G.; Carpi, A. Tumour growth and immune evasion as targets for a new strategy in advanced cancer. Endocr. Relat. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Skora, A.D.; Li, Z.; Liu, Q.; Tam, A.J.; Blosser, R.L.; Diaz, L.A., Jr.; Papadopoulos, N.; Kinzler, K.W.; Vogelstein, B.; et al. Eradication of metastatic mouse cancers resistant to immune checkpoint blockade by suppression of myeloid-derived cells. Proc. Natl. Acad. Sci. USA 2014, 111, 11774–11779. [Google Scholar] [CrossRef] [PubMed]
- Ahmadzadeh, M.; Johnson, L.A.; Heemskerk, B.; Wunderlich, J.R.; Dudley, M.E.; White, D.E.; Rosenberg, S.A. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009, 114, 1537–1544. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.; Strome, S.E.; Salomao, D.R.; Tamura, H.; Hirano, F.; Flies, D.B.; Roche, P.C.; Lu, J.; Zhu, G.; Tamada, K. Tumor-associated B7-H1 promotes T-cell apoptosis: A potential mechanism of immune evasion. Nat. Med. 2002, 8, 793–800. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Lim, S.O.; Xia, W.; Lee, H.H.; Chan, L.C.; Kuo, C.W.; Khoo, K.H.; Chang, S.S.; Cha, J.H.; Kim, T.; et al. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat. Commun. 2016, 7, 12632. [Google Scholar] [CrossRef] [PubMed]
- Betancur, P.A.; Abraham, B.J.; Yiu, Y.Y.; Willingham, S.B.; Khameneh, F.; Zarnegar, M.; Kuo, A.H.; McKenna, K.; Kojima, Y.; Leeper, N.J.; et al. A CD47-associated super-enhancer links pro-inflammatory signalling to CD47 upregulation in breast cancer. Nat. Commun. 2017, 8, 14802. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D.; et al. The CD47-signal regulatory protein alpha (SIRPα) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed]
- Jaiswal, S.; Jamieson, C.H.; Pang, W.W.; Park, C.Y.; Chao, M.P.; Majeti, R.; Traver, D.; van Rooijen, N.; Weissman, I.L. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell 2009, 138, 271–285. [Google Scholar] [CrossRef] [PubMed]
- Davidson, E.H. Emerging properties of animal gene regulatory networks. Nature 2010, 468, 911–920. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Schuijers, J.; Lin, C.Y.; Weintraub, A.S.; Abraham, B.J.; Lee, T.I.; Bradner, J.E.; Young, R.A. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol. Cell 2015, 58, 362–370. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Abraham, B.J.; Lee, T.I.; Lau, A.; Saint-André, V.; Sigova, A.A.; Hoke, H.A.; Young, R.A. Super-enhancers in the control of cell identity and disease. Cell 2013, 155, 934–947. [Google Scholar] [CrossRef] [PubMed]
- Ahn, R.; Sabourin, V.; Bolt, A.M.; Hébert, S.; Totten, S.; De Jay, N.; Festa, M.C.; Young, Y.K.; Im, Y.K.; Pawson, T. The Shc1 adaptor simultaneously balances Stat1 and Stat3 activity to promote breast cancer immune suppression. Nat. Commun. 2017, 8, 14638. [Google Scholar] [CrossRef] [PubMed]
- Akbay, E.A.; Koyama, S.; Carretero, J.; Altabef, A.; Tchaicha, J.H.; Christensen, C.L.; Mikse, O.R.; Cherniack, A.D.; Beauchamp, E.M.; Pugh, T.J. Activation of the PD-1 pathway contributes to immune escape in EGFR-driven lung tumors. Cancer Discov. 2013, 3, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- Eyob, H.; Ekiz, H.A.; Derose, Y.S.; Waltz, S.E.; Williams, M.A.; Welm, A.L. Inhibition of ron kinase blocks conversion of micrometastases to overt metastases by boosting antitumor immunity. Cancer Discov. 2013, 3, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Paolino, M.; Choidas, A.; Wallner, S.; Pranjic, B.; Uribesalgo, I.; Loeser, S.; Jamieson, A.M.; Langdon, W.Y.; Ikeda, F.; Fededa, J.P. The E3 ligase Cbl-b and TAM receptors regulate cancer metastasis via natural killer cells. Nature 2014, 507, 508–512. [Google Scholar] [CrossRef] [PubMed]
- Ursini-Siegel, J.; Cory, S.; Zuo, D.; Hardy, W.R.; Rexhepaj, E.; Lam, S.; Schade, B.; Jirstrom, K.; Bjur, E.; Piccirillo, C.A. Receptor tyrosine kinase signaling favors a protumorigenic state in breast cancer cells by inhibiting the adaptive immune response. Cancer Res. 2010, 70, 7776–7787. [Google Scholar] [CrossRef] [PubMed]
- Ha, J.R.; Siegel, P.M.; Ursini-Siegel, J. The Tyrosine Kinome Dictates Breast Cancer Heterogeneity and Therapeutic Responsiveness. J. Cell. Biochem. 2016, 117, 1971–1990. [Google Scholar] [CrossRef] [PubMed]
- Sonbul, S.N.; Gorringe, K.L.; Aleskandarany, M.A.; Mukherjee, A.; Green, A.R.; Ellis, I.O.; Rakha, E.A. Chemokine (C-C motif) receptor 7 (CCR7) associates with the tumour immune microenvironment but not progression in invasive breast carcinoma. J. Pathol. Clin. Res. 2017, 3, 105–114. [Google Scholar] [CrossRef] [PubMed]
- Tutunea-Fatan, E.; Majumder, M.; Xin, X.; Lala, P.K. The role of CCL21/CCR7 chemokine axis in breast cancer-induced lymphangiogenesis. Mol. Cancer. 2015, 14, 35. [Google Scholar] [CrossRef] [PubMed]
- Chi, B.J.; Du, C.L.; Fu, Y.F.; Zhang, Y.N.; Wang, R.W. Silencing of CCR7 inhibits the growth, invasion and migration of prostate cancer cells induced by VEGFC. Int. J. Clin. Exp. Pathol. 2015, 8, 12533–12540. [Google Scholar] [PubMed]
- Cunningham, H.D.; Shannon, L.A.; Calloway, P.A.; Fassold, B.C.; Dunwiddie, I.; Vielhauer, G.; Zhang, M.; Vines, C.M. Expression of the C-C chemokine receptor 7 mediates metastasis of breast cancer to the lymph nodes in mice. Transl. Oncol. 2010, 3, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Andre, F.; Cabioglu, N.; Assi, H.; Sabourin, J.C.; Delaloge, S.; Sahin, A.; Broglio, K.; Spano, J.P.; Combadiere, C.; Bucana, C.; et al. Expression of chemokine receptors predicts the site of metastatic relapse in patients with axillary node positive primary breast cancer. Ann. Oncol. 2006, 17, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ji, R.; Li, J.; Gu, Q.; Zhao, X.; Sun, T.; Wang, J.; Li, J.; Du, Q.; Sun, B. Correlation effect of EGFR and CXCR4 and CCR7 chemokine receptors in predicting breast cancer metastasis and prognosis. J. Exp. Clin. Cancer Res. 2010, 29, 16. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.L.; Burchell, J.; Grimshaw, M.J. Endothelins induce CCR7 expression by breast tumor cells via endothelin receptor A and hypoxia-inducible factor-1. Cancer Res. 2006, 66, 11802–11807. [Google Scholar] [CrossRef] [PubMed]
- Weitzenfeld, P.; Kossover, O.; Körner, C.; Meshel, T.; Wiemann, S.; Seliktar, D.; Legler, D.F.; Ben-Baruch, A. Chemokine axes in breast cancer: Factors of the tumor microenvironment reshape the CCR7-driven metastatic spread of luminal-A breast tumors. J. Leukoc. Biol. 2016, 99, 1009–1025. [Google Scholar] [CrossRef] [PubMed]
- Xuan, W.; Qu, Q.; Zheng, B.; Xiong, S.; Fan, G.H. The chemotaxis of M1 and M2 macrophages is regulated by different chemokines. J. Leukoc. Biol. 2015, 97, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Na, I.K.; Busse, A.; Scheibenbogen, C.; Ghadjar, P.; Coupland, S.E.; Letsch, A.; Loddenkemper, C.; Stroux, A.; Bauer, S.; Thiel, E.; et al. Identification of truncated chemokine receptor 7 in human colorectal cancer unable to localize to the cell surface and unreactive to external ligands. Int. J. Cancer 2008, 123, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Moraes, L.A.; Kar, S.; Foo, S.L.; Gu, T.; Toh, Y.Q.; Ampomah, P.B.; Sachaphibulkij, K.; Yap, G.; Zharkova, O.; Lukman, H.M. Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 2017, 7, 17925. [Google Scholar] [CrossRef] [PubMed]
- Mussunoor, S.; Murray, G.I. The role of annexins in tumour development and progression. J. Pathol. 2008, 216, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Bist, P.; Leow, S.C.; Phua, Q.H.; Shu, S.; Zhuang, Q.; Loh, W.T.; Nguyen, T.H.; Zhou, J.B.; Hooi, S.C.; Lim, L.H. Annexin-1 interacts with NEMO and RIP1 to constitutively activate IKK complex and NF-κB: Implication in breast cancer metastasis. Oncogene 2011, 30, 3174–3185. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Anbalagan, D.; Lee, L.H.; Samy, R.P.; Shanmugam, M.K.; Kumar, A.P.; Sethi, G.; Lobie, P.E.; Lim, L.H. ANXA1 inhibits miRNA-196a in a negative feedback loop through NF-κB and c-Myc to reduce breast cancer proliferation. Oncotarget 2016, 7, 27007–27020. [Google Scholar] [CrossRef] [PubMed]
- Khau, T.; Langenbach, S.Y.; Schuliga, M.; Harris, T.; Johnstone, C.N.; Anderson, R.L.; Stewart, A.G. Annexin-1 signals mitogen-stimulated breast tumor cell proliferation by activation of the formyl peptide receptors (FPRs) 1 and 2. FASEB J. 2011, 25, 483–496. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investg. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, B.L.; Francis, P.A.; Parker, B.S.; Anderson, R.L. Strategies for the discovery and development of therapies for metastatic breast cancer. Nat. Rev. Drug Discov. 2012, 11, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Drabsch, Y.; Dekker, T.J.; de Vinuesa, A.G.; Li, Y.; Hawinkels, L.J.; Sheppard, K.A.; Goumans, M.J.; Luwor, R.B.; de Vries, C.J. Nuclear receptor NR4A1 promotes breast cancer invasion and metastasis by activating TGF-β signalling. Nat. Commun. 2014, 5, 3388. [Google Scholar] [CrossRef] [PubMed]
- Schmierer, B.; Hill, C.S. TGFβ-SMAD signal transduction: Molecular specificity and functional flexibility. Nat. Rev. Mol. Cell. Biol. 2007, 8, 970–982. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Liu, C.; Derynck, R. New regulatory mechanisms of TGF-β receptor function. Trends Cell Biol. 2009, 19, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Allavena, P.; Mantovani, A. Cancer related inflammation: The macrophage connection. Cancer Lett. 2008, 267, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Okubo, M.; Kioi, M.; Nakashima, H.; Sugiura, K.; Mitsudo, K.; Aoki, I.; Taniguchi, H.; Tohnai, I. M2-polarized macrophages contribute to neovasculogenesis, leading to relapse of oral cancer following radiation. Sci. Rep. 2016, 6, 27548. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Miremadi, A.; Pinder, S.E.; Ellis, I.O.; Caldas, C. An immune response gene expression module identifies a good prognosis subtype in estrogen receptor negative breast cancer. Genome Biol. 2007, 8, R157. [Google Scholar] [CrossRef] [PubMed]
- Lakhani, S.R.; Jacquemier, J.; Sloane, J.P.; Gusterson, B.A.; Anderson, T.J.; van de Vijver, M.J.; Farid, L.M.; Venter, D.; Antoniou, A.; Storfer-Isser, A.; et al. Multifactorial analysis of differences between sporadic breast cancers and cancers involving BRCA1 and BRCA2 mutations. J. Natl. Cancer Inst. 1998, 90, 1138–1145. [Google Scholar] [CrossRef] [PubMed]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. 2007, 9, R65. [Google Scholar] [CrossRef] [PubMed]
- Jézéquel, P.; Loussouarn, D.; Guérin-Charbonnel, C.; Campion, L.; Vanier, A.; Gouraud, W.; Lasla, H.; Guette, C.; Valo, I.; Verrièle, V.; et al. Gene-expression molecular subtyping of triple-negative breast cancer tumours: Importance of immune response. Breast Cancer Res. 2015, 17, 43. [Google Scholar] [CrossRef] [PubMed]
- Buckley, N.E.; Haddock, P.; De Matos Simoes, R.; Parkes, E.; Irwin, G.; Emmert-Streib, F.; McQuaid, S.; Kennedy, R.; Mullan, P. A BRCA1 deficient, NFκB driven immune signal predicts good outcome in triple negative breast cancer. Oncotarget 2016, 7, 19884–19896. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.C.; Zacksenhouse, M.; Eisen, A.; Nofech-Mozes, S.; Zacksenhaus, E. Identification of cell proliferation, immune response and cell migration as critical pathways in a prognostic signature for HER2+:ERα- breast cancer. PLoS ONE 2017, 12, e0179223. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Kong, D.; Chen, Q.; Ping, Y.; Pang, D. Oncogenic long noncoding RNA landscape in breast cancer. Mol. Cancer 2017, 16, 129. [Google Scholar] [CrossRef] [PubMed]
- Foukakis, T.; Lövrot, J.; Matikas, A.; Zerdes, I.; Lorent, J.; Tobin, N.; Suzuki, C.; Brage, S.E.; Carlsson, L.; Einbeigi, Z. Immune gene expression and response to chemotherapy in advanced breast cancer. Br. J. Cancer 2018, 118, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Orthmann, A.; Peiker, L.; Fichtner, I.; Hoffmann, A.; Hilger, R.A.; Zeisig, R. Improved Treatment of MT-3 Breast Cancer and Brain Metastases in a Mouse Xenograft by LRP-Targeted Oxaliplatin Liposomes. J. Biomed. Nanotechnol. 2016, 12, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, A.; Aroldi, F.; Bertocchi, P.; Prochilo, T.; Mutti, S.; Savelli, G.; Fraccon, A.P.; Zaniboni, A. GEMOX: An Active Regimen for the Treatment of Luminal and Human Epidermal Growth Factor Receptor 2-Positive Metastatic Breast Cancer. Chemotherapy 2017, 62, 30–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Wang, Z.; Hu, X.; Wang, B.; Cao, J.; Lv, F.; Zhen, C.; Zhang, S.; Shao, Z. A phase II trial of biweekly vinorelbine and oxaliplatin in second- or third-line metastatic triple-negative breast cancer. Cancer Biol. Ther. 2015, 16, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Xiao, Y.; Wei, W.; Guo, J.X.; Liu, Y.C.; Huang, X.H.; Zhang, R.X.; Wu, Y.J.; Zhou, J. Clinical efficacy of administering oxaliplatin combined with S-1 in the treatment of advanced triple-negative breast cancer. Exp. Ther. Med. 2015, 10, 379–385. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Shalapour, S.; Font-Burgada, J.; Di Caro, G.; Zhong, Z.; Sanchez-Lopez, E.; Dhar, D.; Willimsky, G.; Ammirante, M.; Strasner, A.; Hansel, D.E. Immunosuppressive plasma cells impede T-cell-dependent immunogenic chemotherapy. Nature 2015, 521, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Larkin, J.; Chiarion-Sileni, V.; Gonzalez, R.; Grob, J.J.; Cowey, C.L.; Lao, C.D.; Schadendorf, D.; Dummer, R.; Smylie, M.; Rutkowski, P. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N. Engl. J. Med. 2015, 373, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Brahmer, J.; Reckamp, K.L.; Baas, P.; Crinò, L.; Eberhardt, W.E.; Poddubskaya, E.; Antonia, S.; Pluzanski, A.; Vokes, E.E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Borghaei, H.; Paz-Ares, L.; Horn, L.; Spigel, D.R.; Steins, M.; Ready, N.E.; Chow, L.Q.; Vokes, E.E.; Felip, E.; Holgado, E.; et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 373, 1627–1639. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| IR Factor or Mediator | Mechanism | Result | Perspective | Ref. |
|---|---|---|---|---|
| MDSCs | Hyperactivated Akt-mTOR pathway G-CSF increased expression MDSC mediated Notch stemness-related genes upregulation | TICs mediated MDSCs accumulation Increased TICs frequency | mTOR plus checkpoint inhibitors FGFR or G-CSF inhibitors | [5] |
| INF-α | miR-199a overexpression LCOR repression and modulation of the INF-α mediated suppressive effects | CSCs protected by INF-mediated effects MaSC-enriched basal vs luminal population | INF-α plus miR-199-LCOR targeting as adjuvant therapy | [15] |
| PD-L1 | N192, N200, N219 glycosylation induces PD-L1 stability and antagonizes PD-L1 GSK3-β interactions as well as EGF and other EGFR ligands | Immunesuppression | Targeting PD-L1 stabilization | [18] |
| CD47 | TNF-NF-κB mediated CD47 upregulation by SEs CD47 SIRP α binding on macrophages | Cancer cells protection from phagocytosis | Increased macrophage phagocytosis by TNF-NF-κB inhibition | [19] |
| Y239/Y240-Shc-A phosphory-lation | Antitumor STAT-1 activity decrease STAT-3 mediated immune suppression increase | Immunesuppression | Constitutive binding or specific Y239/Y240-Shc-A inhibitors to sensitize to immunotherapies | [25] |
| IR Factor or Mediator | Mechanism | Result | Perspective | Ref. |
|---|---|---|---|---|
| CCR7 | Membrane CCR7-CCL19/CCL21 interaction; No cytoplasmic CCR7-CCL19/CCL21 interaction | Treg and macrophage attraction to the microenvironment; inversely associated with CD3+ cells in the stroma | Better evaluation of CCR7 role in membrane and cytoplasm | [31] |
| Annexin-1 | FPR2-ERK-NF-κB pathway activation, M2 phenotype macrophages polarization | Angiogenesis, tumor progression, immune suppression | Targeting FPR2-ERK signaling | [41] |
| NR4A1 | NR4A1 hyperexpression T-βRI activation, SMAD 2/3 phosphorylation, intense SMAD signaling | EMT and cell migration, poor prognosis | Targeting TGF-β and NR4A1 | [49] |
| Immune Genes/s or IR Mediator | Mechanism | Result | Perspective | Ref. |
|---|---|---|---|---|
| TNBC with BRCA1 dysfunction | “NF-κB on” signal, M1-type macrophages microenvironment, and CD8+ infiltration | Better outcome | Checkpoint inhibitors in addition to conventional FEC CT | [56] |
| 17-gene HTICs signature | IR, proliferation and migration as critical biological pathways | Worse prognosis and benefit from trastuzumab in HER2+ ER- BC | More appropriate adjuvant therapy in HER2+ ER-BC | [57] |
| LncRNAs | Regulation of the immune system activation by 30 hyper- and 25 hypo- expressed Lnc RNAs | Tumor progression and worse survival | Prognosis and complementation of conventional parameters in specific subtypes | [58] |
| Immune module SCORE | Activated immune microenvironment | Prediction of response to CT in ER+ and Luminal BCs | Better patient selection and design of combined chemo-immunotherapies | [59] |
| Immune suppressive plasma cells expressing IGA, IL-10 and PDL-1 | Inhibition of oxaliplatin tumor directed CTL activation and ICD | Poor response to oxaliplatin | Inhibition of IGA+ plasmocytes in oxaliplatin treated patients | [65] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nicolini, A.; Ferrari, P.; Diodati, L.; Carpi, A. Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management. Int. J. Mol. Sci. 2018, 19, 2733. https://doi.org/10.3390/ijms19092733
Nicolini A, Ferrari P, Diodati L, Carpi A. Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management. International Journal of Molecular Sciences. 2018; 19(9):2733. https://doi.org/10.3390/ijms19092733
Chicago/Turabian StyleNicolini, Andrea, Paola Ferrari, Lucrezia Diodati, and Angelo Carpi. 2018. "Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management" International Journal of Molecular Sciences 19, no. 9: 2733. https://doi.org/10.3390/ijms19092733
APA StyleNicolini, A., Ferrari, P., Diodati, L., & Carpi, A. (2018). Alterations of Signaling Pathways Related to the Immune System in Breast Cancer: New Perspectives in Patient Management. International Journal of Molecular Sciences, 19(9), 2733. https://doi.org/10.3390/ijms19092733