The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections



 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
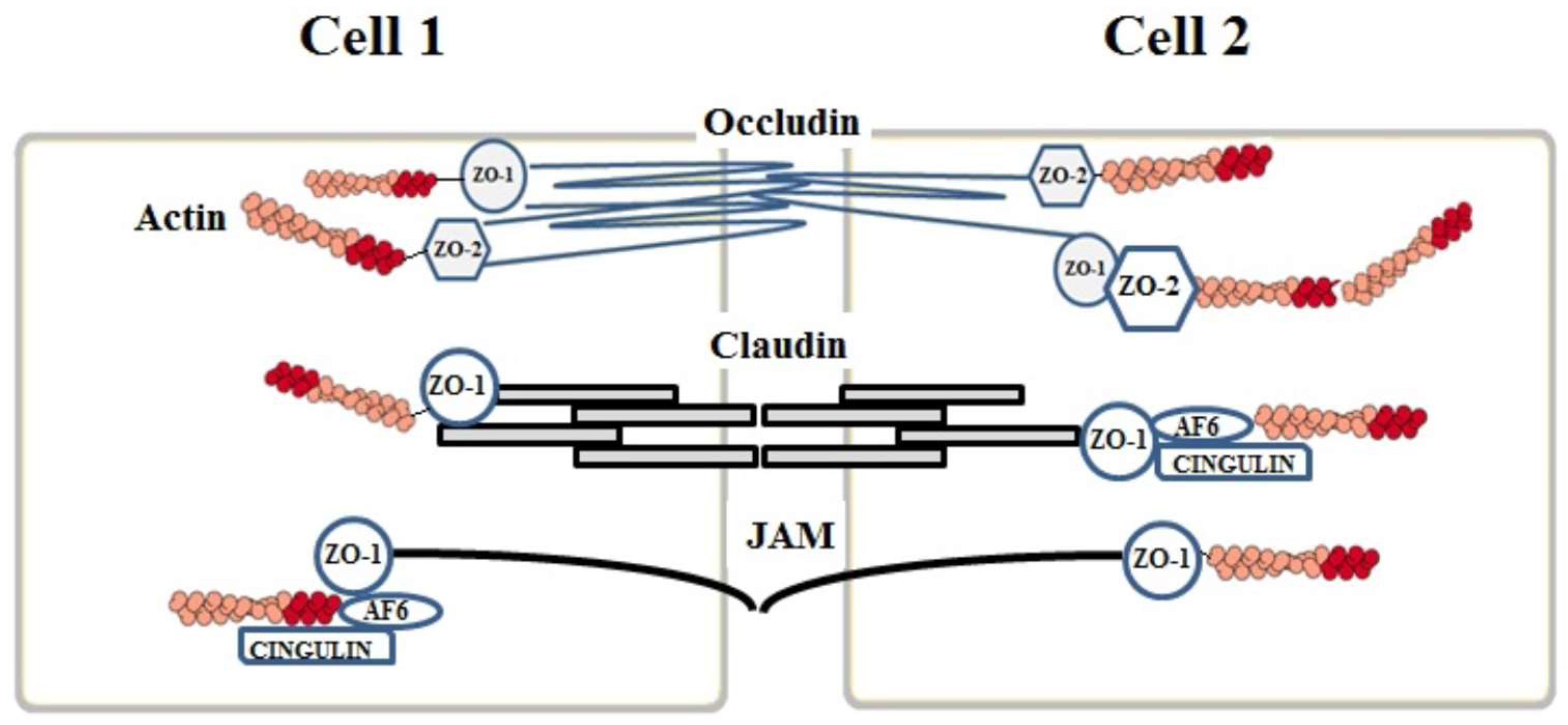
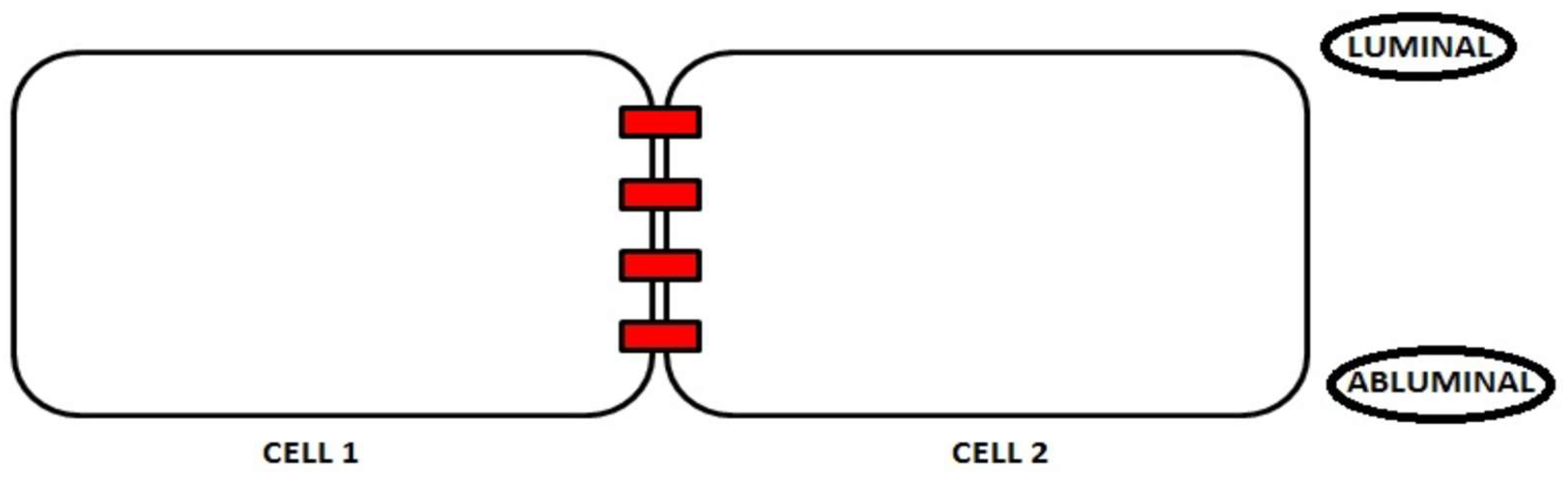
2. Junction Proteins of the Endothelial Cells
2.1. Tight Junctions Complex
2.2. Claudins
2.3. Occludins
2.4. Junctional Adhesion Molecules (JAMs)
3. Auxiliary Cytoplasmic Proteins
4. Adherens Junctions Complex (AJ)
5. The Polarization Process
6. Junction Proteins and Neurodegeneration
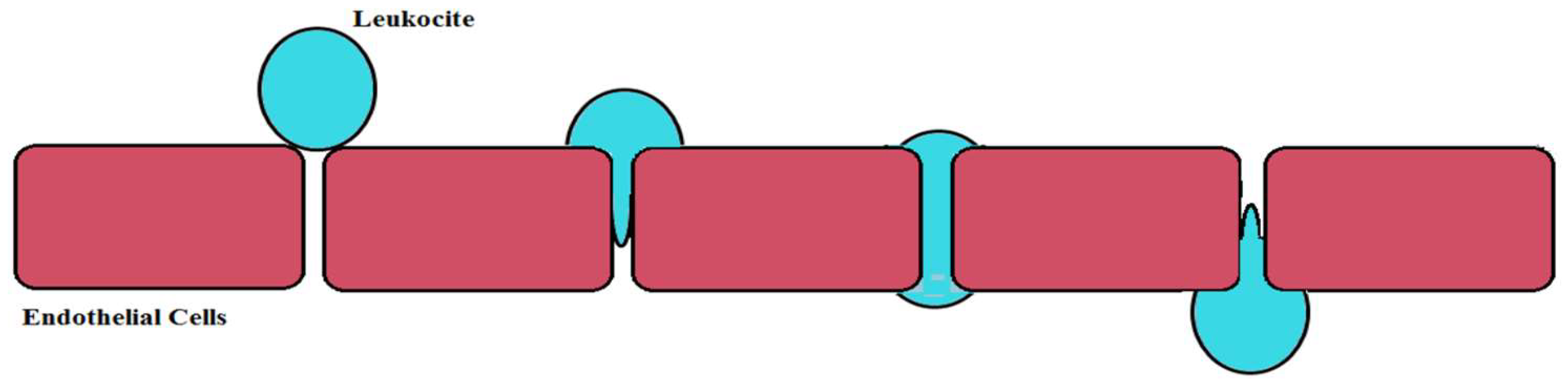
6.1. Brain Ischemia
6.2. Amyotrophic Lateral Sclerosis (ALS)
6.3. Multiple Sclerosis
6.4. Alzheimer’s Disease
6.5. Viral and Bacterial Infections
7. BBB and Nutrients
8. Nutrient Transport and Neurodegeneration
9. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Sweeney, M.D.; Sagare, A.P.; Zlokovic, B.V. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat. Rev. Neurol. 2018, 14, 133–150. [Google Scholar] [CrossRef] [PubMed]
- Begley, D.J.; Brightman, M.W. Structural and functional aspects of the blood-brain barrier. Prog. Drug 2003, 61, 39–78. [Google Scholar]
- Obermeier, B.; Daneman, R.; Ranshoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Saunders, N.R.; Liddelow, S.A.; Dziegielewska, K.M. Barrier mechanisms in the developing brain. Front. Pharmacol. 2012, 29, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Olendof, W.H.; Cornford, M.E.; Brown, W.J. The large apparent work capability of the blood-brain barrier: A study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Ann. Neurol. 1977, 1, 409–417. [Google Scholar] [CrossRef]
- Bazzoni, G.; Dejana, E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol. Rev. 2004, 84, 869–901. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Sato, S.; Yamaguchi, H.; Kamoi, M.; Asashima, T.; Terasaki, T. Exogenous expression of claudin-5 induces barrier properties in cultured rat brain capillary endothelial cells. J. Cell Physiol. 2007, 210, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed]
- Liebner, S.; Corada, M.; Bangsow, T.; Babbage, J.; Taddei, A.; Czupalla, C.J.; Reis, M.; Felici, A.; Wolburg, H.; Fruttiger, M. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J. Cell Biol. 2008, 183, 409–417. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, T.; Agarwal, R.; Morin, P.J. Phosphorylation of claudin-3 at threonine 192 by cAMP-dependent protein kinase regulates tight junction barrier function in ovarian cancer cells. J. Biol. Chem. 2005, 280, 26233–26260. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Tachikawa, M.; Obuchi, W.; Hoshi, Y.; Tomioka, Y.; Ohtsuki, S.; Terasaki, T. A study protocol for quantitative targeted absolute proteomics (QTAP) by LC-MS/MS: Application for inter-strain differences in protein expression levels of transporters, receptors, claudin-5 and marker proteins at the blood-brain barrier in ddY, FVB and C57BL/6J mice. Fluids Barriers CNS 2013, 10, 1–21. [Google Scholar] [CrossRef]
- Luissint, A.C.; Federici, C.; Guillonneau, F.; Chrétien, F.; Camoin, L.; Ganeshamoorthy, K.; Couraud, P.O. Guanine nucleotide-binding protein G alpha i2: A new partner of claudin-5 that regulates tight junction integrity in human brain endothelial cells. J. Cereb. Blood Flow Metab. 2012, 32, 860–873. [Google Scholar] [CrossRef] [PubMed]
- Mandel, I.; Paperna, T.; Volkowich, A.; Merhav, M.; Glass-Marmor, L.; Miller, A. The ubiquitin-proteasome pathway regulates claudin-5 degradation. J. Cell Biochem. 2012, 113, 2415–2423. [Google Scholar] [CrossRef] [PubMed]
- Musolino, P.L.; Gong, Y.; Snyder, J.M.; Jimenez, S.; Lok, J.; Lo, E.H.; Moser, A.B.; Grabowski, E.F.; Frosch, M.P.; Eichler, F.S. Brain endothelial dysfunction in cerebral adrenoleukodystrophy. Brain 2015, 138, 3206–3220. [Google Scholar] [CrossRef] [PubMed]
- Haseloff, R.F.; Dithmer, S.; Winkler, L.; Wolburg, H.; Blasig, I.E. Transmembrane proteins of the tight junctions at the blood-brain barrier: Structural anf functional aspects. Semin. Cell Dev. Biol. 2015, 38, 16–25. [Google Scholar] [CrossRef] [PubMed]
- Furuse, M.; Hirase, T.; Itoh, M.; Nagafuchi, A.; Yonemura, S.; Tsukita, S. Occludin: A novel integral membrane protein localizing at tight junctions. J. Cell Biol. 1993, 123, 1777–1788. [Google Scholar] [CrossRef] [PubMed]
- Dorfel, M.J.; Huber, O. Modulation of tight junction structure and function by kinases and phosphatases targeting occluding. J. Biomed. Biotechnol. 2012, 2012, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Muthusamy, A.; Lin, C.M.; Shanmugam, S.; Lindner, H.M.; Abcouwer, S.F.; Antonetti, D.A. Ischemia-reperfusion injury induces occludin phosphorylation/ubiquitination and retinal vascular permeability in a VEGFR-2-dependent manner. J. Cereb. Blood Flow Metab. 2014, 34, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Garcidueñas, L.; Vojdani, A.; Blaurock-Busch, E.; Busch, Y.; Friedle, A.; Franco-Lira, M.; Sarathi-Mukherjee, P.; Martínez-Aguirre, X.; Park, S.-B.; Torres-Jardón, R. Air pollution and children: Neural and tight junction antibodies and combustion metals, the role of barrier breakdown and brain immunity in neurodegeneration. J. Alzheimers Dis. 2015, 43, 1039–1058. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G. Phatobiology of junctional adhesion molecules. Antioxid. Redox. Signal 2011, 1221–1234. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Furuse, M.; Morita, K.; Kubota, K.; Saitou, M.; Tsukita, S. Direct binding of three tigt junction-associate MAGUKs, ZO-1, ZO-2 and ZO-3 with the COOH termini of claudins. J. Cell Biol. 1999, 147, 1351–1363. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G.; Martı́nez-Estrada, O.M.; Orsenigo, F.; Cordenonsi, M.; Citi, S.; Dejana, E. Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin and occludin. J. Biol. Chem. 2000, 275, 20520–20526. [Google Scholar] [CrossRef] [PubMed]
- Fanning, A.S.; Ma, T.Y.; Anderson, J.M. Isolation and functional characterization of the actin binding region in the tight junction protein ZO-1. FASEB 2002, 16, 1835–1837. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Abbruscato, T.J.; Egleton, R.D.; Brown, R.C.; Huber, J.D.; Campos, C.R.; Davis, T.P. Nicotine increase in vivo blood-brain barrier permeability and alters cerebral microvascular tight junction protein distribution. Brain Res. 2004, 1027, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Betanzos, A.; Huerta, M.; Lopez-Bayghen, E.; Azuara, E.; Amerena, J.; González-Mariscal, L. The tight junction protein ZO-2 associates with Jun, Fos and C/EBP transcription factors in epithelial cells. Exp. Cell Res. 2004, 292, 51–66. [Google Scholar] [CrossRef] [PubMed]
- Bauer, H.C.; Traweger, A.; Bauer, H. Proteins of the tight junction in the blood-brain barrier. In Blood-Spinal Cord and Brain Barriers in Health and Disease; Sharma, H.S., Westman, J., Eds.; Elsevier: New York, NY, USA, 2004; pp. 1–10. [Google Scholar]
- Matter, K.; Balda, M.S. Signalling to and from tight junctions. Nat. Rev. Mol. Cell Biol. 2003, 4, 225–236. [Google Scholar] [CrossRef] [PubMed]
- Worzfeld, T.; Schwainger, M. Apicobasal polariry of brain endothelial cells. J. Cereb. Blood Flow Metab. 2015, 36, 340–362. [Google Scholar] [CrossRef] [PubMed]
- Sigurbjornsdottir, S.; Mathew, R.; Leptin, M. Molecular mechanisms of de novo lumen formation. Nat. Rev. Mol. Cell Biol. 2014, 15, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, M.S.; Colon, F.L. Cellular and molecular mechanisms underlying blood vessel lumen formation. Bioessays 2014, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Wolff, J.; Chao, T.I. Cytoarchitectonics of non-neuronal cells in the central nervous system. Adv. Mol. Cell Biol. 2004, 31, 1–51. [Google Scholar]
- Tewes, B.J.; Galla, H.J. Lipid polarity in brain capillarity endothelial cells. Endothelium 2001, 8, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.C.; Lankheet, N.A.; Poller, B.; Wagenaar, E.; Beijnen, J.H.; Schinkel, A.H. P-Glycoprotein (ABCB1) and breast cancer resistance protein (ABCG2) restrict brain accumulation of the active sunitinib metabolite N-desethyl sunitinib. J. Pharmacol. Exp. Ther 2012, 341, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; Van Zandvoort, M.A.; Oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions and visualization. Pflugers. Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef] [PubMed]
- Bendayan, R.; Ronaldson, P.T.; Gingras, D.; Bendayan, M. In situ localization of P-glycoprotein (ABXB1) in human and rat brain. J. Histochem. Cytochem. 2006, 54, 1159–1167. [Google Scholar] [CrossRef] [PubMed]
- Martin-Belmonte, F.; Gassama, A.; Datta, A.; Yu, W.; Rescher, U.; Gerke, V.; Mostov, K. PTEN mediated apical segregation of phosphoinositides controls epithelial morphogenesis through Cdc42. Cell 2007, 128, 383–397. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Nakayama, A.; Van Lessen, M.; Yamamoto, H.; Hoffmann, S.; Drexler, H.C.; Itoh, N.; Hirose, T.; Breier, G.; Vestweber, D. Spatial regulation of VEGF receptor endocytosis in angiogenesis. Nat. Cell Biol. 2013, 15, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Ebnet, K.; Aurrand-Lions, M.; Kuhn, A.; Kiefer, F.; Butz, S.; Zander, K.; zu Brickwedde, M.-K.M.; Suzuki, A.; Imhof, B.A.; Vestweber, D. The junctional adhesion molecule (JAM) family members JAM-2 and JAM-3 associate with the cell polarity protein PAR- 3: A possible role for JAMs in endothelial cell polarity. J. Cell Sci. 2003, 116, 3879–3891. [Google Scholar] [CrossRef] [PubMed]
- Ngok, S.P.; Geyer, R.; Liu, M.; Kourtidis, A.; Agrawal, S.; Wu, C.; Seerapu, H.R.; Lewis-Tuffin, L.J.; Moodie, K.L.; Huveldt, D. VEGF and Angiopoietin-1 exert opposing effects on cell junctions by regulating the Rho GEF Syx. J. Cell Biol. 2012, 199, 1103–1115. [Google Scholar] [CrossRef] [PubMed]
- Michaelis, U.R.; Chavakis, E.; Kruse, C.; Jungblut, B.; Kaluza, D.; Wandzioch, K.; Manavski, Y.; Heide, H.; Santoni, M.-J.; Potente, M. The polarity protein Scrib is essential for directed endothelial cell migration. Circ. Res. 2013, 112, 924–934. [Google Scholar] [CrossRef] [PubMed]
- Luissint, A.C.; Artus, C.; Glacial, F.; Ganeshamoorthy, K.; Couraud, P.O. Tight junctions at the blood brain barrier:physiological architecture and disease-associated dysregulation. Fluids Barriers CNS 2012, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Farral, A.J.; Wardlaw, J.M. Blood-brain barrier: Ageing and microvascular disease-systematic review and meta-analysis. Neurobiol. Aging 2009, 30, 337–352. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Gonzalez, B.; Hurtado-Alvarado, G.; Esqueda-Leon, E.; Santana-Miranda, R.; Rojas-Zamorano, J.A.; Velazquez-Moctezuma, J. REM sleep loss and recovery regulates blood-brain barrier function. Curr. Neurovasc. Res. 2013, 10, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Czeslawa, V.B.M.A.A.; Miklós, K.F.A.A.A.; Lai, T.A.K.N.B.; Gulyás, G.N.P.K.B.; Nilsson, C.H.K.H.H.; Betty, H.H.B.T.V.; Pettersson, D.S. The gut microbiota influences blood-brain barrier permeability in mice. Sci. Transl. Med. 2014, 6, 263. [Google Scholar] [CrossRef]
- Cummins, P.M. Occludin: One protein, many forms. Mol. Cell Biol. 2012, 32, 242–250. [Google Scholar] [CrossRef] [PubMed]
- Morgan, L.; Shah, B.; Rivers, L.E.; Barden, L.A.; Groom, J.; Chung, R.; Higazi, D.; Desmond, H.; Smith, T.; Staddon, J.M. Inflammation and dephosphorylation of the tight junction protein occludin in an experimental model of multiple sclerosis. Neuroscience 2007, 147, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Ramirez, S.H.; Sato, S.; Kiyota, T.; Cerny, R.L.; Kaibuchi, K.; Persidsky, Y.; Ikezu, T. Phosphorylation of claudin-5 and occludin by rho kinase inbrain endothelial cells. Am. J. Pathol. 2008, 172, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Desai, T.R.; Leeper, N.J.; Hynes, K.L.; Gewertz, B.L. Interleukin-6 causes endothelial barrier dysfunction via the protein kinase C pathway. J. Surg. Res. 2002, 104, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Willis, C.L.; Meske, D.S.; Davis, T.P. Protein kinase C activation modulates reversible increase in cortical blood–brain barrier permeability and tight junction protein expression during hypoxia and posthypoxicreoxygenation. J. Cereb. Blood Flow Metab. 2010, 30, 1847–1859. [Google Scholar] [CrossRef] [PubMed]
- Kanmogne, G.D.; Schall, K.; Leibhart, J.; Knipe, B.; Gendelman, H.E.; Persidsky, Y. HIV-1 gp120 compromises blood–brain barrier integrity and enhancesmonocyte migration across blood–brain barrier: Implication for viral neuropathogenesis. J. Cereb. Blood Flow Metab. 2007, 27, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Sonobe, Y.; Takeuchi, H.; Kataoka, K.; Li, H.; Jin, S.; Mimuro, M.; Hashizume, Y.; Sano, Y.; Kanda, T.; Mizuno, T. Interleukin-25 expressed by braincapillary endothelial cells maintains blood–brain barrier function in aprotein kinase Cepsilon-dependent manner. J. Biol. Chem. 2009, 284, 31834–31842. [Google Scholar] [CrossRef] [PubMed]
- Stamatovic, S.M.; Dimitrijevic, O.B.; Keep, R.F.; Andjelkovic, A.V. Protein kinase C alpha-RhoA cross-talk in CCL2-induced alterations in brain endothelial permeability. J. Biol. Chem. 2006, 281, 8379–8388. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B.; Wolburg, H. Mini-review: Transendothelial migration of leukocytes: Through the front door or around the side of the house. Eur. J. Immunol. 2004, 34, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.; Campbell, M. The dynamic blood brain barrier. FEBS J. 2015, 282, 4067–4079. [Google Scholar] [CrossRef] [PubMed]
- Sandoval, K.E.; Witt, K.A. Blood brain barrier tight junction permeability and ischemic stroke. Neurobiol. Dis. 2008, 32, 200–219. [Google Scholar] [CrossRef] [PubMed]
- Pillai, D.R.; Dittmar, M.S.; Baldaranov, D.; Heidemann, R.M.; Henning, E.C.; Schuierer, G.; Bogdahn, U.; Schlachetzki, F. Cerebral ischemia-reperfusion injury in rats—a3 MRI study on biphasic blood-brain barrier opening and the dynamics of edema formation. J. Cereb. Blood Flow Metab. 2009, 29, 1846–1855. [Google Scholar] [CrossRef] [PubMed]
- Enzmann, G.; Mysiorek, C.; Gorina, R.; Cheng, Y.J.; Ghavampour, S.; Hannocks, M.J.; Prinz, V.; Dirnagl, U.; Endres, M.; Prinz, M.; et al. The neurovascular unit as a selective barrier to polymorphonuclear granulocyte (PMN) infiltration into the brain after ischemic injury. Acta Neuropathol. 2013, 125, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Knowland, D.; Arac, A.; Sekiguchi, K.J.; Hsu, M.; Lutz, S.E.; Perrino, J.; Steinberg, G.K.; Barres, B.A.; Nimmerjahn, A.; Agalliu, D. Stepwise recruitment of transcellular and paracellular pathways underlies blood-brain barrier breakdown in stroke. Neuron 2014, 82, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, K.; Ohta, Y.; Nagai, M.; Morimoto, N.; Kurata, T.; Takehisa, Y.; Ikeda, Y.; Matsuura, T.; Abe, K. Disruption of neurovascular unit prior to motor neuron degeneration in amyotrophic lateral sclerosis. J. Neurosci. Res. 2011, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron 2008, 57, 178–201. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.; Plumb, J.; Mirakhur, M.; McQuaid, S. Tight junctional abnormality in multiple sclerosis white matter affects all calibres of vessel and is associated with blood-brain barrier leakage and active demyelination. J. Pathol. 2003, 201, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Waubant, E. Biomarkers indicative of blood-brain barrier disruption in multiple sclerosis. Dis. Mark. 2006, 22, 235–244. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Kaye, J.A.; Moore, M.; Waichunas, D.; Carlson, N.E.; Quinn, J.F. Blood-brain barrier impairment in Alzheimer disease: Stability and functional significance. Neurology 2007, 68, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.T.; Chu, K.; Jung, K.H.; Park, H.K.; Kim, D.H.; Bahn, J.J.; Kim, J.H.; Oh, M.J.; Lee, S.K.; Kim, M.; et al. Reduced circulating angiogenic cells in Alzheimer disease. Neurology 2009, 72, 1858–1863. [Google Scholar] [CrossRef] [PubMed]
- Solé, M.; Miñano-Molina, A.J.; Unzeta, M. A cross-talk between Abeta and endothelial SSAO/VAP-1 accelerates vascular damage and Abeta aggregation related to CAA-AD. Neurobiol. Aging 2015, 36, 762–775. [Google Scholar] [CrossRef] [PubMed]
- Stuertz, K.; Merx, I.; Eiffert, H.; Schmutzhard, E.; Mader, M.; Nau, R. Enzyme immunoassay detecting teichoic and lipoteichoic acids versus cerebrospinal fluid culture and latex agglutination for diagnosis of Streptococcus pneumoniae meningitis. J. Clin. Microbiol. 1998, 36, 2346–2348. [Google Scholar] [PubMed]
- Doulet, N.; Donnadieu, E.; Laran-Chich, M.P.; Niedergang, F.; Nassif, X.; Couraud, P.O.; Bourdoulous, S. Neisseiria meningitidis infection of human endothelial cells interferes with leukocyte transmigration by preventing the formation of endothelial docking structures. J. Cell Biol. 2006, 173, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.J.; Hancock, B.M.; Bermudez, A.; Del Cid, N.; Reyes, E.; van Sorge, N.M.; Lauth, X.; Smurthwaite, C.A.; Hilton, B.J.; Stotland, A. Bacterial induction of Snail1 contributes to blood-brain barrier disruption. Clin. Investig. 2015, 125, 2373–2383. [Google Scholar] [CrossRef] [PubMed]
- Roe, K.; Kumar, M.; Lum, S.; Orillo, B.; Nerurkar, V.R.; Verma, S. West Nile virus-induced disruption of the blood-brain barrier in mice is characterized by the degradation of the junctional complex proteins and increase in multiple matrix metalloproteinases. J. Gen. Virol. 2012, 93, 1193–1203. [Google Scholar] [CrossRef] [PubMed]
- Brew, B.J.; Chan, P. Update on HIV dementia and HIV-associated neurocognitive disorders. Curr. Neurol. Neurosci. Rep. 2014, 14, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Yao, H.; Guo, M.; Mori, T.; Mathias-Costa, B.; Singh, V.; Seth, P.; Wang, J.; Su, T.P. Cocaine and HIV-1 interplay in CNS: Cellular and molecular mechanisms. Curr. HIV Res. 2012, 10, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Kim, K.; Duan, M.; Hayashi, T.; Guo, M.; Morgello, S.; Prat, A.; Wang, J.; Su, T.P.; Buch, S. Cocaine hijacks sigma l receptor to initiate induction of activated leukocyte cell adhesion molecule: Implication for increased monocyte adhesion and migration in the CNS. J. Neurosci. 2011, 31, 5942–5955. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Zhang, X.; Manda, K.R.; Banks, W.A.; Ercal, N. HIV proteins (gp120 and Tat) and methamphetamine in oxidative stress-induced damage in the brain: Potential role of the antioxidant N-aceylcysteine amide. Free Radic. Biol. Med. 2010, 48, 1388–1398. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, N.; Saiyed, Z.M.; Napuri, J.; Samikkannu, T.; Reddy, P.V.; Agudelo, M.; Khatavkar, P.; Saxena, S.K.; Nair, M.P. Interactive role of human immunodeficiency virus type (HIV-1) clade-specific Tat protein and cocaine in blood-brain barrier dysfunction: Implications for HIV-associated neurocognitive disorder. J. Neurovirol. 2010, 16, 294–305. [Google Scholar] [CrossRef] [PubMed]
- De Bock, M.; Van Haver, V.; Vandenbroucke, R.E.; Decrock, E.; Wang, N.; Leybaert, L. Into Rather Unexplored Terrain—Transcellular Transport Across the Blood–Brain Barrier. GLIA 2016, 64, 1097–1123. [Google Scholar] [CrossRef] [PubMed]
- Mangas-Sanjuan, V.; González-Alvarez, M.; Gonzalez-Alvarez, I.; Bermejo, M. Drug penetration across the blood-brain barrier: An overview. Ther. Deliv. 2010, 1, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Nau, R.; Sorgel, F.; Eiffert, H. Penetration of drugs through the blood-cerebrospinal fluid/blood-brain barrier for treatment of central nervous system infections. Clin. Microbiol. Rev. 2010, 23, 858–883. [Google Scholar] [CrossRef] [PubMed]
- Bazzoni, G. Endothelial tight junctions: Permeable barriers of the vessel wall. Thromb Haemost. 2006, 95, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W.; van Nieuw Amerongen, G.P. Intracellular signalling involved in modulating human endothelial barrier function. J. Anat. 2006, 200, 549–560. [Google Scholar] [CrossRef]
- Strazielle, N.; Ghersi-Egea, J.F. Physiology of blood-brain interfaces inrelation to brain disposition of small compounds and macromolecules. Mol. Pharm. 2013, 10, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Hediger, M.A.; Clémençon, B.; Burrier, R.E.; Bruford, E.A. The ABCs of membrane transporters in health and disease (SLC series): Introduction. Mol. Aspect Med. 2013, 34, 95–107. [Google Scholar] [CrossRef] [PubMed]
- Uchida, Y.; Ohtsuki, S.; Katsukura, Y.; Ikeda, C.; Suzuki, T.; Kamiie, J.; Terasaki, T. Quantitative targeted absolute proteomics of human blood-brain barrier transporters and receptors. J. Neurochem. 2011, 117, 333–345. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S.; Terasaki, T. Contribution of carrier-mediated transport systems to the blood-brain barrier as a supporting and protecting interface for the brain; importance for CNS drug discovery and development. Pharm. Res. 2007, 24, 1745–1758. [Google Scholar] [CrossRef] [PubMed]
- Enerson, B.E.; Drewes, L.R. The rat blood-brain barrier transcriptome. J. Cereb. Blood Flow Metab. 2006, 26, 959–973. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.S.; Lee, K.E.; Lee, N.Y.; Terasaki, T. Donepezil, tacrine and α-phenil-n-tert-butyl nitone (PBN) inhibit choline transport by conditionally immortalized rat brain capillary endothelial cell lines (TR-BBB). Arch. Pharm. Res. 2005, 28, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Shawahna, R.; Uchida, Y.; Decleves, X.; Ohtsuki, S.; Yousif, S.; Dauchy, S.; Jacob, A.; Chassoux, F.; Daumas-Duport, C.; Couraud, P.-O. Transcriptomic and quantitative proteomic analysis of transporters and drug metabolizing enzymes in freshly isolated human brain microvessels. Mol. Pharm. 2011, 8, 1332–1341. [Google Scholar] [CrossRef] [PubMed]
- Ohtsuki, S. New aspects of the blood-brain barrier transporters; its physiological roles in the central nervous system. Biol. Pharm. Bull. 2004, 27, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Johanson, C.E. Vitamin transport and homeostasis in mammalian brain: Focus on Vitamins B and E. J. Neurochem. 2007, 103, 425–438. [Google Scholar] [CrossRef] [PubMed]
- Montalbetti, N.; Simonin, A.; Kovacs, G.; Hediger, M.A. Mammalian iron transporters: Families SLC11 and SLC40. Mol. Aspects Med. 2013, 34, 270–287. [Google Scholar] [CrossRef] [PubMed]
- Agre, P.; King, L.S.; Yasui, M.; Guggino, W.B.; Ottersen, O.P.; Fujiyoshi, Y.; Engel, A.; Nielsen, S. Aquaporin water channels—From atomic structure to clinical medicine. J. Physiol. 2002, 542, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Campos-Bedolla, P.; Walter, F.R.; Veszelka, S.; and Deli, M.A. Role of the BloodeBrain Barrier in the Nutrition of the Central Nervous System. Arch. Med. Res. 2014, 45, 610–638. [Google Scholar] [CrossRef] [PubMed]
- Speake, T.; Freeman, L.J.; Brown, P.D. Expression of aquaporin 1 and aquaporin 4 water channels in rat choroid plexus. Biochim. Biophys. Acta 2003, 1609, 80–86. [Google Scholar] [CrossRef]
- Dean, M.; Yannick, H.; Chimini, G. The human ATP-binding cassette (ABC) transporter superfamily. J. Lipid Res. 2001, 42, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed]
- Moitra, K.; Dean, M. Evolution of ABC transporters by gene duplication and their role in human disease. Biol. Chem. 2011, 392, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Hartz, A.M.; Bauer, B. ABC transporters in the CNS—An inventory. Curr. Pharm. Biotechnol. 2011, 12, 656–673. [Google Scholar] [CrossRef] [PubMed]
- Stefkovà, J.; Poledne, R.; Hubacek, J.A. ATP-binding cassette (ABC) transporters in human metabolism and diseases. Physiol. Res. 2004, 53, 235–243. [Google Scholar] [PubMed]
- Yang, Y.; Bai, L.; Li, X.; Xiong, J.; Xu, P.; Guo, C.; Xue, M. Transport of active flavonoids, based oncytotoxicity and lipophilicity: An evaluation using the blood-brain barrier cell and Caco-2 cell models. Toxicol. In Vitro 2014, 28, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Hanley, M.J.; Cancalon, P.; Widmer, W.W.; Greenblatt, D.J. The effect of grapefruit juice on drug disposition. Expert. Opin. Drug Metab. Toxicol. 2011, 7, 267–286. [Google Scholar] [CrossRef] [PubMed]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef] [PubMed]
- Mikitsh, J.L.; Chacko, A.M. Pathways for small molecule delivery to the central nervous system across the blood–brain barrier. Perspect. Med. Chem. 2014, 6, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Deli, M.A. The role of blood-brain barrier in neurodegenerative diseases. In Molecular Bases of Neurodegeneration; Di Liegro, I., Savettieri, G., Eds.; Research Signpost: Kerala, India, 2005; pp. 137–161. [Google Scholar]
- Arsov, T.; Mullen, S.A.; Rogers, S.; Phillips, A.M.; Lawrence, K.M.; Damiano, J.A.; Goldberg-Stern, H.; Afawi, Z.; Kivity, S.; Trager, C. Glucose transporter deficiency in the idiopathic generalized epilepsies. Ann. Neurol. 2012, 72, 807–815. [Google Scholar] [CrossRef] [PubMed]
- Tzadok, M.; Nissenkorn, A.; Porper, K.; Matot, I.; Marcu, S.; Anikster, Y.; Menascu, S.; Bercovich, D.; Zeev, B.B. The many faces of Glut1 deficiency syndrome. J. Child. Neurol. 2014, 29, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, M.; Ghosh, P.M.; Madison, C.; Laforce, R., Jr.; Corbetta-Rastelli, C.; Weiner, M.W.; Greicius, M.D.; Seeley, W.W.; Gorno-Tempini, M.L.; Rosen, H.J. Diverging patterns of amyloid deposition and hypometabolism in clinical variants of probable Alzheimer’s disease. Brain 2013, 136, 844–858. [Google Scholar] [CrossRef] [PubMed]
- Warskulat, U.; Heller-Stilb, B.; Oermann, E.; Zilles, K.; Haas, H.; Lang, F.; Häussinger, D. Phenotype of the taurine transporter knock-out mouse. Methods Enzymol. 2007, 428, 439–458. [Google Scholar] [CrossRef] [PubMed]
- Molina, J.A.; Jiménez-Jiménez, F.J.; Gómez, P.; Vargas, C.; Navarro, J.A.; Ortı́-Pareja, M.; Gasalla, T.; Benito-León, J.; Bermejo, F.; Arenas, J.N. Decreased cerebrospinal fluid levels of neutral and basic amino acids in patients with Parkinson’s disease. J. Neurol. Sci. 2007, 150, 123–127. [Google Scholar] [CrossRef]
- Gerards, M.; Kamps, R.; van Oevelen, J.; Boesten, I.; Jongen, E.; de Koning, B.; Scholte, H.R.; de Angst, I.; Schoonderwoerd, K.; Sefiani, A. Exome sequencing reveals a novel Moroccan founder mutation in SLC19A3 as a new cause of early-childhood fatal Leigh syndrome. Brain 2013, 136, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Skjorringe, T.; Moller, L.B.; Moos, T. Impairment of interrelated iron and copper homeostatic mechanisms in brain contributes to the pathogenesis of neurodegenerative disorders. Front. Pharmacol. 2012, 3, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Tuumer, Z.; Moller, L.B. Menkes disease. Eur. J. Hum. Genet. 2010, 18, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Bucossi, S.; Polimanti, R.; Mariani, S.; Ventriglia, M.; Bonvicini, C.; Migliore, S.; Manfellotto, D.; Salustri, C.; Vernieri, F.; Rossini, P.M. Association of K832R and R952K SNPs of Wilson disease gene with Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 913–919. [Google Scholar] [CrossRef] [PubMed]
- Jablonski, M.R.; Jacob, D.A.; Campos, C.; Miller, D.S.; Maragakis, N.J.; Pasinelli, P.; Trotti, D. Selective increase of two ABC drug efflux transporters at the blood-spinal cord barrier suggests induced pharmacoresistance in ALS. Neurobiol. Dis. 2012, 47, 194–200. [Google Scholar] [CrossRef] [PubMed]
- Potschka, H.; Luna-Munguia, H. CNS transporters and drug delivery in epilepsy. Curr. Pharm. Des. 2014, 20, 1534–1542. [Google Scholar] [CrossRef] [PubMed]
- Vogelgesang, S.; Warzok, R.W.; Cascorbi, I.; Kunert-Keil, C.; Schroeder, E.; Kroemer, H.K.; Siegmund, W.; Walker, L.C.; Pahnke, J. The role of P-glycoprotein in cerebral amyloid angiopathy; implications for the early pathogenesis of Alzheimer’s disease. Curr. Alzheimer Res. 2004, 1, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Ramasamy, I. Update on the molecular biology of dyslipidemias. Clin. Chim. Acta 2016, 454, 143–185. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.; Hartig, W.; Reichenbach, A.; Bechmann, I.; Michalski, D. Blood–brain barrier breakdown after embolic stroke in rats occurs without ultrastructural evidence for disrupting tight junctions. PLoS ONE 2013, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Krueger, M.; Bechmann, I.; Immig, K.; Reichenbach, A.; Hartig, W.; Michalski, D. Blood–brain barrier breakdown involves four distinct stages of vascular damage in various models of experimental focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2015, 35, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Klaassen, I.; Van Noorden, C.J.; Schlingemann, R.O. Molecular basis of the inner blood-retinal barrier and its breakdown in diabetic macular edema and other pathological conditions. Prog. Retin. Eye Res. 2013, 34, 19–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.W.; Gumbleton, M. Endocytosis at the blood–brain barrier: From basic understanding to drug delivery strategies. J. Drug Target 2006, 14, 191–214. [Google Scholar] [CrossRef] [PubMed]
- Errede, M.; Girolamo, F.; Ferrara, G.; Strippoli, M.; Morando, S.; Boldrin, V.; Rizzi, M.; Uccelli, A.; Perris, R.; Bendotti, C.; et al. Blood–brain barrier alterations in the cerebral cortex in experimental autoimmune encephalomyelitis. J. Neuropathol. Exp. Neurol. 2012, 71, 840–854. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maiuolo, J.; Gliozzi, M.; Musolino, V.; Scicchitano, M.; Carresi, C.; Scarano, F.; Bosco, F.; Nucera, S.; Ruga, S.; Zito, M.C.; et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. Int. J. Mol. Sci. 2018, 19, 2693. https://doi.org/10.3390/ijms19092693
Maiuolo J, Gliozzi M, Musolino V, Scicchitano M, Carresi C, Scarano F, Bosco F, Nucera S, Ruga S, Zito MC, et al. The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. International Journal of Molecular Sciences. 2018; 19(9):2693. https://doi.org/10.3390/ijms19092693
Chicago/Turabian StyleMaiuolo, Jessica, Micaela Gliozzi, Vincenzo Musolino, Miriam Scicchitano, Cristina Carresi, Federica Scarano, Francesca Bosco, Saverio Nucera, Stefano Ruga, Maria Caterina Zito, and et al. 2018. "The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections" International Journal of Molecular Sciences 19, no. 9: 2693. https://doi.org/10.3390/ijms19092693
APA StyleMaiuolo, J., Gliozzi, M., Musolino, V., Scicchitano, M., Carresi, C., Scarano, F., Bosco, F., Nucera, S., Ruga, S., Zito, M. C., Mollace, R., Palma, E., Fini, M., Muscoli, C., & Mollace, V. (2018). The “Frail” Brain Blood Barrier in Neurodegenerative Diseases: Role of Early Disruption of Endothelial Cell-to-Cell Connections. International Journal of Molecular Sciences, 19(9), 2693. https://doi.org/10.3390/ijms19092693