Roles of the TGF-β–VEGF-C Pathway in Fibrosis-Related Lymphangiogenesis


Abstract
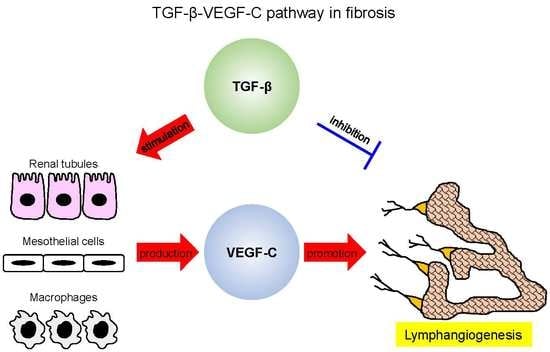
1. Introduction
2. Induction of Peritoneal Fibrosis and Neoangiogenesis by TGF-β
3. Roles of Renal and Peritoneal Lymphatics
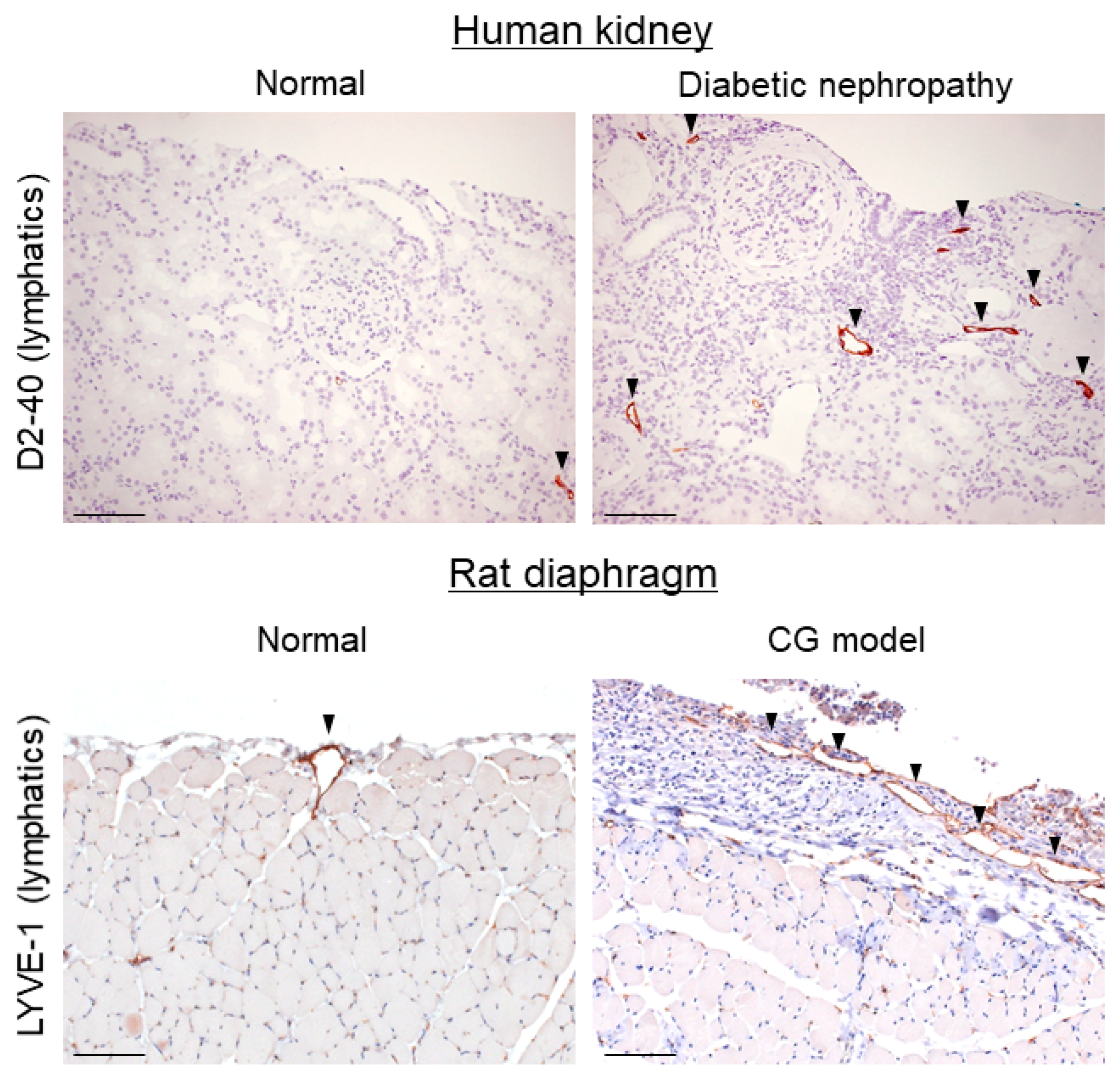
4. Lymphangiogenesis Occurs during Tissue Fibrosis
5. TGF-β Mediates Lymphangiogenesis during Fibrosis
6. Roles of CTGF in Fibrosis and Lymphangiogenesis
7. Conclusions
Conflicts of Interest
References
- Norrmen, C.; Tammela, T.; Petrova, T.V.; Alitalo, K. Biological basis of therapeutic lymphangiogenesis. Circulation 2011, 123, 1335–1351. [Google Scholar] [CrossRef] [PubMed]
- Alitalo, A.; Detmar, M. Interaction of tumor cells and lymphatic vessels in cancer progression. Oncogene 2012, 31, 4499–4508. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kataru, R.P.; Koh, G.Y. Inflammation-associated lymphangiogenesis: A double-edged sword? J. Clin. Investig. 2014, 124, 936–942. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Asai, J.; Ii, M.; Thorne, T.; Losordo, D.W.; D’Amore, P.A. Decreased macrophage number and activation lead to reduced lymphatic vessel formation and contribute to impaired diabetic wound healing. Am. J. Pathol. 2007, 170, 1178–1191. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Liu, K.; Monzon-Medina, M.E.; Padera, R.F.; Wang, H.; George, G.; Toprak, D.; Abdelnour, E.; D’Agostino, E.; Goldberg, H.J.; et al. Therapeutic lymphangiogenesis ameliorates established acute lung allograft rejection. J. Clin. Investig. 2015, 125, 4255–4268. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Aspelund, A.; Alitalo, K. Lymphangiogenic factors, mechanisms, and applications. J. Clin. Investig. 2014, 124, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Coso, S.; Bovay, E.; Petrova, T.V. Pressing the right buttons: Signaling in lymphangiogenesis. Blood 2014, 123, 2614–2624. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Ito, Y.; Mizuno, M.; Kinashi, H.; Sawai, A.; Noda, Y.; Mizuno, T.; Shimizu, H.; Fujita, Y.; Matsui, K.; et al. Transforming growth factor-β induces vascular endothelial growth factor-C expression leading to lymphangiogenesis in rat unilateral ureteral obstruction. Kidney Int. 2012, 81, 865–879. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.S.; Lee, J.E.; Jung, Y.J.; Kim, D.H.; Kang, K.P.; Lee, S.; Park, S.K.; Lee, S.Y.; Kang, M.J.; Moon, W.S.; et al. Vascular endothelial growth factor-C and -D are involved in lymphangiogenesis in mouse unilateral ureteral obstruction. Kidney Int. 2013, 83, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, H.; Ito, Y.; Mizuno, M.; Suzuki, Y.; Terabayashi, T.; Nagura, F.; Hattori, R.; Matsukawa, Y.; Mizuno, T.; Noda, Y.; et al. TGF-β 1 Promotes Lymphangiogenesis during Peritoneal Fibrosis. J. Am. Soc. Nephrol. 2013, 24, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Oka, M.; Iwata, C.; Suzuki, H.I.; Kiyono, K.; Morishita, Y.; Watabe, T.; Komuro, A.; Kano, M.R.; Miyazono, K. Inhibition of endogenous TGF-β signaling enhances lymphangiogenesis. Blood 2008, 111, 4571–4579. [Google Scholar] [CrossRef] [PubMed]
- Clavin, N.W.; Avraham, T.; Fernandez, J.; Daluvoy, S.V.; Soares, M.A.; Chaudhry, A.; Mehrara, B.J. TGF-β 1 is a negative regulator of lymphatic regeneration during wound repair. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2113–H2127. [Google Scholar] [CrossRef] [PubMed]
- Avraham, T.; Daluvoy, S.; Zampell, J.; Yan, A.; Haviv, Y.S.; Rockson, S.G.; Mehrara, B.J. Blockade of transforming growth factor-β 1 accelerates lymphatic regeneration during wound repair. Am. J. Pathol. 2010, 177, 3202–3214. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Li, L.; Zhang, X.X.; Wan, D.Y.; Xi, B.X.; Hu, Z.; Ding, W.C.; Zhu, D.; Wang, X.L.; Wang, W.; et al. SIX1 promotes tumor lymphangiogenesis by coordinating TGF-β signals that increase expression of VEGF-C. Cancer Res. 2014, 74, 5597–5607. [Google Scholar] [CrossRef] [PubMed]
- Kinashi, H.; Falke, L.L.; Nguyen, T.Q.; Bovenschen, N.; Aten, J.; Leask, A.; Ito, Y.; Goldschmeding, R. Connective tissue growth factor regulates fibrosis-associated renal lymphangiogenesis. Kidney Int. 2017, 92, 850–863. [Google Scholar] [CrossRef] [PubMed]
- Yung, S.; Chan, T.M. Intrinsic cells: Mesothelial cells—central players in regulating inflammation and resolution. Perit. Dial. Int. 2009, 29, S21–S27. [Google Scholar] [PubMed]
- Yung, S.; Chan, T.M. Pathophysiological changes to the peritoneal membrane during PD-related peritonitis: The role of mesothelial cells. Mediat. Inflamm. 2012, 2012, 484167. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.D.; Craig, K.J.; Topley, N.; Von Ruhland, C.; Fallon, M.; Newman, G.R.; Mackenzie, R.K.; Williams, G.T. Morphologic changes in the peritoneal membrane of patients with renal disease. J. Am. Soc. Nephrol. 2002, 13, 470–479. [Google Scholar] [PubMed]
- Ciszewicz, M.; Wu, G.; Tam, P.; Polubinska, A.; Breborowicz, A. Changes in peritoneal mesothelial cells phenotype after chronic exposure to glucose or N-acetylglucosamine. Transl. Res. 2007, 150, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Ishibashi, Y.; Sugimoto, T.; Ichikawa, Y.; Akatsuka, A.; Miyata, T.; Nangaku, M.; Tagawa, H.; Kurokawa, K. Glucose dialysate induces mitochondrial DNA damage in peritoneal mesothelial cells. Perit. Dial. Int. 2002, 22, 11–21. [Google Scholar] [PubMed]
- Boulanger, E.; Wautier, M.P.; Gane, P.; Mariette, C.; Devuyst, O.; Wautier, J.L. The triggering of human peritoneal mesothelial cell apoptosis and oncosis by glucose and glycoxydation products. Nephrol. Dial. Transplant. 2004, 19, 2208–2216. [Google Scholar] [CrossRef] [PubMed]
- Witowski, J.; Jorres, A. Glucose degradation products: Relationship with cell damage. Perit. Dial. Int. 2000, 20, S31–S36. [Google Scholar] [PubMed]
- Haslinger, B.; Mandl-Weber, S.; Sellmayer, A.; Lederer, S.R.; Sitter, T. Effect of high glucose concentration on the synthesis of monocyte chemoattractant protein-1 in human peritoneal mesothelial cells: Involvement of protein kinase C. Nephron 2001, 87, 346–351. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.K.; Kim, B.S.; Yang, W.S.; Kim, S.B.; Park, S.K.; Park, J.S. High glucose induces MCP-1 expression partly via tyrosine kinase-AP-1 pathway in peritoneal mesothelial cells. Kidney Int. 2001, 60, 55–64. [Google Scholar] [CrossRef] [PubMed]
- Wong, T.Y.; Phillips, A.O.; Witowski, J.; Topley, N. Glucose-mediated induction of TGF-β 1 and MCP-1 in mesothelial cells in vitro is osmolality and polyol pathway dependent. Kidney Int. 2003, 63, 1404–1416. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.H.; Hong, Y.S.; Lim, H.J.; Choi, J.H.; Han, D.S.; Yoon, K.I. High glucose solution and spent dialysate stimulate the synthesis of transforming growth factor-β 1 of human peritoneal mesothelial cells: Effect of cytokine costimulation. Perit. Dial. Int. 1999, 19, 221–230. [Google Scholar] [PubMed]
- Inagi, R.; Miyata, T.; Yamamoto, T.; Suzuki, D.; Urakami, K.; Saito, A.; van Ypersele de Strihou, C.; Kurokawa, K. Glucose degradation product methylglyoxal enhances the production of vascular endothelial growth factor in peritoneal cells: Role in the functional and morphological alterations of peritoneal membranes in peritoneal dialysis. FEBS Lett. 1999, 463, 260–264. [Google Scholar] [CrossRef]
- Yanez-Mo, M.; Lara-Pezzi, E.; Selgas, R.; Ramirez-Huesca, M.; Dominguez-Jimenez, C.; Jimenez-Heffernan, J.A.; Aguilera, A.; Sanchez-Tomero, J.A.; Bajo, M.A.; Alvarez, V.; et al. Peritoneal dialysis and epithelial-to-mesenchymal transition of mesothelial cells. N. Engl. J. Med. 2003, 348, 403–413. [Google Scholar] [CrossRef] [PubMed]
- Verger, C.; Luger, A.; Moore, H.L.; Nolph, K.D. Acute Changes in Peritoneal Morphology and Transport-Properties with Infectious Peritonitis and Mechanical Injury. Kidney Int. 1983, 23, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Tawada, M.; Ito, Y.; Hamada, C.; Honda, K.; Mizuno, M.; Suzuki, Y.; Sakata, F.; Terabayashi, T.; Matsukawa, Y.; Maruyama, S.; et al. Vascular Endothelial Cell Injury Is an Important Factor in the Development of Encapsulating Peritoneal Sclerosis in Long-Term Peritoneal Dialysis Patients. PLoS ONE 2016, 11, e0154644. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.N.; Lai, K.B.; Lam, C.W.K.; Chan, T.M.; Li, F.K.; Leung, J.C.K. Changes of cytokine profiles during peritonitis in patients on continuous ambulatory peritoneal dialysis. Am. J. Kidney Dis. 2000, 35, 644–652. [Google Scholar] [CrossRef]
- Loureiro, J.; Aguilera, A.; Selgas, R.; Sandoval, P.; Albar-Vizcaino, P.; Perez-Lozano, M.L.; Ruiz-Carpio, V.; Majano, P.L.; Lamas, S.; Rodriguez-Pascual, F.; et al. Blocking TGF-β 1 Protects the Peritoneal Membrane from Dialysate-Induced Damage. J. Am. Soc. Nephrol. 2011, 22, 1682–1695. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, C.; Sanaka, T.; Sato, T.; Omata, M.; Watanabe, M.; Mine, S.; Inuzuka, N.; Nihei, H. The effect of glucose on the proliferation of peritoneal fibroblasts. Adv. Perit. D. 1997, 13, 253–256. [Google Scholar]
- Higuchi, C.; Nihei, H. The role of protein kinase C activity in the proliferation of peritoneal fibroblasts. Perit. Dial. Int. 1999, 19, S353–S357. [Google Scholar] [PubMed]
- Breborowicz, A.; Wisniewska, J.; Polubinska, A.; Wieczorowska-Tobis, K.; Martis, L.; Oreopoulos, D.G. Role of peritoneal mesothelial cells and fibroblasts in the synthesis of hyaluronan during peritoneal dialysis. Perit. Dial. Int. 1998, 18, 382–386. [Google Scholar] [PubMed]
- Duan, W.J.; Yu, X.Q.; Huang, X.R.; Yu, J.W.; Lan, H.Y. Opposing Roles for Smad2 and Smad3 in Peritoneal Fibrosis in Vivo and in Vitro. Am. J. Pathol. 2014, 184, 2275–2284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Bajo, M.A.; del Peso, G.; Yu, X.Q.; Selgas, R. Preventing peritoneal membrane fibrosis in peritoneal dialysis patients. Kidney Int. 2016, 90, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Nessim, S.J.; Perl, J.; Bargman, J.M. The renin-angiotensin-aldosterone system in peritoneal dialysis: Is what is good for the kidney also good for the peritoneum? Kidney Int. 2010, 78, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Honda, K.; Nitta, K.; Horita, S.; Yumura, W.; Nihei, H.; Nagai, R.; Ikeda, K.; Horiuchi, S. Accumulation of advanced glycation end products in the peritoneal vasculature of continuous ambulatory peritoneal dialysis patients with low ultra-filtration. Nephrol. Dial. Transplant. 1999, 14, 1541–1549. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M. Vascular endothelial growth factor and its receptor system: Physiological functions in angiogenesis and pathological roles in various diseases. J. Biochem. 2013, 153, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Zweers, M.M.; de Waart, D.R.; Smit, W.; Struijk, D.G.; Krediet, R.T. Growth factors VEGF and TGF-β 1 in peritoneal dialysis. J. Lab. Clin. Med. 1999, 134, 124–132. [Google Scholar] [CrossRef]
- Yamagishi, S.; Yonekura, H.; Yamamoto, Y.; Katsuno, K.; Sato, F.; Mita, I.; Ooka, H.; Satozawa, N.; Kawakami, T.; Nomura, M.; et al. Advanced glycation end products-driven angiogenesis in vitro. Induction of the growth and tube formation of human microvascular endothelial cells through autocrine vascular endothelial growth factor. J. Biol. Chem. 1997, 272, 8723–8730. [Google Scholar] [CrossRef] [PubMed]
- Aroeira, L.S.; Aguilera, A.; Selgas, R.; Ramirez-Huesca, M.; Perez-Lozano, M.L.; Cirugeda, A.; Bajo, M.A.; del Peso, G.; Sanchez-Tomero, J.A.; Jimenez-Heffernan, J.A.; et al. Mesenchymal conversion of mesothelial cells as a mechanism responsible for high solute transport rate in peritoneal dialysis: Role of vascular endothelial growth factor. Am. J. Kidney Dis. 2005, 46, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Margetts, P.J.; Kolb, M.; Galt, T.; Hoff, C.M.; Shockley, T.R.; Gauldie, J. Gene transfer of transforming growth factor-β to the rat peritoneum: Effects on membrane function. J. Am. Soc. Nephrol. 2001, 12, 2029–2039. [Google Scholar] [PubMed]
- Kariya, T.; Nishimura, H.; Mizuno, M.; Suzuki, Y.; Matsukawa, Y.; Sakata, F.; Maruyama, S.; Takei, Y.; Ito, Y. TGF-β1-VEGF-A pathway induces neoangiogenesis with peritoneal fibrosis in patients undergoing peritoneal dialysis. Am. J. Physiol. Ren. Physiol. 2018, 314, F167–F180. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.K.; Ranjan, P.; Sakhuja, V.; Jha, V. The Case: Milky urine. Kidney Int. 2008, 74, 1100–1101. [Google Scholar] [CrossRef] [PubMed]
- Kerjaschki, D.; Regele, H.M.; Moosberger, I.; Nagy-Bojarski, K.; Watschinger, B.; Soleiman, A.; Birner, P.; Krieger, S.; Hovorka, A.; Silberhumer, G.; et al. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J. Am. Soc. Nephrol. 2004, 15, 603–612. [Google Scholar] [CrossRef] [PubMed]
- Sakamoto, I.; Ito, Y.; Mizuno, M.; Suzuki, Y.; Sawai, A.; Tanaka, A.; Maruyama, S.; Takei, Y.; Yuzawa, Y.; Matsuo, S. Lymphatic vessels develop during tubulointerstitial fibrosis. Kidney Int. 2009, 75, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Kerjaschki, D.; Huttary, N.; Raab, I.; Regele, H.; Bojarski-Nagy, K.; Bartel, G.; Krober, S.M.; Greinix, H.; Rosenmaier, A.; Karlhofer, F.; et al. Lymphatic endothelial progenitor cells contribute to de novo lymphangiogenesis in human renal transplants. Nat. Med. 2006, 12, 230–234. [Google Scholar] [CrossRef] [PubMed]
- Matsui, K.; Nagy-Bojarsky, K.; Laakkonen, P.; Krieger, S.; Mechtler, K.; Uchida, S.; Geleff, S.; Kang, D.H.; Johnson, R.J.; Kerjaschki, D. Lymphatic microvessels in the rat remnant kidney model of renal fibrosis: Aminopeptidase p and podoplanin are discriminatory markers for endothelial cells of blood and lymphatic vessels. J. Am. Soc. Nephrol. 2003, 14, 1981–1989. [Google Scholar] [CrossRef] [PubMed]
- Huber, S.; Bruns, C.J.; Schmid, G.; Hermann, P.C.; Conrad, C.; Niess, H.; Huss, R.; Graeb, C.; Jauch, K.W.; Heeschen, C.; et al. Inhibition of the mammalian target of rapamycin impedes lymphangiogenesis. Kidney Int. 2007, 71, 771–777. [Google Scholar] [CrossRef] [PubMed]
- Palin, N.K.; Savikko, J.; Koskinen, P.K. Sirolimus inhibits lymphangiogenesis in rat renal allografts, a novel mechanism to prevent chronic kidney allograft injury. Transpl. Int. 2013, 26, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Yazdani, S.; Hijmans, R.S.; Poosti, F.; Dam, W.; Navis, G.; van Goor, H.; van den Born, J. Targeting tubulointerstitial remodeling in proteinuric nephropathy in rats. Dis. Model. Mech. 2015, 8, 919–930. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, S.; Nakano, T.; Torisu, K.; Tsuchimoto, A.; Eriguchi, M.; Haruyama, N.; Masutani, K.; Tsuruya, K.; Kitazono, T. Vascular endothelial growth factor-C ameliorates renal interstitial fibrosis through lymphangiogenesis in mouse unilateral ureteral obstruction. Lab. Investig. 2017, 97, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.L.; Woolf, A.S.; Kolatsi-Joannou, M.; Baluk, P.; Sandford, R.N.; Peters, D.J.; McDonald, D.M.; Price, K.L.; Winyard, P.J.; Long, D.A. Vascular Endothelial Growth Factor C for Polycystic Kidney Diseases. J. Am. Soc. Nephrol. 2016, 27, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Mactier, R.A.; Khanna, R.; Twardowski, Z.; Moore, H.; Nolph, K.D. Contribution of lymphatic absorption to loss of ultrafiltration and solute clearances in continuous ambulatory peritoneal dialysis. J. Clin. Investig. 1987, 80, 1311–1316. [Google Scholar] [CrossRef] [PubMed]
- Abuhijleh, M.F.; Habbal, O.A.; Moqattash, S.T. The Role of the Diaphragm in Lymphatic Absorption from the Peritoneal-Cavity. J. Anat. 1995, 186, 453–467. [Google Scholar]
- Fussholler, A.; zur Nieden, S.; Grabensee, B.; Plum, J. Peritoneal fluid and solute transport: Influence of treatment time, peritoneal dialysis modality, and peritonitis incidence. J. Am. Soc. Nephrol. 2002, 13, 1055–1060. [Google Scholar] [PubMed]
- Smit, W.; Schouten, N.; van den Berg, N.; Langedijk, M.J.; Struijk, D.G.; Krediet, R.T. Netherlands Ultrafiltration Failure Study, G., Analysis of the prevalence and causes of ultrafiltration failure during long-term peritoneal dialysis: A cross-sectional study. Perit. Dial. Int. 2004, 24, 562–570. [Google Scholar] [PubMed]
- Krediet, R.T. The effective lymphatic absorption rate is an accurate and useful concept in the physiology of peritoneal dialysis. Perit. Dial. Int. 2004, 24, 309–313. [Google Scholar] [PubMed]
- Flessner, M. Effective lymphatic absorption rate is not a useful or accurate term to use in the physiology of peritoneal dialysis. Perit. Dial. Int. 2004, 24, 313–316. [Google Scholar] [PubMed]
- Yang, W.S.; Tsai, T.J.; Shih, C.L.; Huang, J.W.; Chuang, H.F.; Chen, M.H.; Fang, C.C. Intraperitoneal Vascular Endothelial Growth Factor C Level Is Related to Peritoneal Dialysis Ultrafiltration. Blood Purif. 2009, 28, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Fabbrini, P.; Schilte, M.N.; Zareie, M.; ter Wee, P.M.; Keuning, E.D.; Beelen, R.H.J.; van den Born, J. Celecoxib treatment reduces peritoneal fibrosis and angiogenesis and prevents ultrafiltration failure in experimental peritoneal dialysis. Nephrol. Dial. Transplant. 2009, 24, 3669–3676. [Google Scholar] [CrossRef] [PubMed]
- Iwata, C.; Kano, M.R.; Komuro, A.; Oka, M.; Kiyono, K.; Johansson, E.; Morishita, Y.; Yashiro, M.; Hirakawa, K.; Kaminishi, M.; et al. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res. 2007, 67, 10181–10189. [Google Scholar] [CrossRef] [PubMed]
- Vlahu, C.A.; de Graaff, M.; Aten, J.; Struijk, D.G.; Krediet, R.T. Lymphangiogenesis and Lymphatic Absorption Are Related and Increased in Chronic Kidney Failure, Independent of Exposure to Dialysis Solutions. Adv. Perit. Dial. 2015, 31, 21–25. [Google Scholar] [PubMed]
- Garcia-Lopez, E.; Lindholm, B.; Davies, S. An update on peritoneal dialysis solutions. Nat. Rev. Nephrol. 2012, 8, 224–233. [Google Scholar] [CrossRef] [PubMed]
- Thodis, E.; Passadakis, P.; Panagoutsos, S.; Marinopoulos, D.; Vargemezis, V. Failure of icodextrin to provide adequate ultrafiltration in continuous ambulatory peritoneal dialysis patients. Adv. Perit. Dial. 1999, 15, 171–174. [Google Scholar] [PubMed]
- Terabayashi, T.; Ito, Y.; Mizuno, M.; Suzuki, Y.; Kinashi, H.; Sakata, F.; Tomita, T.; Iguchi, D.; Tawada, M.; Nishio, R.; et al. Vascular endothelial growth factor receptor-3 is a novel target to improve net ultrafiltration in methylglyoxal-induced peritoneal injury. Lab. Investig. 2015, 95, 1029–1043. [Google Scholar] [CrossRef] [PubMed]
- Nykanen, A.I.; Sandelin, H.; Krebs, R.; Keranen, M.A.; Tuuminen, R.; Karpanen, T.; Wu, Y.; Pytowski, B.; Koskinen, P.K.; Ylä-Herttuala, S.; et al. Targeting lymphatic vessel activation and CCL21 production by vascular endothelial growth factor receptor-3 inhibition has novel immunomodulatory and antiarteriosclerotic effects in cardiac allografts. Circulation 2010, 121, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Akishima-Fukasawa, Y.; Ito, K.; Akasaka, Y.; Tanaka, M.; Shimokawa, R.; Kimura-Matsumoto, M.; Morita, H.; Sato, S.; Kamata, I.; et al. Lymphangiogenesis in myocardial remodelling after infarction. Histopathology 2007, 51, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Henri, O.; Pouehe, C.; Houssari, M.; Galas, L.; Nicol, L.; Edwards-Levy, F.; Henry, J.P.; Dumesnil, A.; Boukhalfa, I.; Banquet, S.; et al. Selective Stimulation of Cardiac Lymphangiogenesis Reduces Myocardial Edema and Fibrosis Leading to Improved Cardiac Function Following Myocardial Infarction. Circulation 2016, 133, 1484–1497. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Yoon, J.Y.; Ko, S.M.; Jin, S.A.; Kim, J.H.; Cho, C.H.; Kim, J.M.; Lee, J.H.; Choi, S.W.; Seong, I.W.; et al. Endothelial progenitor cell transplantation decreases lymphangiogenesis and adverse myocardial remodeling in a mouse model of acute myocardial infarction. Exp. Mol. Med. 2011, 43, 479–485. [Google Scholar] [CrossRef] [PubMed]
- El-Chemaly, S.; Malide, D.; Zudaire, E.; Ikeda, Y.; Weinberg, B.A.; Pacheco-Rodriguez, G.; Rosas, I.O.; Aparicio, M.; Ren, P.; MacDonald, S.D.; et al. Abnormal lymphangiogenesis in idiopathic pulmonary fibrosis with insights into cellular and molecular mechanisms. Proc. Natl. Acad. Sci. USA 2009, 106, 3958–3963. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Corral, I.; Olmeda, D.; Dieguez-Hurtado, R.; Tammela, T.; Alitalo, K.; Ortega, S. In vivo imaging of lymphatic vessels in development, wound healing, inflammation, and tumor metastasis. Proc. Natl. Acad. Sci. USA 2012, 109, 6223–6228. [Google Scholar] [CrossRef] [PubMed]
- Paavonen, K.; Puolakkainen, P.; Jussila, L.; Jahkola, T.; Alitalo, K. Vascular endothelial growth factor receptor-3 in lymphangiogenesis in wound healing. Am. J. Pathol. 2000, 156, 1499–1504. [Google Scholar] [CrossRef]
- Okudera, K.; Kamata, Y.; Takanashi, S.; Hasegawa, Y.; Tsushima, T.; Ogura, Y.; Nakanishi, K.; Sato, H.; Okumura, K. Small adenocarcinoma of the lung: Prognostic significance of central fibrosis chiefly because of its association with angiogenesis and lymphangiogenesis. Pathol. Int. 2006, 56, 494–502. [Google Scholar] [CrossRef] [PubMed]
- Van den Eynden, G.G.; Van der Auwera, I.; Van Laere, S.J.; Colpaert, C.G.; van Dam, P.; Dirix, L.Y.; Vermeulen, P.B.; Van Marck, E.A. Distinguishing blood and lymph vessel invasion in breast cancer: A prospective immunohistochemical study. Br. J. Cancer 2006, 94, 1643–1649. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.; Du, Y.; Liu, Y.; He, Y.; Yang, C.; Wang, W.; Gao, F. Low molecular weight hyaluronan induces lymphangiogenesis through LYVE-1-mediated signaling pathways. PLoS ONE 2014, 9, e92857. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.J.; Lee, A.S.; Nguyen-Thanh, T.; Kang, K.P.; Lee, S.; Jang, K.Y.; Kim, M.K.; Kim, S.H.; Park, S.K.; Kim, W. Hyaluronan-induced VEGF-C promotes fibrosis-induced lymphangiogenesis via Toll-like receptor 4-dependent signal pathway. Biochem. Biophys. Res. Commun. 2015, 466, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Bazigou, E.; Xie, S.; Chen, C.; Weston, A.; Miura, N.; Sorokin, L.; Adams, R.; Muro, A.F.; Sheppard, D.; Makinen, T. Integrin-α9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 2009, 17, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Ou, J.; Li, J.; Pan, F.; Xie, G.; Zhou, Q.; Huang, H.; Liang, H. Endostatin suppresses colorectal tumor-induced lymphangiogenesis by inhibiting expression of fibronectin extra domain A and integrin α9. J. Cell. Biochem. 2011, 112, 2106–2114. [Google Scholar] [CrossRef] [PubMed]
- Meens, M.J.; Sabine, A.; Petrova, T.V.; Kwak, B.R. Connexins in lymphatic vessel physiology and disease. FEBS Lett. 2014, 588, 1271–1277. [Google Scholar] [CrossRef] [PubMed]
- Avraham, T.; Clavin, N.W.; Daluvoy, S.V.; Fernandez, J.; Soares, M.A.; Cordeiro, A.P.; Mehrara, B.J. Fibrosis is a key inhibitor of lymphatic regeneration. Plast. Reconstr. Surg. 2009, 124, 438–450. [Google Scholar] [CrossRef] [PubMed]
- Veikkola, T.; Jussila, L.; Makinen, T.; Karpanen, T.; Jeltsch, M.; Petrova, T.V.; Kubo, H.; Thurston, G.; McDonald, D.M.; Achen, M.G.; et al. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. EMBO J. 2001, 20, 1223–1231. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Alitalo, K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell 2010, 140, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Dumont, D.J.; Jussila, L.; Taipale, J.; Lymboussaki, A.; Mustonen, T.; Pajusola, K.; Breitman, M.; Alitalo, K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 1998, 282, 946–949. [Google Scholar] [CrossRef] [PubMed]
- Partanen, T.A.; Arola, J.; Saaristo, A.; Jussila, L.; Ora, A.; Miettinen, M.; Stacker, S.A.; Achen, M.G.; Alitalo, K. VEGF-C and VEGF-D expression in neuroendocrine cells and their receptor, VEGFR-3, in fenestrated blood vessels in human tissues. FASEB J. 2000, 14, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Makinen, T.; Veikkola, T.; Mustjoki, S.; Karpanen, T.; Catimel, B.; Nice, E.C.; Wise, L.; Mercer, A.; Kowalski, H.; Kerjaschki, D.; et al. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. EMBO J. 2001, 4762–4773. [Google Scholar] [CrossRef] [PubMed]
- Bui, H.M.; Enis, D.; Robciuc, M.R.; Nurmi, H.J.; Cohen, J.; Chen, M.; Yang, Y.; Dhillon, V.; Johnson, K.; Zhang, H.; et al. Proteolytic activation defines distinct lymphangiogenic mechanisms for VEGFC and VEGFD. J. Clin. Investig. 2016, 126, 2167–2180. [Google Scholar] [CrossRef] [PubMed]
- Tammela, T.; Saaristo, A.; Holopainen, T.; Lyytikka, J.; Kotronen, A.; Pitkonen, M.; Abo-Ramadan, U.; Yla-Herttuala, S.; Petrova, T.V.; Alitalo, K. Therapeutic differentiation and maturation of lymphatic vessels after lymph node dissection and transplantation. Nat. Med. 2007, 13, 1458–1466. [Google Scholar] [CrossRef] [PubMed]
- Karkkainen, M.J.; Haiko, P.; Sainio, K.; Partanen, J.; Taipale, J.; Petrova, T.V.; Jeltsch, M.; Jackson, D.G.; Talikka, M.; Rauvala, H.; et al. Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat. Immunol. 2004, 5, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, M.E.; Halford, M.M.; Roufail, S.; Williams, R.A.; Hibbs, M.L.; Grail, D.; Kubo, H.; Stacker, S.A.; Achen, M.G. Vascular endothelial growth factor D is dispensable for development of the lymphatic system. Mol. Cell. Biol. 2005, 25, 2441–2449. [Google Scholar] [CrossRef] [PubMed]
- Jeltsch, M.; Jha, S.K.; Tvorogov, D.; Anisimov, A.; Leppanen, V.M.; Holopainen, T.; Kivela, R.; Ortega, S.; Karpanen, T.; Alitalo, K. CCBE1 enhances lymphangiogenesis via A disintegrin and metalloprotease with thrombospondin motifs-3-mediated vascular endothelial growth factor-C activation. Circulation 2014, 129, 1962–1971. [Google Scholar] [CrossRef] [PubMed]
- Ghanta, S.; Cuzzone, D.A.; Torrisi, J.S.; Albano, N.J.; Joseph, W.J.; Savetsky, I.L.; Gardenier, J.C.; Chang, D.; Zampell, J.C.; Mehrara, B.J. Regulation of inflammation and fibrosis by macrophages in lymphedema. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H1065–H1077. [Google Scholar] [CrossRef] [PubMed]
- Baluk, P.; Tammela, T.; Ator, E.; Lyubynska, N.; Achen, M.G.; Hicklin, D.J.; Jeltsch, M.; Petrova, T.V.; Pytowski, B.; Stacker, S.A.; et al. Pathogenesis of persistent lymphatic vessel hyperplasia in chronic airway inflammation. J. Clin. Investig. 2005, 115, 247–257. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, S.; Correale, C.; Tacconi, C.; Gandelli, A.; Pietrogrande, G.; Vetrano, S.; Genua, M.; Arena, V.; Spinelli, A.; Peyrin-Biroulet, L.; et al. VEGF-C-dependent stimulation of lymphatic function ameliorates experimental inflammatory bowel disease. J. Clin. Investig. 2014, 124, 3863–3878. [Google Scholar] [CrossRef] [PubMed]
- Enholm, B.; Karpanen, T.; Jeltsch, M.; Kubo, H.; Stenback, F.; Prevo, R.; Jackson, D.G.; Yla-Herttuala, S.; Alitalo, K. Adenoviral expression of vascular endothelial growth factor-C induces lymphangiogenesis in the skin. Circ. Res. 2001, 88, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Saaristo, A.; Veikkola, T.; Enholm, B.; Hytonen, M.; Arola, J.; Pajusola, K.; Turunen, P.; Jeltsch, M.; Karkkainen, M.J.; Kerjaschki, D.; et al. Adenoviral VEGF-C overexpression induces blood vessel enlargement, tortuosity, and leakiness but no sprouting angiogenesis in the skin or mucous membranes. FASEB J. 2002, 16, 1041–1049. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, Y.; Chosa, N.; Sawada, S.; Kondo, H.; Yaegashi, T.; Ishisaki, A. VEGF-C and TGF-β reciprocally regulate mesenchymal stem cell commitment to differentiation into lymphatic endothelial or osteoblastic phenotypes. Int. J. Mol. Med. 2016, 37, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Yan, A.; Avraham, T.; Zampell, J.C.; Haviv, Y.S.; Weitman, E.; Mehrara, B.J. Adipose-derived stem cells promote lymphangiogenesis in response to VEGF-C stimulation or TGF-β 1 inhibition. Future Oncol. 2011, 7, 1457–1473. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Osorio, J.C.; Risquez, C.; Wang, H.; Shi, Y.; Gochuico, B.R.; Morse, D.; Rosas, I.O.; El-Chemaly, S. Transforming growth factor-β 1 downregulates vascular endothelial growth factor-D expression in human lung fibroblasts via the Jun NH2-terminal kinase signaling pathway. Mol. Med. 2014, 20, 120–134. [Google Scholar] [CrossRef] [PubMed]
- Stuhlmeier, K.M.; Pollaschek, C. Differential effect of transforming growth factor-β (TGF-β) on the genes encoding hyaluronan synthases and utilization of the p38 MAPK pathway in TGF-β-induced hyaluronan synthase 1 activation. J. Biol. Chem. 2004, 279, 8753–8760. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Tung, W.H.; Tang, K.T.; Wong, Y.K.; Huang, G.J.; Wu, J.C.; Guo, Y.J.; Chen, C.C. TGF-β induced hyaluronan synthesis in orbital fibroblasts involves protein kinase C βII activation in vitro. J. Cell. Biochem. 2005, 95, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.F. Cell surface receptors for CCN proteins. J. Cell Commun. Signal. 2016, 10, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef]
- Abreu, J.G.; Ketpura, N.I.; Reversade, B.; De Robertis, E.M. Connective-tissue growth factor (CTGF) modulates cell signalling by BMP and TGF-β. Nat. Cell Biol. 2002, 4, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.H.; Li, P.; Liu, M.T.; Liu, C.; Sun, Z.M.; Guo, X.; Zhang, Y.L. CCN2 and CCN5 exerts opposing effect on fibroblast proliferation and transdifferentiation induced by TGF-β. Clin. Exp. Pharmacol. Physiol. 2015, 42, 1207–1219. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.C.; Chuang, S.M.; Hsu, C.J.; Tsai, C.H.; Wang, S.W.; Tang, C.H. CTGF increases vascular endothelial growth factor-dependent angiogenesis in human synovial fibroblasts by increasing miR-210 expression. Cell Death Dis. 2014, 5, e1485. [Google Scholar] [CrossRef] [PubMed]
- Pi, L.Y.; Shenoy, A.K.; Liu, J.W.; Kim, S.; Nelson, N.; Xia, H.M.; Hauswirth, W.W.; Petersen, B.E.; Schultz, G.S.; Scott, E.W. CCN2/CTGF regulates neovessel formation via targeting structurally conserved cystine knot motifs in multiple angiogenic regulators. FASEB J. 2012, 26, 3365–3379. [Google Scholar] [CrossRef] [PubMed]
- Inoki, I.; Shiomi, T.; Hashimoto, G.; Enomoto, H.; Nakamura, H.; Makino, K.; Ikeda, E.; Takata, S.; Kobayashi, K.; Okada, Y. Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J. 2001, 15, 219–221. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, G.; Inoki, I.; Fujii, Y.; Aoki, T.; Ikeda, E.; Okada, Y. Matrix metalloproteinases cleave connective tissue growth factor and reactivate angiogenic activity of vascular endothelial growth factor 165. J. Biol. Chem. 2002, 277, 36288–36295. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Ito, Y.; Mizuno, M.; Nishimura, H.; Suzuki, Y.; Hattori, R.; Matsukawa, Y.; Imai, M.; Oliver, N.; Goldschmeding, R.; et al. Connective tissue growth factor (CTGF/CCN2) is increased in peritoneal dialysis patients with high peritoneal solute transport rate. Am. J. Physiol.-Ren. Physiol. 2010, 298, F721–F733. [Google Scholar] [CrossRef] [PubMed]
- Zarrinkalam, K.H.; Stanley, J.M.; Gray, J.; Oliver, N.; Faull, R.J. Connective tissue growth factor and its regulation in the peritoneal cavity of peritoneal dialysis patients. Kidney Int. 2003, 64, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.C.K.; Chan, L.Y.Y.; Tam, K.Y.; Tang, S.C.W.; Lam, M.F.; Cheng, A.S.; Chu, K.M.; Lai, K.N. Regulation of CCN2/CTGF and related cytokines in cultured peritoneal cells under conditions simulating peritoneal dialysis. Nephrol. Dial. Transplant. 2009, 24, 458–469. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Nakamura, M.; Lipson, K.E.; Miyake, T.; Kamikawa, Y.; Sagara, A.; Shinozaki, Y.; Kitajima, S.; Toyama, T.; Hara, A.; et al. Inhibition of CTGF ameliorates peritoneal fibrosis through suppression of fibroblast and myofibroblast accumulation and angiogenesis. Sci. Rep. 2017, 7, 5392. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Mori, K.; Kasahara, M.; Koga, K.; Ishii, A.; Mori, K.P.; Osaki, K.; Mukoyama, M.; Yanagita, M.; Yokoi, H. Deletion of connective tissue growth factor ameliorates peritoneal fibrosis by inhibiting angiogenesis and inflammation. Nephrol. Dial. Transplant. 2018, 33, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Falke, L.L.; Goldschmeding, R.; Nguyen, T.Q. A perspective on anti-CCN2 therapy for chronic kidney disease. Nephrol. Dial. Transplant. 2014, 29, I30–I37. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Aten, J.; Bende, R.J.; Oemar, B.S.; Rabelink, T.J.; Weening, J.J.; Goldschmeding, R. Expression of connective tissue growth factor in human renal fibrosis. Kidney Int. 1998, 53, 853–861. [Google Scholar] [CrossRef] [PubMed]
- Ito, Y.; Aten, J.; Nguyen, T.Q.; Joles, J.A.; Matsuo, S.; Weening, J.J.; Goldschmeding, R. Involvement of Connective Tissue Growth Factor in Human and Experimental Hypertensive Nephrosclerosis. Nephron Exp. Nephrol. 2011, 117, E9–E20. [Google Scholar] [CrossRef] [PubMed]
- Kanemoto, K.; Usui, J.; Nitta, K.; Horita, S.; Harada, A.; Koyama, A.; Aten, J.; Nagata, M. In situ expression of connective tissue growth factor in human crescentic glomerulonephritis. Virchows Arch. 2004, 444, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Metalidis, C.; van Vuuren, S.H.; Broekhuizen, R.; Lerut, E.; Naesens, M.; Bakker, S.J.L.; Wetzels, J.F.M.; Goldschmeding, R.; Kuypers, D.R.J. Urinary Connective Tissue Growth Factor Is Associated with Human Renal Allograft Fibrogenesis. Transplantation 2013, 96, 494–500. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Sugawara, A.; Mukoyama, M.; Mori, K.; Makino, H.; Suganami, T.; Nagae, T.; Yahata, K.; Fujinaga, Y.; Tanaka, I.; et al. Role of connective tissue growth factor in profibrotic action of transforming growth factor-β: A potential target for preventing renal fibrosis. Am. J. Kidney Dis. 2001, 38, S134–S138. [Google Scholar] [CrossRef] [PubMed]
- Castro, N.E.; Kato, M.; Park, J.T.; Natarajan, R. Transforming Growth Factor beta 1 (TGF-β 1) Enhances Expression of Profibrotic Genes through a Novel Signaling Cascade and MicroRNAs in Renal Mesangial Cells. J. Biol. Chem. 2014, 289, 29001–29013. [Google Scholar] [CrossRef] [PubMed]
- Luo, D.D.; Phillips, A.; Fraser, D. Bone Morphogenetic Protein-7 Inhibits Proximal Tubular Epithelial Cell Smad3 Signaling via Increased SnoN Expression. Am. J. Pathol. 2010, 176, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Roestenberg, P.; van Nieuwenhoven, F.A.; Wieten, L.; Boer, P.; Diekman, T.; Tiller, A.M.; Wiersinga, W.M.; Oliver, N.; Usinger, W.; Weitz, S.; et al. Connective tissue growth factor is increased in plasma of type 1 diabetic patients with nephropathy. Diabetes Care 2004, 27, 1164–1170. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.Q.; Tarnow, L.; Andersen, S.; Hovind, P.; Parving, H.H.; Goldschmeding, R.; Van Nieuwenhoven, F.A. Urinary connective tissue growth factor excretion correlates with clinical markers of renal disease in a large population of type 1 diabetic patients with diabetic nephropathy. Diabetes Care 2006, 29, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Tam, F.W.K.; Riser, B.L.; Meeran, K.; Rambow, J.; Pusey, C.D.; Frankel, A.H. Urinary monocyte chemoattractant protein-1 (MCP-1) and connective tissue growth factor (CCN2) as prognostic markers for progression of diabetic nephropathy. Cytokine 2009, 47, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Vanhove, T.; Kinashi, H.; Nguyen, T.Q.; Metalidis, C.; Poesen, K.; Naesens, M.; Lerut, E.; Goldschmeding, R.; Kuypers, D.R.J. Tubulointerstitial expression and urinary excretion of connective tissue growth factor 3 months after renal transplantation predict interstitial fibrosis and tubular atrophy at 5 years in a retrospective cohort analysis. Transpl. Int. 2017, 30, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, H.; Mukoyama, M.; Nagae, T.; Mori, K.; Suganami, T.; Sawai, K.; Yoshioka, T.; Koshikawa, M.; Nishida, T.; Takigawa, M.; et al. Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2004, 15, 1430–1440. [Google Scholar] [CrossRef] [PubMed]
- Guha, M.; Xu, Z.G.; Tung, D.; Lanting, L.; Natarajan, R. Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J. 2007, 21, 3355–3368. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.H.; Lu, Y.P.; Song, J.; Yang, L.; Shi, Y.J.; Li, Y.P. Inhibition of connective tissue growth factor by small interfering RNA prevents renal fibrosis in rats undergoing chronic allograft nephropathy. Transpl. Proc. 2008, 40, 2365–2369. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kikuta, T.; Kobayashi, T.; Inoue, T.; Kanno, Y.; Takigawa, M.; Sugaya, T.; Kopp, J.B.; Suzuki, H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J. Am. Soc. Nephrol. 2005, 16, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Adler, S.G.; Schwartz, S.; Williams, M.E.; Arauz-Pacheco, C.; Bolton, W.K.; Lee, T.; Li, D.X.; Neff, T.B.; Urquilla, P.R.; Sewell, K.L. Phase 1 Study of Anti-CTGF Monoclonal Antibody in Patients with Diabetes and Microalbuminuria. Clin. J. Am. Soc. Nephrol. 2010, 5, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Raghu, G.; Scholand, M.B.; de Andrade, J.; Lancaster, L.; Mageto, Y.; Goldin, J.; Brown, K.K.; Flaherty, K.R.; Wencel, M.; Wanger, J.; et al. FG-3019 anti-connective tissue growth factor monoclonal antibody: Results of an open-label clinical trial in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016, 47, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.C.; Chen, P.C.; Lein, M.Y.; Tsao, C.W.; Huang, C.C.; Wang, S.W.; Tang, C.H.; Tung, K.C. WISP-1 promotes VEGF-C-dependent lymphangiogenesis by inhibiting miR-300 in human oral squamous cell carcinoma cells. Oncotarget 2016, 7, 9993–10005. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Organs | Research Methods | Findings | References |
|---|---|---|---|
| Heart | Autopsied hearts after MI | ・ Lymphangiogenesis was observed in healing stages with fibrosis. | [71] |
| Rat models of MI | ・ Administration of VEGF-C accereated lymphangiogenesis, leading to reducing cardiac inflammation, fibrosis, and dysfunction. | [72] | |
| Lung | Human lung tissues and BALF | ・ Areas of lymphatic vessels were correlated with the severeity of IPF. ・ Short-fragments of hyaluronic acid in BALF might mediate lymphatic endothelial cell growth. ・ CD11b+ alveolar macrophages in IPF could differenciate into lymphatic endothelial cells. | [74] |
| Kidney | Human kidney biopsies | ・ The number of lymphatic vessels was correlated with the degree of tubulointerstitial fibrosis. | [49] |
| Rat models of UUO | ・ TGF-β1 promoted VEGF-C production in proximal tubular cells, collecting duct cells, and macrophages, leading to fibrosis-associated renal lymphangiogenesis. | [8] | |
| Cultured renal tubular cells, macrophages | |||
| Mouse models of UUO | ・ TGF-β1 and TNF-α induced VEGF-C production in proximal tubular cells and macrophages. ・ VEGF-D prevented direct inhibitory effects on lymphatic endothelial cell growth by TGF-β1. | [9] | |
| Cultured renal tubular cells, macrophages, lymphatic endothelial cells | |||
| Rat models of proteinuric nephropathy | ・ Suppression of lymphangiogenesis by anti-VEGFR3 antibody did not affect inflammation, fibrosis, and proteinuria. | [54] | |
| Mouse models of UUO, IRI | ・ CTGF induced VEGF-C production in proximal tubular cells, leading to fibrosis-associated renal lymphangiogenesis. ・ CTGF bound to VEGF-C and inhibited VEGF-C-induced lymphatic endothelial cell growth. | [16] | |
| Cultured renal tubular cells, lymphatic endothelial cells | |||
| Mouse models of UUO | ・ Administration of VEGF-C accereated lymphangiogenesis, leading to reducing inflammation, TGF-β1 expression, and fibrosis. | [55] | |
| Peritoneum | Human peritoneal biopsies | ・ Expression of lymphatic vessels was correlated with the degree of peritoneal fibrosis. ・ TGF-β1 promoted VEGF-C production in mesothelial cells, leading to fibrosis-associated peritoneal lymphangiogenesis. | [10] |
| Rat PF models induced by CG | |||
| Cultured mesothelial cells | |||
| Mouse PF models induced by MGO | ・ Suppression of lymphangiogenesis by soluble VEGFR-3 improved deteriorated net ultrafiltration. | [69] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kinashi, H.; Ito, Y.; Sun, T.; Katsuno, T.; Takei, Y. Roles of the TGF-β–VEGF-C Pathway in Fibrosis-Related Lymphangiogenesis. Int. J. Mol. Sci. 2018, 19, 2487. https://doi.org/10.3390/ijms19092487
Kinashi H, Ito Y, Sun T, Katsuno T, Takei Y. Roles of the TGF-β–VEGF-C Pathway in Fibrosis-Related Lymphangiogenesis. International Journal of Molecular Sciences. 2018; 19(9):2487. https://doi.org/10.3390/ijms19092487
Chicago/Turabian StyleKinashi, Hiroshi, Yasuhiko Ito, Ting Sun, Takayuki Katsuno, and Yoshifumi Takei. 2018. "Roles of the TGF-β–VEGF-C Pathway in Fibrosis-Related Lymphangiogenesis" International Journal of Molecular Sciences 19, no. 9: 2487. https://doi.org/10.3390/ijms19092487
APA StyleKinashi, H., Ito, Y., Sun, T., Katsuno, T., & Takei, Y. (2018). Roles of the TGF-β–VEGF-C Pathway in Fibrosis-Related Lymphangiogenesis. International Journal of Molecular Sciences, 19(9), 2487. https://doi.org/10.3390/ijms19092487