TGF-β Signaling in Lung Health and Disease

Abstract

1. Introduction
2. TGF-β Activation in the Extracellular Milieu
3. Context-Dependency of TGF-β Signaling
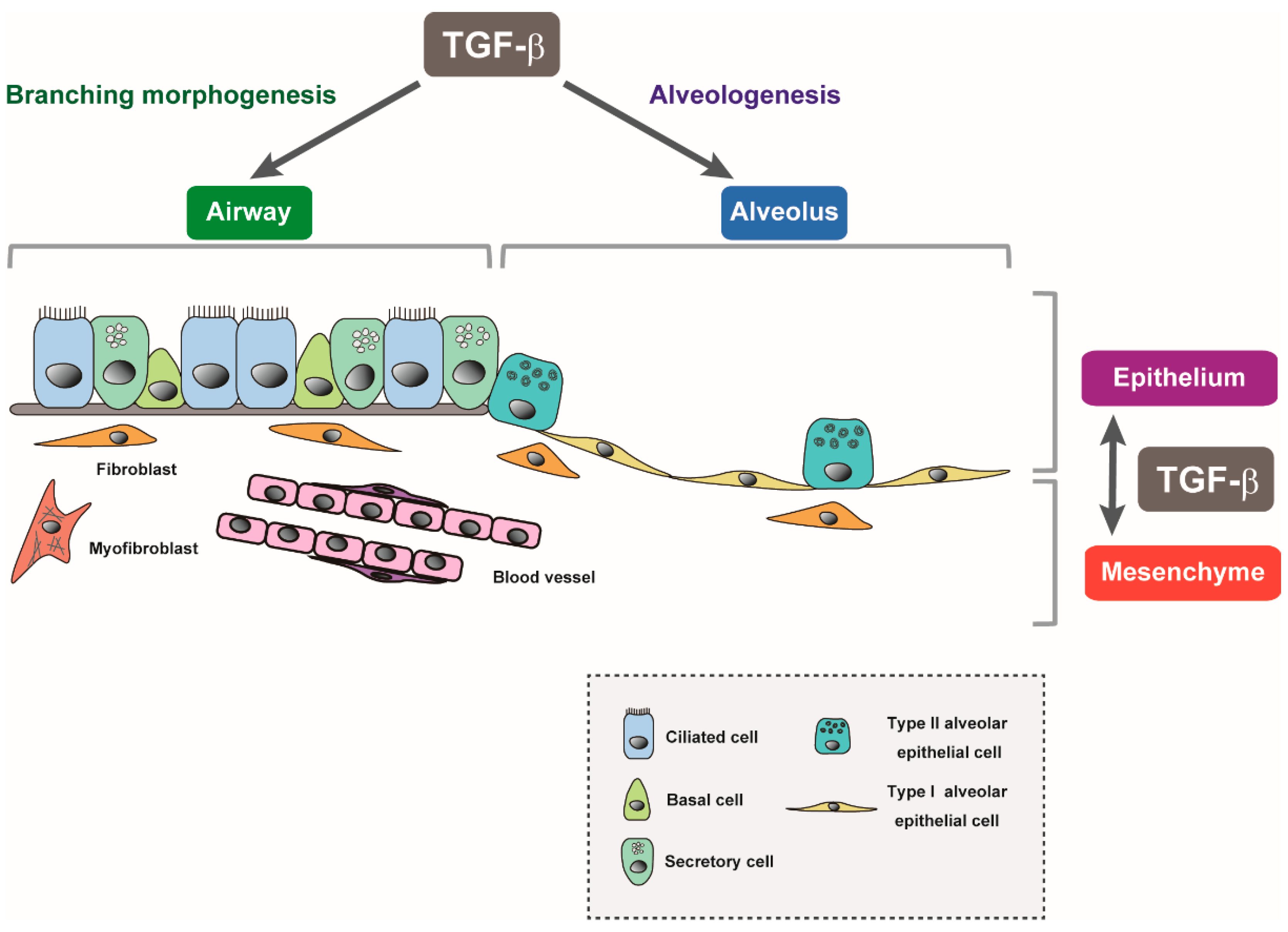
4. TGF-β Signaling in Lung Organogenesis
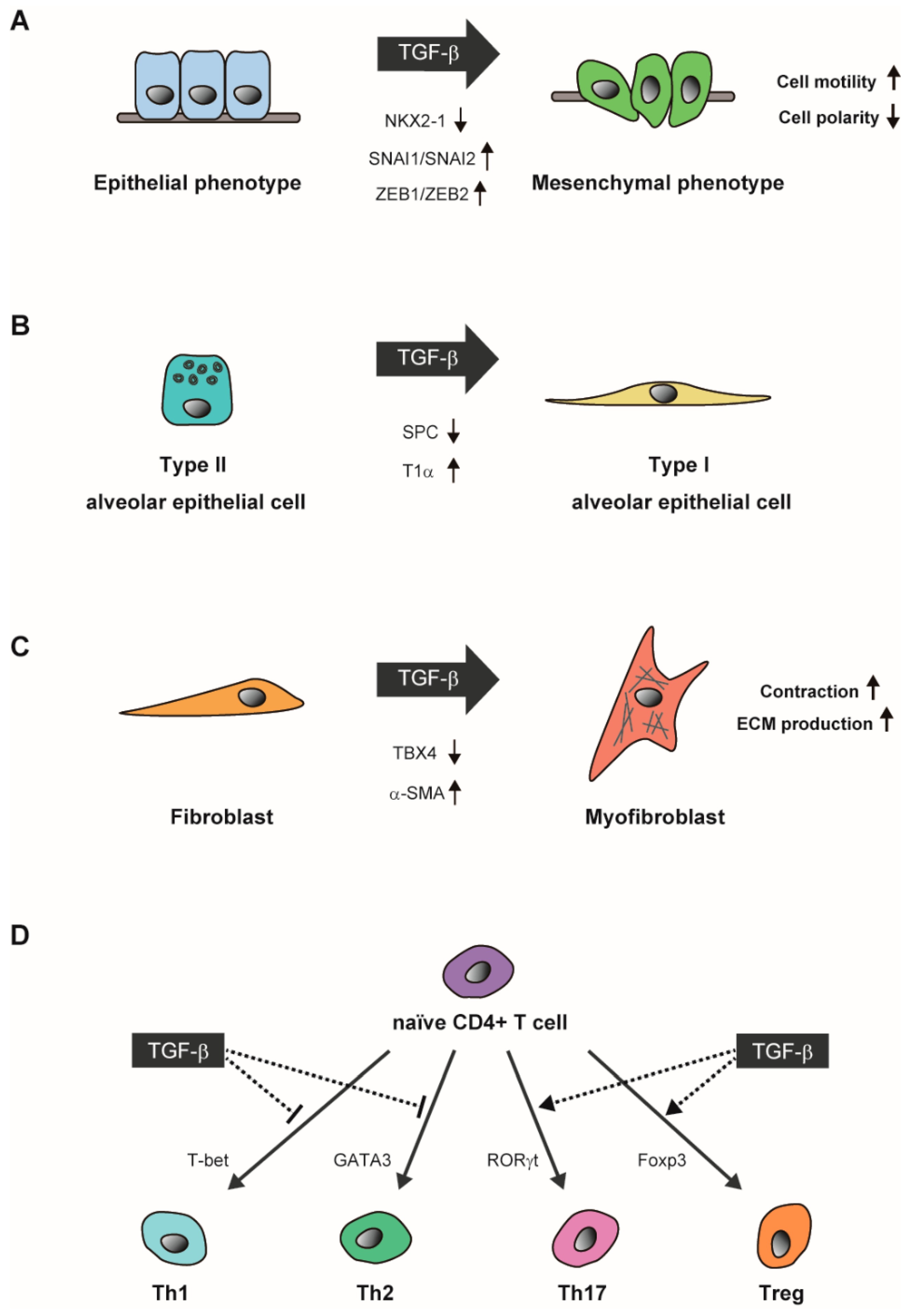
5. TGF-β Signaling in Lung Alveolar Epithelial Growth and Differentiation
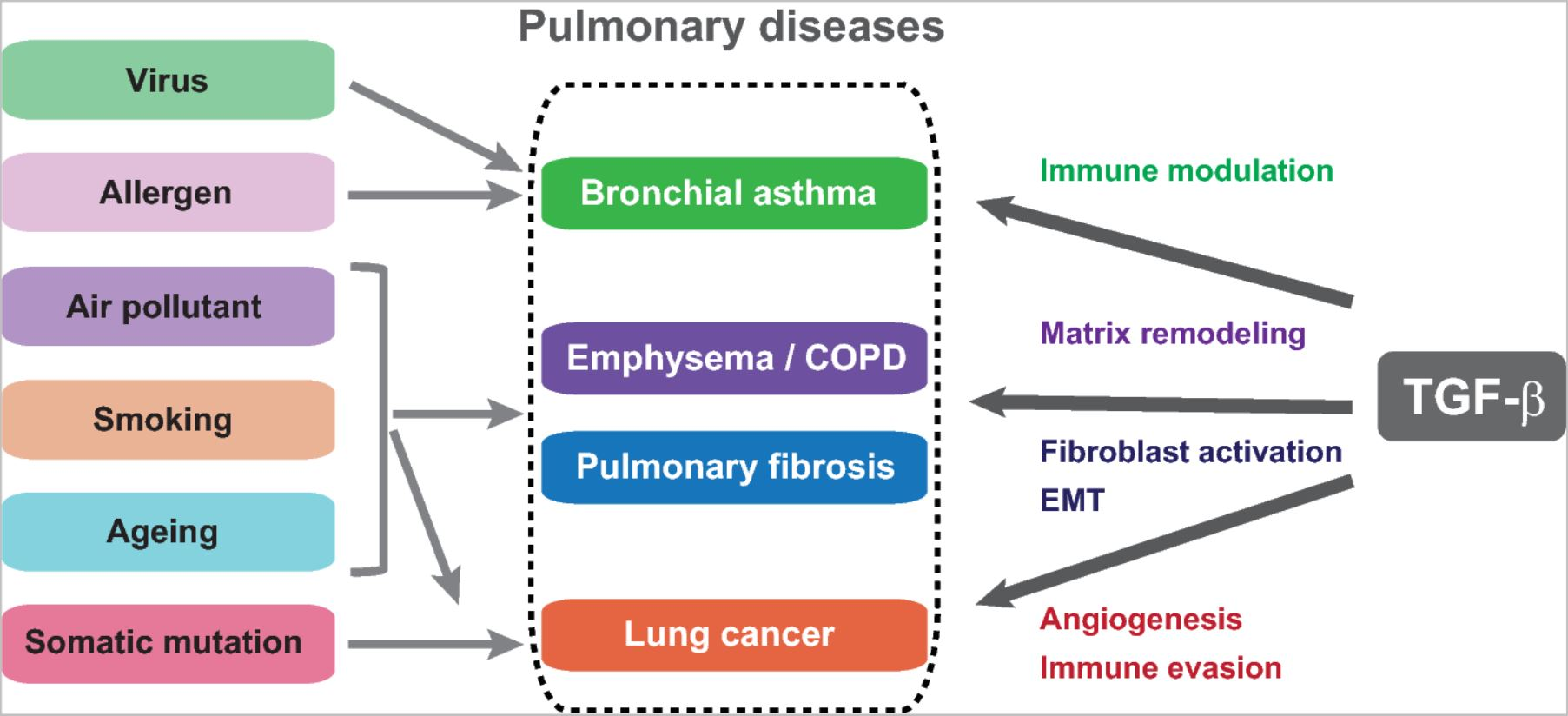
6. TGF-β Signaling in Pulmonary Diseases
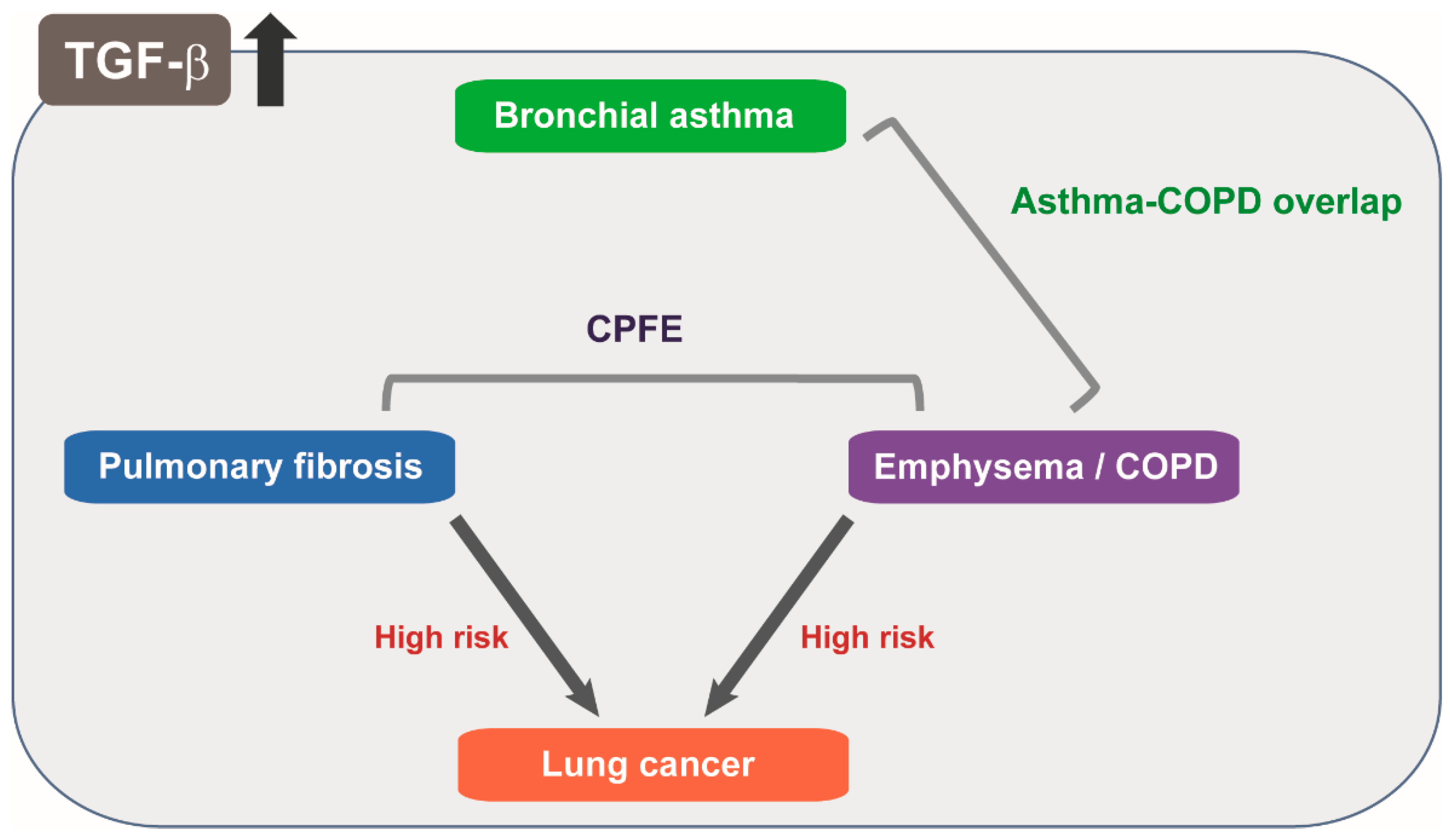
6.1. TGF-β Signaling in Pulmonary Fibrosis and Emphysema
6.2. Dual Roles of TGF-β in Asthma Involving Immune Response and Airway Remodeling
6.3. TGF-β Mediates EMT in Non-Small Cell Lung Cancer (NSCLC)
6.4. TGF-β Orchestrates the Tumor Microenvironment
6.5. TGF-β Signaling in Small Cell Lung Cancer (SCLC)
7. Gene Regulatory Networks Responsible for the TGF-β-Induced Cellular Response
8. Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Jarrett, J.A.; Chen, E.Y.; Eaton, D.H.; Bell, J.R.; Assoian, R.K.; Roberts, A.B.; Sporn, M.B.; Goeddel, D.V. Human transforming growth factor-beta complementary DNA sequence and expression in normal and transformed cells. Nature 1985, 316, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Franzén, P.; ten Dijke, P.; Ichijo, H.; Yamashita, H.; Schulz, P.; Heldin, C.H.; Miyazono, K. Cloning of a TGF beta type I receptor that forms a heteromeric complex with the TGF beta type II receptor. Cell 1993, 75, 681–692. [Google Scholar] [CrossRef]
- Aschner, Y.; Downey, G.P. Transforming Growth Factor-β: Master Regulator of the Respiratory System in Health and Disease. Am. J. Respir. Cell Mol. Biol. 2016, 54, 647–655. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Stellmach, V.; Murphy-Ullrich, J.E.; Ribeiro, S.M.; Lawler, J.; Hynes, R.O.; Boivin, G.P.; Bouck, N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 1998, 93, 1159–1170. [Google Scholar] [CrossRef]
- Miyazono, K.; Heldin, C.H. Role for carbohydrate structures in TGF-beta 1 latency. Nature 1989, 338, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, T.; Olofsson, A.; Morén, A.; Wernstedt, C.; Hellman, U.; Miyazono, K.; Claesson-Welsh, L.; Heldin, C.H. TGF-beta 1 binding protein: A component of the large latent complex of TGF-beta 1 with multiple repeat sequences. Cell 1990, 61, 1051–1061. [Google Scholar] [CrossRef]
- Colarossi, C.; Chen, Y.; Obata, H.; Jurukovski, V.; Fontana, L.; Dabovic, B.; Rifkin, D.B. Lung alveolar septation defects in Ltbp-3-null mice. Am. J. Pathol. 2005, 167, 419–428. [Google Scholar] [CrossRef]
- Dabovic, B.; Chen, Y.; Choi, J.; Davis, E.C.; Sakai, L.Y.; Todorovic, V.; Vassallo, M.; Zilberberg, L.; Singh, A.; Rifkin, D.B. Control of lung development by latent TGF-β binding proteins. J. Cell. Physiol. 2011, 226, 1499–1509. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Dabovic, B.; Takagi, K.; Inoue, T.; Horiguchi, M.; Hirai, M.; Fujikawa, Y.; Akama, T.O.; Kusumoto, K.; Zilberberg, L.; et al. Latent TGF-β binding protein 4 promotes elastic fiber assembly by interacting with fibulin-5. Proc. Natl. Acad. Sci. USA 2013, 110, 2852–2857. [Google Scholar] [CrossRef] [PubMed]
- Sterner-Kock, A.; Thorey, I.S.; Koli, K.; Wempe, F.; Otte, J.; Bangsow, T.; Kuhlmeier, K.; Kirchner, T.; Jin, S.; Keski-Oja, J.; et al. Disruption of the gene encoding the latent transforming growth factor-beta binding protein 4 (LTBP-4) causes abnormal lung development, cardiomyopathy, and colorectal cancer. Genes Dev. 2002, 16, 2264–2273. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Ramonet, D.; Klingberg, F.; Formey, A.; Smith-Clerc, J.; Meister, J.J.; Hinz, B. The single-molecule mechanics of the latent TGF-β1 complex. Curr. Biol. 2011, 21, 2046–2054. [Google Scholar] [CrossRef] [PubMed]
- Saito, A.; Suzuki, H.I.; Horie, M.; Ohshima, M.; Morishita, Y.; Abiko, Y.; Nagase, T. An integrated expression profiling reveals target genes of TGF-β and TNF-α possibly mediated by microRNAs in lung cancer cells. PLoS ONE 2013, 8, e56587. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Noguchi, S.; Yamaguchi, Y.; Ohshima, M.; Morishita, Y.; Suzuki, H.I.; Kohyama, T.; Nagase, T. Differential knockdown of TGF-β ligands in a three-dimensional co-culture tumor-stromal interaction model of lung cancer. BMC Cancer 2014, 14, 580. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Miyazono, K.; ten Dijke, P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 1997, 390, 465–471. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Mullen, A.C.; Orlando, D.A.; Newman, J.J.; Lovén, J.; Kumar, R.M.; Bilodeau, S.; Reddy, J.; Guenther, M.G.; DeKoter, R.P.; Young, R.A. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell 2011, 147, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGFβ signalling in context. Nat. Rev. Mol. Cell Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Miyazawa, K.; Miyazono, K. Regulation of TGF-β Family Signaling by Inhibitory Smads. Cold Spring Harb. Perspect. Biol. 2017, 9, a022095. [Google Scholar] [CrossRef] [PubMed]
- Nakao, A.; Afrakhte, M.; Morén, A.; Nakayama, T.; Christian, J.L.; Heuchel, R.; Itoh, S.; Kawabata, M.; Heldin, N.E.; Heldin, C.H.; et al. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature 1997, 389, 631–635. [Google Scholar] [PubMed]
- Ebisawa, T.; Fukuchi, M.; Murakami, G.; Chiba, T.; Tanaka, K.; Imamura, T.; Miyazono, K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem. 2001, 276, 12477–12480. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Integration of Smad and MAPK pathways: A link and a linker revisited. Genes Dev. 2003, 17, 2993–2997. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.S.; Saunier, E.F.; Akhurst, R.J.; Derynck, R. The type I TGF-beta receptor is covalently modified and regulated by sumoylation. Nat. Cell Biol. 2008, 10, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Du, Y.; Deng, H. Direct lineage reprogramming: Strategies, mechanisms, and applications. Cell Stem Cell 2015, 16, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Minoo, P.; Su, G.; Drum, H.; Bringas, P.; Kimura, S. Defects in tracheoesophageal and lung morphogenesis in Nkx2.1(−/−) mouse embryos. Dev. Biol. 1999, 209, 60–71. [Google Scholar] [CrossRef] [PubMed]
- Bohinski, R.J.; Di Lauro, R.; Whitsett, J.A. The lung-specific surfactant protein B gene promoter is a target for thyroid transcription factor 1 and hepatocyte nuclear factor 3, indicating common factors for organ-specific gene expression along the foregut axis. Mol. Cell. Biol. 1994, 14, 5671–5681. [Google Scholar] [CrossRef] [PubMed]
- Minoo, P.; Hu, L.; Zhu, N.; Borok, Z.; Bellusci, S.; Groffen, J.; Kardassis, D.; Li, C. SMAD3 prevents binding of NKX2.1 and FOXA1 to the SpB promoter through its MH1 and MH2 domains. Nucleic Acids Res. 2008, 36, 179–188. [Google Scholar] [CrossRef] [PubMed]
- Isogaya, K.; Koinuma, D.; Tsutsumi, S.; Saito, R.A.; Miyazawa, K.; Aburatani, H.; Miyazono, K. A Smad3 and TTF-1/NKX2-1 complex regulates Smad4-independent gene expression. Cell Res. 2014, 24, 994–1008. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.A.; Watabe, T.; Horiguchi, K.; Kohyama, T.; Saitoh, M.; Nagase, T.; Miyazono, K. Thyroid transcription factor-1 inhibits transforming growth factor-beta-mediated epithelial-to-mesenchymal transition in lung adenocarcinoma cells. Cancer Res. 2009, 69, 2783–2791. [Google Scholar] [CrossRef] [PubMed]
- Herriges, M.; Morrisey, E.E. Lung development: Orchestrating the generation and regeneration of a complex organ. Development 2014, 141, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Bragg, A.D.; Moses, H.L.; Serra, R. Signaling to the epithelium is not sufficient to mediate all of the effects of transforming growth factor beta and bone morphogenetic protein 4 on murine embryonic lung development. Mech. Dev. 2001, 109, 13–26. [Google Scholar] [CrossRef]
- Shull, M.M.; Ormsby, I.; Kier, A.B.; Pawlowski, S.; Diebold, R.J.; Yin, M.; Allen, R.; Sidman, C.; Proetzel, G.; Calvin, D.; et al. Targeted disruption of the mouse transforming growth factor-beta 1 gene results in multifocal inflammatory disease. Nature 1992, 359, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.B.; Huh, C.G.; Becker, D.; Geiser, A.; Lyght, M.; Flanders, K.C.; Roberts, A.B.; Sporn, M.B.; Ward, J.M.; Karlsson, S. Transforming growth factor beta 1 null mutation in mice causes excessive inflammatory response and early death. Proc. Natl. Acad. Sci. USA 1993, 90, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Sanford, L.P.; Ormsby, I.; Gittenberger-de Groot, A.C.; Sariola, H.; Friedman, R.; Boivin, G.P.; Cardell, E.L.; Doetschman, T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development 1997, 124, 2659–2670. [Google Scholar] [PubMed]
- Kaartinen, V.; Voncken, J.W.; Shuler, C.; Warburton, D.; Bu, D.; Heisterkamp, N.; Groffen, J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 1995, 11, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhuang, F.; Liu, Y.H.; Xu, B.; Del Moral, P.; Deng, W.; Chai, Y.; Kolb, M.; Gauldie, J.; Warburton, D.; et al. TGF-beta receptor II in epithelia versus mesenchyme plays distinct roles in the developing lung. Eur. Respir. J. 2008, 32, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Krishnaveni, M.S.; Li, C.; Zhou, B.; Xing, Y.; Banfalvi, A.; Li, A.; Lombardi, V.; Akbari, O.; Borok, Z.; et al. Epithelium-specific deletion of TGF-β receptor type II protects mice from bleomycin-induced pulmonary fibrosis. J. Clin. Investig. 2011, 121, 277–287. [Google Scholar] [CrossRef] [PubMed]
- Bonniaud, P.; Kolb, M.; Galt, T.; Robertson, J.; Robbins, C.; Stampfli, M.; Lavery, C.; Margetts, P.J.; Roberts, A.B.; Gauldie, J. Smad3 null mice develop airspace enlargement and are resistant to TGF-beta-mediated pulmonary fibrosis. J. Immunol. 2004, 173, 2099–2108. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Sun, J.; Buckley, S.; Chen, C.; Warburton, D.; Wang, X.F.; Shi, W. Abnormal mouse lung alveolarization caused by Smad3 deficiency is a developmental antecedent of centrilobular emphysema. Am. J. Physiol. Lung Cell. Mol. Physiol. 2005, 288, L683–L691. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, C.; Li, A.; Sridurongrit, S.; Tiozzo, C.; Bellusci, S.; Borok, Z.; Kaartinen, V.; Minoo, P. Signaling via Alk5 controls the ontogeny of lung Clara cells. Development 2010, 137, 825–833. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Li, C.; Liu, Y.H.; Xing, Y.; Hu, L.; Borok, Z.; Kwong, K.Y.; Minoo, P. Mesodermal deletion of transforming growth factor-beta receptor II disrupts lung epithelial morphogenesis: Cross-talk between TGF-beta and Sonic hedgehog pathways. J. Biol. Chem. 2008, 283, 36257–36264. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Ma, S.; Smith, S.M.; Lee, M.K.; Fischer, A.; Borok, Z.; Bellusci, S.; Li, C.; Minoo, P. Mesodermal ALK5 controls lung myofibroblast versus lipofibroblast cell fate. BMC Biol. 2016, 14, 19. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Dey, C.R.; Wert, S.E.; Whitsett, J.A. Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-beta 1 chimeric gene. Dev. Biol. 1996, 175, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Li, C.; Hu, L.; Tiozzo, C.; Li, M.; Chai, Y.; Bellusci, S.; Anderson, S.; Minoo, P. Mechanisms of TGFbeta inhibition of LUNG endodermal morphogenesis: The role of TbetaRII, Smads, Nkx2.1 and Pten. Dev. Biol. 2008, 320, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Hogan, B.L.; Barkauskas, C.E.; Chapman, H.A.; Epstein, J.A.; Jain, R.; Hsia, C.C.; Niklason, L.; Calle, E.; Le, A.; Randell, S.H.; et al. Repair and regeneration of the respiratory system: Complexity, plasticity, and mechanisms of lung stem cell function. Cell Stem Cell 2014, 15, 123–138. [Google Scholar] [CrossRef] [PubMed]
- Ryan, R.M.; Mineo-Kuhn, M.M.; Kramer, C.M.; Finkelstein, J.N. Growth factors alter neonatal type II alveolar epithelial cell proliferation. Am. J. Physiol. 1994, 266, L17–L22. [Google Scholar] [CrossRef] [PubMed]
- Saitoh, M. Epithelial-mesenchymal transition is regulated at post-transcriptional levels by transforming growth factor-β signaling during tumor progression. Cancer Sci. 2015, 106, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Shirakihara, T.; Saitoh, M.; Miyazono, K. Differential regulation of epithelial and mesenchymal markers by deltaEF1 proteins in epithelial mesenchymal transition induced by TGF-beta. Mol. Biol. Cell 2007, 18, 3533–3544. [Google Scholar] [CrossRef] [PubMed]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; Liebler, J.M.; Luby-Phelps, K.; Nicholson, A.G.; Crandall, E.D.; du Bois, R.M.; Borok, Z. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: Potential role in idiopathic pulmonary fibrosis. Am. J. Pathol. 2005, 166, 1321–1332. [Google Scholar] [CrossRef]
- Kage, H.; Borok, Z. EMT and interstitial lung disease: A mysterious relationship. Curr. Opin. Pulm. Med. 2012, 18, 517–523. [Google Scholar] [CrossRef] [PubMed]
- Borok, Z.; Hami, A.; Danto, S.I.; Zabski, S.M.; Crandall, E.D. Rat serum inhibits progression of alveolar epithelial cells toward the type I cell phenotype in vitro. Am. J. Respir. Cell Mol. Biol. 1995, 12, 50–55. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yee, M.; O’Reilly, M.A. Transdifferentiation of alveolar epithelial type II to type I cells is controlled by opposing TGF-β and BMP signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 305, L409–L418. [Google Scholar] [CrossRef] [PubMed]
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025–3036. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Barkauskas, C.E.; Takeda, N.; Bowie, E.J.; Aghajanian, H.; Wang, Q.; Padmanabhan, A.; Manderfield, L.J.; Gupta, M.; Li, D.; et al. Plasticity of Hopx(+) type I alveolar cells to regenerate type II cells in the lung. Nat. Commun. 2015, 6, 6727. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Morley, M.; Hawkins, F.; McCauley, K.B.; Jean, J.C.; Heins, H.; Na, C.L.; Weaver, T.E.; Vedaie, M.; Hurley, K.; et al. Differentiation of Human Pluripotent Stem Cells into Functional Lung Alveolar Epithelial Cells. Cell Stem Cell 2017, 21, 472–488. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Gotoh, S.; Korogi, Y.; Seki, M.; Konishi, S.; Ikeo, S.; Sone, N.; Nagasaki, T.; Matsumoto, H.; Muro, S.; et al. Long-term expansion of alveolar stem cells derived from human iPS cells in organoids. Nat. Methods 2017, 14, 1097–1106. [Google Scholar] [CrossRef] [PubMed]
- Green, M.D.; Chen, A.; Nostro, M.C.; d’Souza, S.L.; Schaniel, C.; Lemischka, I.R.; Gouon-Evans, V.; Keller, G.; Snoeck, H.W. Generation of anterior foregut endoderm from human embryonic and induced pluripotent stem cells. Nat. Biotechnol. 2011, 29, 267–272. [Google Scholar] [CrossRef] [PubMed]
- King, T.E., Jr.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef]
- Coker, R.K.; Laurent, G.J.; Jeffery, P.K.; du Bois, R.M.; Black, C.M.; McAnulty, R.J. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax 2001, 56, 549–556. [Google Scholar] [CrossRef] [PubMed]
- Broekelmann, T.J.; Limper, A.H.; Colby, T.V.; McDonald, J.A. Transforming growth factor beta 1 is present at sites of extracellular matrix gene expression in human pulmonary fibrosis. Proc. Natl. Acad. Sci. USA 1991, 88, 6642–6646. [Google Scholar] [CrossRef] [PubMed]
- Garrison, G.; Huang, S.K.; Okunishi, K.; Scott, J.P.; Kumar Penke, L.R.; Scruggs, A.M.; Peters-Golden, M. Reversal of myofibroblast differentiation by prostaglandin E(2). Am. J. Respir. Cell Mol. Biol. 2013, 48, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Bauman, K.A.; Wettlaufer, S.H.; Okunishi, K.; Vannella, K.M.; Stoolman, J.S.; Huang, S.K.; Courey, A.J.; White, E.S.; Hogaboam, C.M.; Simon, R.H.; et al. The antifibrotic effects of plasminogen activation occur via prostaglandin E2 synthesis in humans and mice. J. Clin. Investig. 2010, 120, 1950–1960. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.H.; McDonald, M.L.; Zhou, X.; Mattheisen, M.; Castaldi, P.J.; Hersh, C.P.; Demeo, D.L.; Sylvia, J.S.; Ziniti, J.; Laird, N.M.; et al. Risk loci for chronic obstructive pulmonary disease: A genome-wide association study and meta-analysis. Lancet Respir. Med. 2014, 2, 214–225. [Google Scholar] [CrossRef]
- Takizawa, H.; Tanaka, M.; Takami, K.; Ohtoshi, T.; Ito, K.; Satoh, M.; Okada, Y.; Yamasawa, F.; Nakahara, K.; Umeda, A. Increased expression of transforming growth factor-beta1 in small airway epithelium from tobacco smokers and patients with chronic obstructive pulmonary disease (COPD). Am. J. Respir. Crit. Care Med. 2001, 163, 1476–1483. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Shi, W.; Wang, Y.L.; Chen, H.; Bringas, P., Jr.; Datto, M.B.; Frederick, J.P.; Wang, X.F.; Warburton, D. Smad3 deficiency attenuates bleomycin-induced pulmonary fibrosis in mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L585–L593. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.G.; Huang, X.; Kaminski, N.; Wang, Y.; Shapiro, S.D.; Dolganov, G.; Glick, A.; Sheppard, D. Loss of integrin alpha(v)beta6-mediated TGF-beta activation causes Mmp12-dependent emphysema. Nature 2003, 422, 169–173. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.S.; Huang, X.; Kawakatsu, H.; Griffiths, M.J.; Dalton, S.L.; Wu, J.; Pittet, J.F.; Kaminski, N.; Garat, C.; Matthay, M.A.; et al. The integrin alpha v beta 6 binds and activates latent TGF beta 1: A mechanism for regulating pulmonary inflammation and fibrosis. Cell 1999, 96, 319–328. [Google Scholar] [CrossRef]
- Horan, G.S.; Wood, S.; Ona, V.; Li, D.J.; Lukashev, M.E.; Weinreb, P.H.; Simon, K.J.; Hahm, K.; Allaire, N.E.; Rinaldi, N.J.; et al. Partial inhibition of integrin alpha(v)beta6 prevents pulmonary fibrosis without exacerbating inflammation. Am. J. Respir. Crit. Care Med. 2008, 177, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Jankowich, M.D.; Roundsm, S.I.S. Combined pulmonary fibrosis and emphysema syndrome: A review. Chest 2012, 141, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Brightling, C.; Pedersen, S.E.; Reddel, H.K. Asthma. Lancet 2018, 391, 783–800. [Google Scholar] [CrossRef]
- Hirota, N.; Martin, J.G. Mechanisms of airway remodeling. Chest 2013, 144, 1026–1032. [Google Scholar] [CrossRef] [PubMed]
- James, A.L.; Bai, T.R.; Mauad, T.; Abramson, M.J.; Dolhnikoff, M.; McKay, K.O.; Maxwell, P.S.; Elliot, J.G.; Green, F.H. Airway smooth muscle thickness in asthma is related to severity but not duration of asthma. Eur. Respir. J. 2009, 34, 1040–1045. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, M.F.; Gut, I.G.; Demenais, F.; Strachan, D.P.; Bouzigon, E.; Heath, S.; von Mutius, E.; Farrall, M.; Lathrop, M.; Cookson, W.O. A large-scale, consortium-based genomewide association study of asthma. N. Engl. J. Med. 2010, 363, 1211–1221. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.S.; Chang, W.W.; He, L.P.; Jin, Y.L.; Li, C.P. An updated meta-analysis of transforming growth factor-β1 gene: Three well-characterized polymorphisms with asthma. Hum. Immunol. 2016, 77, 1291–1299. [Google Scholar] [CrossRef] [PubMed]
- Frischmeyer-Guerrerio, P.A.; Guerrerio, A.L.; Oswald, G.; Chichester, K.; Myers, L.; Halushka, M.K.; Oliva-Hemker, M.; Wood, R.A.; Dietz, H.C. TGFβ receptor mutations impose a strong predisposition for human allergic disease. Sci. Transl. Med. 2013, 5, 195ra94. [Google Scholar] [CrossRef] [PubMed]
- Redington, A.E.; Madden, J.; Frew, A.J.; Djukanovic, R.; Roche, W.R.; Holgate, S.T.; Howarth, P.H. Transforming growth factor-beta 1 in asthma. Measurement in bronchoalveolar lavage fluid. Am. J. Respir. Crit. Care Med. 1997, 156, 642–647. [Google Scholar] [CrossRef] [PubMed]
- Vignola, A.M.; Chanez, P.; Chiappara, G.; Merendino, A.; Pace, E.; Rizzo, A.; la Rocca, A.M.; Bellia, V.; Bonsignore, G.; Bousquet, J. Transforming growth factor-beta expression in mucosal biopsies in asthma and chronic bronchitis. Am. J. Respir. Crit. Care Med. 1997, 156, 591–599. [Google Scholar] [CrossRef] [PubMed]
- Ohno, I.; Nitta, Y.; Yamauchi, K.; Hoshi, H.; Honma, M.; Woolley, K.; O’Byrne, P.; Tamura, G.; Jordana, M.; Shirato, K. Transforming growth factor beta 1 (TGF beta 1) gene expression by eosinophils in asthmatic airway inflammation. Am. J. Respir. Cell Mol. Biol. 1996, 15, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Ojiaku, C.A.; Cao, G.; Zhu, W.; Yoo, E.J.; Shumyatcher, M.; Himes, B.E.; An, S.S.; Panettieri, R.A., Jr. TGF-β1 Evokes Human Airway Smooth Muscle Cell Shortening and Hyperresponsiveness via Smad3. Am. J. Respir. Cell Mol. Biol. 2018, 58, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Jin, W.; Hardegen, N.; Lei, K.J.; Li, L.; Marinos, N.; McGrady, G.; Wahl, S.M. Conversion of peripheral CD4 + CD25 − naive T cells to CD4 + CD25 + regulatory T cells by TGF-beta induction of transcription factor Foxp3. J. Exp. Med. 2003, 198, 1875–1886. [Google Scholar] [CrossRef] [PubMed]
- Marie, J.C.; Letterio, J.J.; Gavin, M.; Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4 + CD25 + regulatory T cells. J. Exp. Med. 2005, 201, 1061–1067. [Google Scholar] [CrossRef] [PubMed]
- Kearley, J.; Robinson, D.S.; Lloyd, C.M. CD4 + CD25 + regulatory T cells reverse established allergic airway inflammation and prevent airway remodeling. J. Allergy Clin. Immunol. 2008, 122, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Sanjabi, S.; Flavell, R.A. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006, 25, 455–471. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; ten Dijke, P. Immunoregulation by members of the TGFβ superfamily. Nat. Rev. Immunol. 2016, 16, 723–740. [Google Scholar] [CrossRef] [PubMed]
- Herbst, R.S.; Heymach, J.V.; Lippman, S.M. Lung cancer. N. Engl. J. Med. 2008, 359, 1367–1380. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, Y.; Takanashi, S.; Kanehira, Y.; Tsushima, T.; Imai, T.; Okumura, K. Transforming growth factor β1 level correlates with angiogenesis, tumor progression, and prognosis in patients with nonsmall cell lung carcinoma. Cancer 2001, 91, 964–971. [Google Scholar] [CrossRef]
- Massague, J. TGFbeta in Cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, S.; Wang, J.; Myeroff, L.; Parsons, R.; Sun, L.; Lutterbaugh, J.; Fan, R.S.; Zborowska, E.; Kinzler, K.W.; Vogelstein, B.; et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science 1995, 268, 1336–1338. [Google Scholar] [CrossRef] [PubMed]
- Hahn, S.A.; Schutte, M.; Hoque, A.T.; Moskaluk, C.A.; da Costa, L.T.; Rozenblum, E.; Weinstein, C.L.; Fischer, A.; Yeo, C.J.; Hruban, R.H.; et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science 1996, 271, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Nakaoku, T.; Tsuta, K.; Tsuchihara, K.; Matsumoto, S.; Yoh, K.; Goto, K. Beyond ALK-RET, ROS1 and other oncogene fusions in lung cancer. Transl. Lung Cancer Res. 2015, 4, 156–164. [Google Scholar] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Yauch, R.L.; Januario, T.; Eberhard, D.A.; Cavet, G.; Zhu, W.; Fu, L.; Pham, T.Q.; Soriano, R.; Stinson, J.; Seshagiri, S.; et al. Epithelial versus mesenchymal phenotype determines in vitro sensitivity and predicts clinical activity of erlotinib in lung cancer patients. Clin. Cancer Res. 2005, 11, 8686–8698. [Google Scholar] [CrossRef] [PubMed]
- Schliekelman, M.J.; Taguchi, A.; Zhu, J.; Dai, X.; Rodriguez, J.; Celiktas, M.; Zhang, Q.; Chin, A.; Wong, C.H.; Wang, H.; et al. Molecular portraits of epithelial, mesenchymal, and hybrid States in lung adenocarcinoma and their relevance to survival. Cancer Res. 2015, 75, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Horiguchi, K.; Shirakihara, T.; Nakano, A.; Imamura, T.; Miyazono, K.; Saitoh, M. Role of Ras signaling in the induction of snail by transforming growth factor-beta. J. Biol. Chem. 2009, 284, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Carstens, J.L.; Kim, J.; Scheible, M.; Kaye, J.; Sugimoto, H.; Wu, C.C.; LeBleu, V.S.; Kalluri, R. Epithelial-to-mesenchymal transition is dispensable for metastasis but induces chemoresistance in pancreatic cancer. Nature 2015, 527, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Bierie, B.; Moses, H.L. Tumour microenvironment: TGFbeta: The molecular Jekyll and Hyde of cancer. Nat. Rev. Cancer 2006, 6, 506–520. [Google Scholar] [CrossRef] [PubMed]
- Ikushima, H.; Miyazono, K. TGFbeta signalling: A complex web in cancer progression. Nat. Rev. Cancer 2010, 10, 415–424. [Google Scholar] [CrossRef] [PubMed]
- Pickup, M.; Novitskiy, S.; Moses, H.L. The roles of TGFβ in the tumour microenvironment. Nat. Rev. Cancer 2013, 13, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, M.; Morikawa, A.; Kawasaki, M.; Matsuno, Y.; Yamada, T.; Hirohashi, S.; Kondo, H.; Shimosato, Y. Small adenocarcinoma of the lung. Histologic characteristics and prognosis. Cancer 1995, 75, 2844–2852. [Google Scholar] [CrossRef]
- Micke, P.; Ostman, A. Tumour-stroma interaction: Cancer-associated fibroblasts as novel targets in anti-cancer therapy? Lung Cancer 2004, 45 (Suppl. 2), S163–S175. [Google Scholar] [CrossRef] [PubMed]
- Saito, R.A.; Micke, P.; Paulsson, J.; Augsten, M.; Peña, C.; Jönsson, P.; Botling, J.; Edlund, K.; Johansson, L.; Carlsson, P.; et al. Forkhead box F1 regulates tumor-promoting properties of cancer-associated fibroblasts in lung cancer. Cancer Res. 2010, 70, 2644–2654. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Mikami, Y.; Ohshima, M.; Morishita, Y.; Nakajima, J.; Kohyama, T.; Nagase, T. Characterization of human lung cancer-associated fibroblasts in three-dimensional in vitro co-culture model. Biochem. Biophys. Res. Commun. 2012, 423, 158–163. [Google Scholar] [CrossRef] [PubMed]
- Navab, R.; Strumpf, D.; Bandarchi, B.; Zhu, C.Q.; Pintilie, M.; Ramnarine, V.R.; Ibrahimov, E.; Radulovich, N.; Leung, L.; Barczyk, M.; et al. Prognostic gene-expression signature of carcinoma-associated fibroblasts in non-small cell lung cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 7160–7165. [Google Scholar] [CrossRef] [PubMed]
- Heldin, C.H.; Rubin, K.; Pietras, K.; Ostman, A. High interstitial fluid pressure—An obstacle in cancer therapy. Nat. Rev. Cancer 2004, 4, 806–813. [Google Scholar] [CrossRef] [PubMed]
- Yoshimatsu, Y.; Miyazaki, H.; Watabe, T. Roles of signaling and transcriptional networks in pathological lymphangiogenesis. Adv. Drug Deliv. Rev. 2016, 99 Pt B, 161–171. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFbeta. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of tumor-associated neutrophil phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Tauriello, D.V.F.; Palomo-Ponce, S.; Stork, D.; Berenguer-Llergo, A.; Badia-Ramentol, J.; Iglesias, M.; Sevillano, M.; Ibiza, S.; Cañellas, A.; Hernando-Momblona, X.; et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018, 554, 538–543. [Google Scholar] [CrossRef] [PubMed]
- Mariathasan, S.; Turley, S.J.; Nickles, D.; Castiglioni, A.; Yuen, K.; Wang, Y.; Kadel Iii, E.E.; Koeppen, H.; Astarita, J.L.; Cubas, R.; et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018, 554, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Micke, P.; Faldum, A.; Metz, T.; Beeh, K.M.; Bittinger, F.; Hengstler, J.G.; Buhl, R. Staging small cell lung cancer: Veterans Administration Lung Study Group versus International Association for the Study of Lung Cancer—What limits limited disease? Lung Cancer 2002, 37, 271–276. [Google Scholar] [CrossRef]
- Borges, M.; Linnoila, R.I.; van de Velde, H.J.; Chen, H.; Nelkin, B.D.; Mabry, M.; Baylin, S.B.; Ball, D.W. An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 1997, 386, 852–855. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Saito, A.; Ohshima, M.; Suzuki, H.I.; Nagase, T. YAP and TAZ modulate cell phenotype in a subset of small cell lung cancer. Cancer Sci. 2016, 107, 1755–1766. [Google Scholar] [CrossRef] [PubMed]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef] [PubMed]
- Hougaard, S.; Norgaard, P.; Abrahamsen, N.; Moses, H.L.; Spang-Thomsen, M.; Poulsen, H.S. Inactivation of the transforming growth factor β type II receptor in human small cell lung cancer cell lines. Br. J. Cancer 1999, 79, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Murai, F.; Koinuma, D.; Shinozaki-Ushiku, A.; Fukayama, M.; Miyaozono, K.; Ehata, S. EZH2 promotes progression of small cell lung cancer by suppressing the TGF-β-Smad-ASCL1 pathway. Cell Discov. 2015, 1, 15026. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Kaczkowski, B.; Ohshima, M.; Matsuzaki, H.; Noguchi, S.; Mikami, Y.; Lizio, M.; Itoh, M.; Kawaji, H.; Lassmann, T.; et al. Integrative CAGE and DNA Methylation Profiling Identify Epigenetically Regulated Genes in NSCLC. Mol. Cancer Res. 2017, 15, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Koinuma, D.; Miyazono, K.; Heldin, C.H. Genome-wide mechanisms of Smad binding. Oncogene 2013, 32, 1609–1615. [Google Scholar] [CrossRef] [PubMed]
- Whyte, W.A.; Orlando, D.A.; Hnisz, D.; Abraham, B.J.; Lin, C.Y.; Kagey, M.H.; Rahl, P.B.; Lee, T.I.; Young, R.A. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 2013, 153, 307–319. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.I.; Young, R.A.; Sharp, P.A. Super-Enhancer-Mediated RNA Processing Revealed by Integrative MicroRNA Network Analysis. Cell 2017, 168, 1000–1014. [Google Scholar] [CrossRef] [PubMed]
- Horie, M.; Miyashita, N.; Mikami, Y.; Noguchi, S.; Yamauchi, Y.; Suzukawa, M.; Fukami, T.; Ohta, K.; Asano, Y.; Sato, S.; et al. TBX4 is involved in the super-enhancer-driven transcriptional programs underlying features specific to lung fibroblasts. Am. J. Physiol. Lung Cell. Mol. Physiol. 2018, 314, L177–L191. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.; Liu, Y.; Xue, M.; Liu, H.; Du, S.; Zhang, L.; Wang, P. Synergistic action of master transcription factors controls epithelial-to-mesenchymal transition. Nucleic Acids Res. 2016, 44, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Akhurst, R.J.; Hata, A. Targeting the TGFb signalling pathway in disease. Nat. Rev. Drug Discov. 2012, 11, 790–811. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, A.N.; Brownfield, D.G.; Harbury, P.B.; Krasnow, M.A.; Desai, T.J. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science 2018, 359, 1118–1123. [Google Scholar] [CrossRef] [PubMed]
- Treutlein, B.; Brownfield, D.G.; Wu, A.R.; Neff, N.F.; Mantalas, G.L.; Espinoza, F.H.; Desai, T.J.; Krasnow, M.A.; Quake, S.R. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature 2014, 509, 371–375. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mouse Model | Induction | Target Cell | Phenotype in the Lungs | Phenotype in Other Organs | Reference |
|---|---|---|---|---|---|
| Tgfb1 (−/−) | perivasculitis with lymphocytic and plasmacytic infiltration | systemic inflammation | [33] | ||
| Tgfb1 (−/−) | perivasculitis with lymphocytic and plasmacytic infiltration; interstitial pneumonia | systemic inflammation | [34] | ||
| Tgfb2 (−/−) | collapsed distal airways with dilated conducting airways | Cardiac, craniofacial, limb, spinal column, eye, inner ear, and urogenital defects | [35] | ||
| Tgfb3 (−/−) | atelectatic, pseudograndular histology with alveolar hypoplasia; mesenchymal thickening; extensive intrapulmonary and pleural hemorrhage; dilated conducting airways | cleft palate | [36] | ||
| Smad3 (−/−) | progressive lung airspace enlargement and emphysematous changes | defects in immune function with inflammatory lesions (originally reported by Yang X et al. in 1999) | [39] | ||
| Smad3 (−/−) | reduced pulmonary alveolarization and subsequent centrilobular emphysema | decreased growth rate (originally reported by Datto MB et al. in 1999) | [40] | ||
| Tgfbr2flox/flox | crossed with SPC-rtTA/TetO-Cre mice | lung epithelial cells | retardation of postnatal lung alveolarization with markedly decreased type I alveolar epithelial cells | [37] | |
| Tgfbr2flox/flox | crossed with Dermo1-Cre mice | mesoderm-derived tissue including lung mesenchyme | abnormal lung branching and reduced cell proliferation | defective secondary ventral body wall formation, congenital diaphragmatic hernia, and abnormal cardiac development | [37] |
| Tgfbr2flox/flox | crossed with Dermo1-Cre mice | mesoderm-derived tissue including lung mesenchyme | failure in branching morphogenesis and cystic airway malformations | abnormalities in multiple organs | [42] |
| Tgfbr2flox/flox | crossed with Nkx2-1-Cre mice | lung epithelial cells | alveolar enlargement and non-progressive emphysema; resistance to TGF-β-mediated, bleomycin-induced lung injury | [38] | |
| Tgfbr1flox/flox | crossed with GATA5-Cre mice | embryonic lung epithelium | immature alveoli and formation of a disorganized and multi-layered epithelium in the proximal airways; marked reduction in the number of club cells | [41] | |
| Tgfbr1flox/flox | crossed with Dermo1-Cre mice | mesoderm-derived tissue including lung mesenchyme | reduced submesothelial mesenchyme; restricted α-SMA-positive cell fate and promoted lipofibroblast differentiation; defective epithelial differentiation; disrupted pulmonary vasculogenesis | [43] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saito, A.; Horie, M.; Nagase, T. TGF-β Signaling in Lung Health and Disease. Int. J. Mol. Sci. 2018, 19, 2460. https://doi.org/10.3390/ijms19082460
Saito A, Horie M, Nagase T. TGF-β Signaling in Lung Health and Disease. International Journal of Molecular Sciences. 2018; 19(8):2460. https://doi.org/10.3390/ijms19082460
Chicago/Turabian StyleSaito, Akira, Masafumi Horie, and Takahide Nagase. 2018. "TGF-β Signaling in Lung Health and Disease" International Journal of Molecular Sciences 19, no. 8: 2460. https://doi.org/10.3390/ijms19082460
APA StyleSaito, A., Horie, M., & Nagase, T. (2018). TGF-β Signaling in Lung Health and Disease. International Journal of Molecular Sciences, 19(8), 2460. https://doi.org/10.3390/ijms19082460