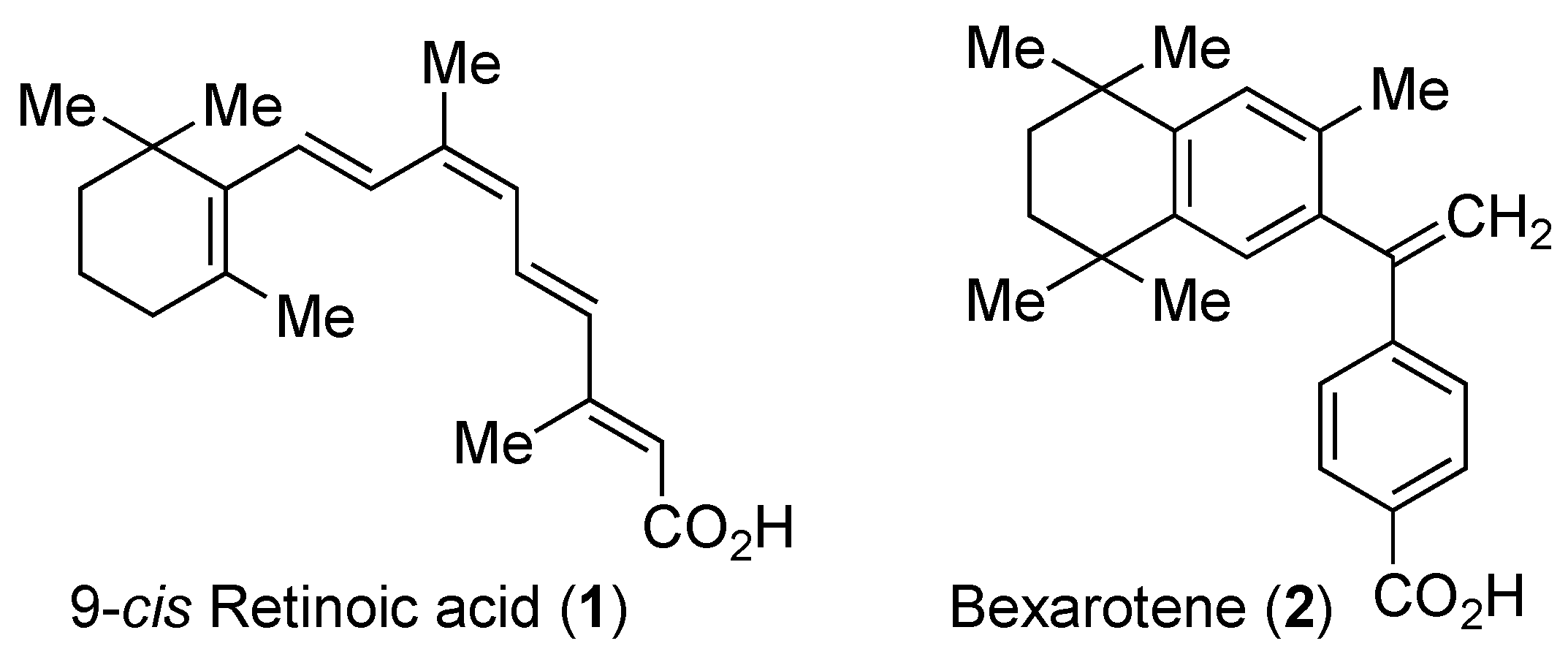
Retinoid X Receptor Antagonists


Abstract
1. Introduction
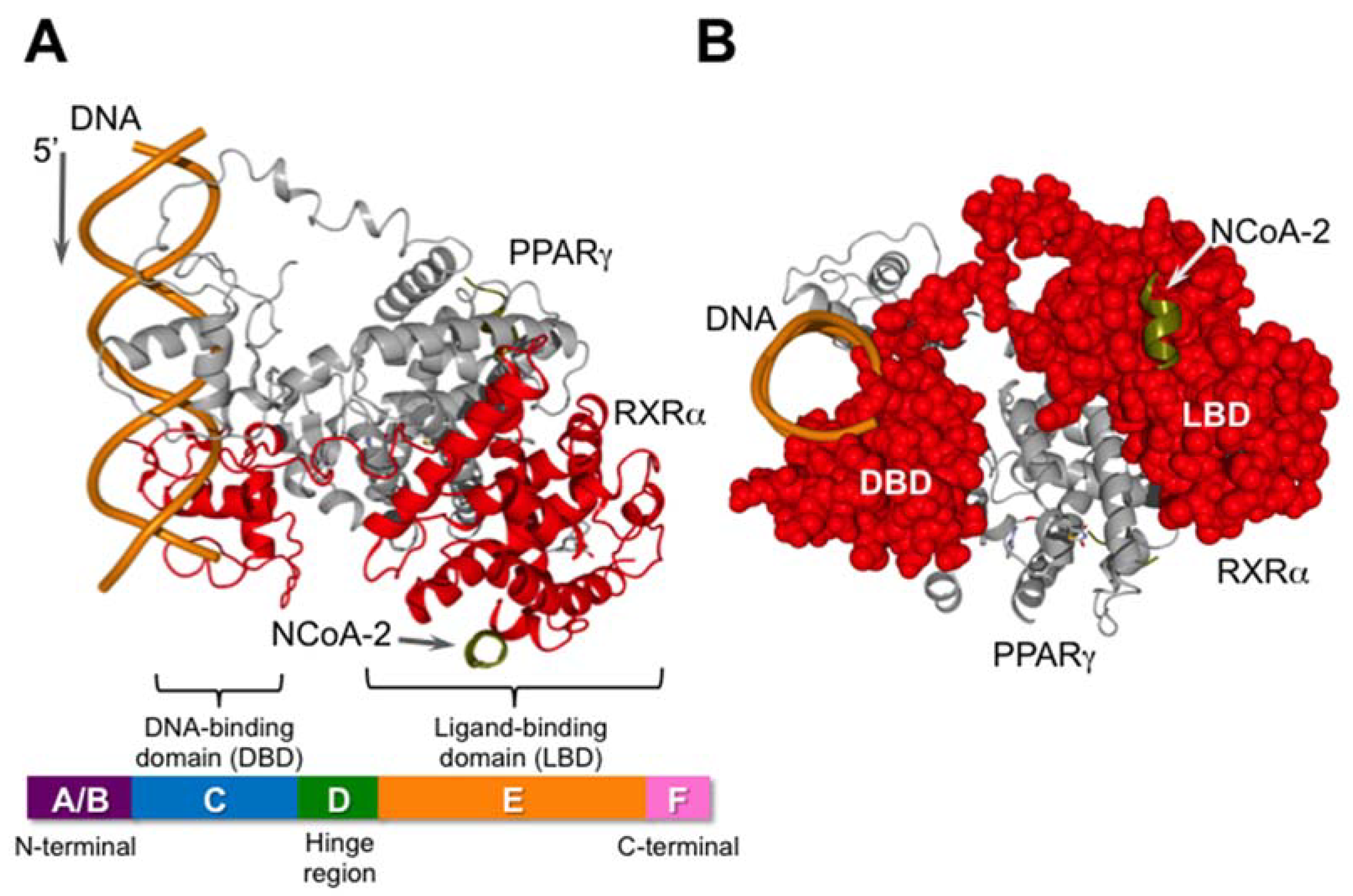
2. The RXRs
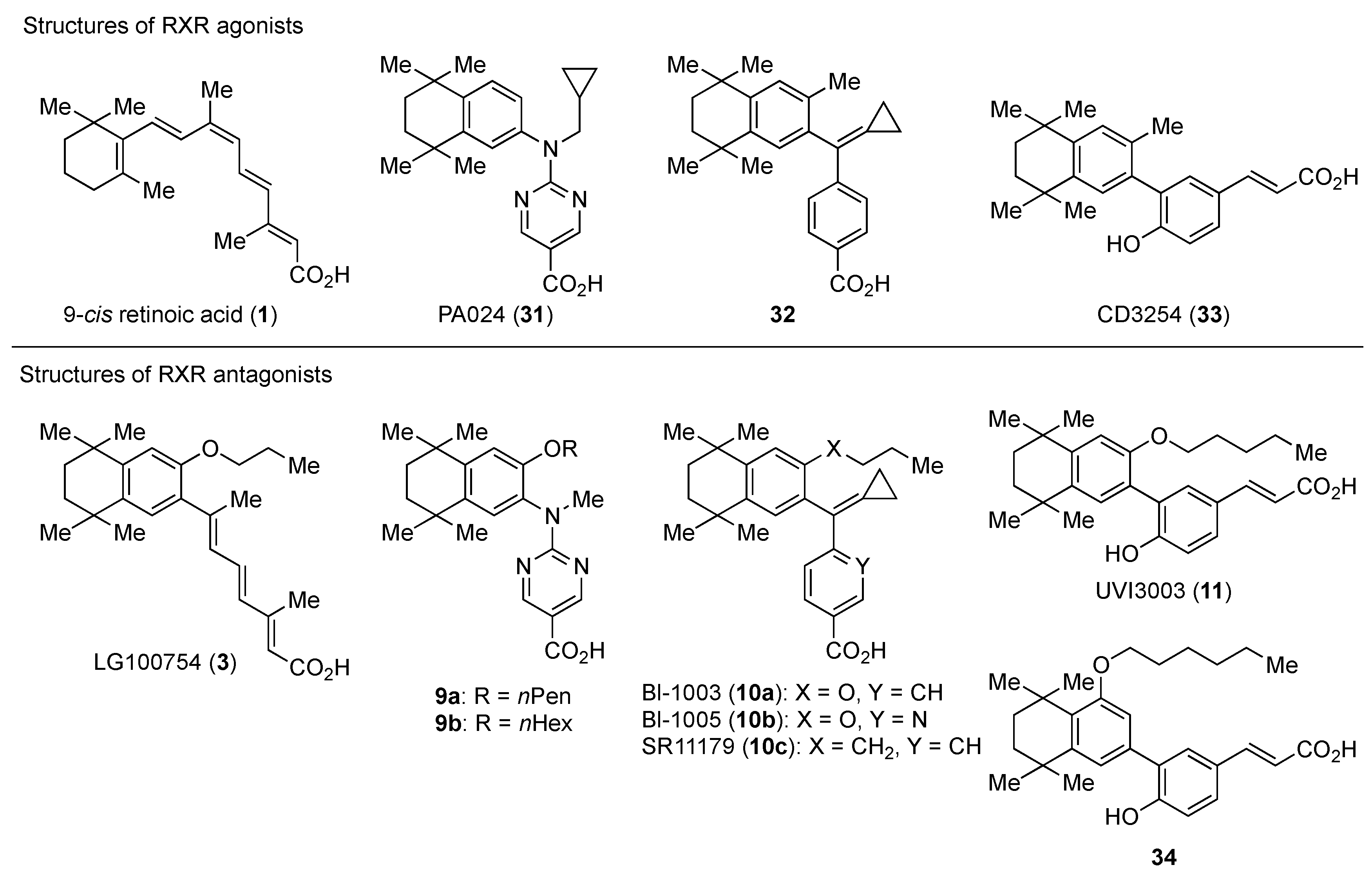
3. Representative RXR Antagonists
3.1. RXR Antagonists Having a Long-Chain Alkoxy Group
3.2. RXR Antagonists Possessing Another Side Group
3.3. RXR Antagonists Discovered among Natural Products or by Docking Simulation or High-Throughput Screening
4. Evaluation of RXR Antagonistic Activity
5. Latest Research on RXR Antagonists
6. Important Points in the Use of RXR Antagonists
7. Conclusions
Funding
Conflicts of Interest
Abbreviations
| i.p. | Intraperitoneal injection |
| NFκB | Nuclear factor-kappa B |
| NR | Nuclear receptor |
| Th2 | T helper type 2 |
| TNFα | Tumor necrosis factor alpha |
| TR | Thyroid hormone receptor |
| VDR | Vitamin D receptor |
| PXR | Pregnane X receptor |
References
- Mangelsdorf, D.J.; Thummel, C.; Beato, M.; Herrlich, P.; Schütz, G.; Umesono, K.; Blumberg, B.; Kastner, P.; Mark, M.; Chambon, P.; et al. The nuclear receptor superfamily: The second decade. Cell 1995, 83, 835–839. [Google Scholar] [CrossRef]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Germain, P.; Staels, B.; Dacquet, C.; Spedding, M.; Laudet, V. Overview of nomenclature of nuclear receptors. Pharmacol. Rev. 2006, 58, 685–704. [Google Scholar] [CrossRef] [PubMed]
- Forman, B.M.; Umesono, K.; Chen, J.; Evans, R.M. Unique response pathways are established by allosteric interactions among nuclear hormone receptors. Cell 1995, 81, 541–550. [Google Scholar] [CrossRef]
- Allenby, G.; Bocquel, M.T.; Saunders, M.; Kazmer, S.; Speck, J.; Rosenberger, M.; Lovey, A.; Kastner, P.; Grippo, J.F.; Chambon, P. Retinoic acid receptors and retinoid X receptors: Interactions with endogenous retinoic acids. Proc. Natl. Acad. Sci. USA 1993, 90, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Pileri, A.; Delfino, C.; Grandi, V.; Pimpinelli, N. Role of bexarotene in the treatment of cutaneous T-cell lymphoma: The clinical and immunological sides. Immunotherapy 2013, 5, 427–433. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, T.; Waki, H.; Kamon, J.; Murakami, K.; Motojima, K.; Komeda, K.; Miki, H.; Kubota, N.; Terauchi, Y.; Tsuchida, A.; et al. Inhibition of RXR and PPARγ ameliorates diet-induced obesity and type 2 diabetes. J. Clin. Investig. 2001, 108, 1001–1013. [Google Scholar] [CrossRef] [PubMed]
- Grenningloh, R.; Gho, A.; di Lucia, P.; Klaus, M.; Bollag, W.; Ho, I.C.; Sinigaglia, F.; Panina-Bordignon, P. Cutting Edge: Inhibition of the Retinoid X Receptor (RXR) Blocks T Helper 2 Differentiation and Prevents Allergic Lung Inflammation. J. Immunol. 2006, 176, 5161–5166. [Google Scholar] [CrossRef] [PubMed]
- Nuclear Receptor Nomenclature Committee. A unified nomenclature system for the nuclear receptor superfamily. Cell 1999, 97, 161–163. [Google Scholar] [CrossRef]
- Germain, P.; Chambon, P.; Eichele, G.; Evans, R.M.; Lazar, M.A.; Leid, M.; De Lera, A.R.; Lotan, R.; Mangelsdorf, D.J.; Gronemeyer, H. International Union of Pharmacology. LXIII. Retinoid X receptors. Pharmacol. Rev. 2006, 58, 760–772. [Google Scholar] [CrossRef] [PubMed]
- Bourguet, W.; Ruff, M.; Chambon, P.; Gronemeyer, H.; Moras, D. Crystal structure of the ligand-binding domain of the human nuclear receptor RXR-alpha. Nature 1995, 375, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Claessens, F.; Gewirth, D.T. DNA recognition by nuclear receptors. Essays Biochem. 2004, 40, 59–72. [Google Scholar] [CrossRef] [PubMed]
- Chandra, V.; Huang, P.; Hamuro, Y.; Raghuram, S.; Wang, Y.; Burris, T.P.; Rastinejad, F. Structure of the intact PPAR-gamma-RXR-nuclear receptor complex on DNA. Nature 2008, 456, 350–356. [Google Scholar] [CrossRef] [PubMed]
- Lou, X.; Toresson, G.; Benod, C.; Suh, J.H.; Philips, K.J.; Webb, P.; Gustafsson, J.A. Structure of the retinoid X receptor α-liver X receptor β (RXRα-LXRβ) heterodimer on DNA. Nat. Struct. Mol. Biol. 2014, 21, 277–281. [Google Scholar] [CrossRef] [PubMed]
- Osz, J.; McEwen, A.G.; Poussin-Courmontagne, P.; Moutier, E.; Birck, C.; Davidson, I.; Moras, D.; Rochel, N. Structural basis of natural promoter recognition by the retinoid X nuclear receptor. Sci. Rep. 2015, 5, 8216. [Google Scholar] [CrossRef] [PubMed]
- Kersten, S.; Kelleher, D.; Chambon, P.; Gronemeyer, H.; Noy, N. Retinoid X receptor α forms tetramers in solution. Proc. Natl. Acad. Sci. USA 1995, 92, 8645–8649. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, R.; Yeung, K.T.; Chung, R.H.; Gaczynska, M.E.; Osmulski, P.A.; Noy, N. DNA-looping by RXR tetramers permits transcriptional regulation “at a distance”. J. Mol. Biol. 2004, 343, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Yasmin, R.; Kannan-Thulasiraman, P.; Kagechika, H.; Dawson, M.I.; Noy, N. Inhibition of mammary carcinoma cell growth by RXR is mediated by the receptor’s oligomeric switch. J. Mol. Biol. 2010, 397, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, L.; Moutier, E.; Altobelli, G.; Legras, S.; Poch, O.; Choukrallah, M.A.; Bertin, I.; Jost, B.; Davidson, I. Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol. Cell. Biol. 2010, 30, 231–244. [Google Scholar] [CrossRef] [PubMed]
- IJpenberg, A.; Tan, N.S.; Gelman, L.; Kersten, S.; Seydoux, J.; Xu, J.; Metzger, D.; Canaple, L.; Chambon, P.; Wahli, W.; et al. In vivo activation of PPAR target genes by RXR homodimers. EMBO J. 2004, 23, 2083–2091. [Google Scholar] [CrossRef] [PubMed]
- Bourguet, W.; Germain, P.; Gronemeyer, H. Nuclear receptor ligand-binding domains: Three-dimensional structures, molecular interactions and pharmacological implications. Trends Pharmacol. Sci. 2000, 21, 381–388. [Google Scholar] [CrossRef]
- Roeder, R.G. The role of general initiation factors in transcription by RNA polymerase II. Trends Biochem. Sci. 1996, 21, 327–335. [Google Scholar] [CrossRef]
- Shang, Y.; Hu, X.; DiRenzo, J.; Lazar, M.A.; Brown, M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell 2000, 103, 843–852. [Google Scholar] [CrossRef]
- Hu, X.; Lazar, M.A. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature 1999, 402, 93–96. [Google Scholar] [CrossRef] [PubMed]
- DiRenzo, J.; Söderstrom, M.; Kurokawa, R.; Ogliastro, M.H.; Ricote, M.; Ingrey, S.; Hörlein, A.; Rosenfeld, M.G.; Glass, C.K. Peroxisome proliferator-activated receptors and retinoic acid receptors differentially control the interactions of retinoid X receptor heterodimers with ligands, coactivators, and corepressors. Mol. Cell. Biol. 1997, 17, 2166–2176. [Google Scholar] [CrossRef] [PubMed]
- Burris, T.P.; Solt, L.A.; Wang, Y.; Crumbley, C.; Banerjee, S.; Griffett, K.; Lundasen, T.; Hughes, T.; Kojetin, D.J. Nuclear receptors and their selective pharmacologic modulators. Pharmacol. Rev. 2013, 65, 710–778. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, K.; Takano, A.; Yakushiji, N.; Morohashi, K.; Morishita, K.; Matsuura, N.; Makishima, M.; Tai, A.; Sasaki, K.; Kakuta, H. The First Potent Subtype-Selective Retinoid X Receptor (RXR) Agonist Possessing a 3-Isopropoxy-4-isopropylphenylamino Moiety, NEt-3IP (RXRα/β-dual agonist). ChemMedChem 2008, 3, 780–787. [Google Scholar] [CrossRef] [PubMed]
- Takamatsu, K.; Takano, A.; Yakushiji, N.; Morishita, K.; Matsuura, N.; Makishima, M.; Ali, H.I.; Akaho, E.; Tai, A.; Sasaki, K.; et al. Reduction of lipophilicity at the lipophilic domain of RXR agonists enables production of subtype preference: RXRalpha-preferential agonist possessing a sulfonamide moiety. ChemMedChem 2008, 3, 454–460. [Google Scholar] [CrossRef] [PubMed]
- Morishita, K.; Yakushiji, N.; Ohsawa, F.; Takamatsu, K.; Matsuura, N.; Makishima, M.; Kawahata, M.; Yamaguchi, K.; Tai, A.; Sasaki, K.; et al. Replacing alkyl sulfonamide with aromatic sulfonamide in sulfonamide-type RXR agonists favors switch towards antagonist activity. Bioorg. Med. Chem. Lett. 2009, 19, 1001–1003. [Google Scholar] [CrossRef] [PubMed]
- Pérez, E.; Bourguet, W.; Gronemeyer, H.; de Lera, A.R. Modulation of RXR function through ligand design. Biochim. Biophys. Acta 2012, 1821, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Wijayaratne, A.L.; McDonnell, D.P. The human estrogen receptor-alpha is a ubiquitinated protein whose stability is affected differentially by agonists, antagonists, and selective estrogen receptor modulators. J. Biol. Chem. 2001, 276, 35684–35692. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T.; Menéndez-Gutiérrez, M.P.; Cedenilla, M.; Ricote, M. Retinoid X receptors in macrophage biology. Trends Endocrinol. Metab. 2013, 24, 460–468. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Ricote, M. Nuclear receptor signaling in macrophages. Biochem. Pharmacol. 2004, 67, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Széles, L.; Póliska, S.; Nagy, G.; Szatmari, I.; Szanto, A.; Pap, A.; Lindstedt, M.; Santegoets, S.J.; Rühl, R.; Dezsö, B.; et al. Research resource: Transcriptome profiling of genes regulated by RXR and its permissive and nonpermissive partners in differentiating monocyte-derived dendritic cells. Mol. Endocrinol. 2010, 24, 2218–2231. [Google Scholar] [CrossRef] [PubMed]
- Nagy, L.; Szanto, A.; Szatmari, I.; Széles, L. Nuclear hormone receptors enable macrophages and dendritic cells to sense their lipid environment and shape their immune response. Physiol. Rev. 2012, 92, 739–789. [Google Scholar] [CrossRef] [PubMed]
- Saijo, K.; Crotti, A.; Glass, C.K. Regulation of microglia activation and deactivation by nuclear receptors. Glia 2013, 61, 104–111. [Google Scholar] [CrossRef] [PubMed]
- Wallace, K.; Cowie, D.E.; Konstantinou, D.K.; Hill, S.J.; Tjelle, T.E.; Axon, A.; Koruth, M.; White, S.A.; Carlsen, H.; Mann, D.A.; et al. The PXR is a drug target for chronic inflammatory liver disease. J. Steroid Biochem. Mol. Biol. 2010, 120, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Vavassori, P.; Mencarelli, A.; Renga, B.; Distrutti, E.; Fiorucci, S. The bile acid receptor FXR is a modulator of intestinal innate immunity. J. Immunol. 2009, 183, 6251–6261. [Google Scholar] [CrossRef] [PubMed]
- Lefebvre, P.; Chinetti, G.; Staels, B. Nur77turing macrophages in atherosclerosis. Circ. Res. 2012, 110, 375–377. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.C.; Soto, E.; Hong, C.; Ito, A.; Pei, L.; Chawla, A.; Conneely, O.M.; Tangirala, R.K.; Evans, R.M.; Tontonoz, P. Bone marrow NR4A expression is not a dominant factor in the development of atherosclerosis or macrophage polarization in mice. J. Lipid Res. 2013, 54, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Hanna, R.N.; Shaked, I.; Hubbeling, H.G.; Punt, J.A.; Wu, R.; Herrley, E.; Zaugg, C.; Pei, H.; Geissmann, F.; Ley, K.; et al. NR4A1 (Nur77) deletion polarizes macrophages toward an inflammatory phenotype and increases atherosclerosis. Circ. Res. 2012, 110, 416–427. [Google Scholar] [CrossRef] [PubMed]
- Canan Koch, S.S.; Dardashti, L.J.; Hebert, J.J.; White, S.K.; Croston, G.E.; Flatten, K.S.; Heyman, R.A.; Nadzan, A.M. Identification of the First Retinoid X Receptor Homodimer Antagonist. J. Med. Chem. 1996, 39, 3229–3234. [Google Scholar] [CrossRef] [PubMed]
- WO2001019770 RXR Modulators with Improved Pharmacologic Profile. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2001019770 (accessed on 24 May 2018).
- WO2000053562 Retinoid Antagonists and Use Thereof. Available online: https://patentscope.wipo.int/search/en/detail.jsf?docId=WO2000053562 (accessed on 24 May 2018).
- Michellys, P.Y.; Ardecky, R.J.; Chen, J.H.; Crombie, D.L.; Etgen, G.J.; Faul, M.M.; Falkner, A.L.; Grese, T.A.; Heyman, R.A.; Karanewsky, D.S.; et al. Novel (2E,4E,6Z)-7-(2-Alkoxy-3,5-dialkylbenzene)-3-methylocta-2,4,6-trienoic Acid Retinoid X Receptor Modulators Are Active in Models of Type 2 Diabetes. J. Med. Chem. 2003, 46, 2683–2696. [Google Scholar] [CrossRef] [PubMed]
- Michellys, P.Y.; Ardecky, R.J.; Chen, J.H.; D’Arrigo, J.; Grese, T.A.; Karanewsky, D.S.; Leibowitz, M.D.; Liu, S.; Mais, D.A.; Mapes, C.M.; et al. Design, Synthesis, and Structure-Activity Relationship Studies of Novel 6,7-Locked-[7-(2-alkoxy-3,5-dialkylbenzene)-3-methylocta]-2,4,6-trienoic Acids. J. Med. Chem. 2003, 46, 4087–4103. [Google Scholar] [CrossRef] [PubMed]
- Michellys, P.Y.; D’Arrigo, J.; Grese, T.A.; Karanewsky, D.S.; Leibowitz, M.D.; Mais, D.A.; Mapes, C.M.; Reifel-Miller, A.; Rungta, D.; Boehm, M.F. Design, synthesis and structure–activity relationship of novel RXR-selective modulators. Bioorg. Med. Chem. Lett. 2004, 14, 1593–1598. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, B.; Ohta, K.; Kawachi, E.; Fukasawa, H.; Hashimoto, Y.; Kagechika, H. Novel Retinoid X Receptor Antagonists: Specific Inhibition of Retinoid Synergism in RXR-RAR Heterodimer Actions. J. Med. Chem. 2002, 45, 3327–3330. [Google Scholar] [CrossRef] [PubMed]
- Fujii, S.; Ohsawa, F.; Yamada, S.; Shinozaki, R.; Fukai, R.; Makishima, M.; Enomoto, S.; Tai, A.; Kakuta, H. Modification at the acidic domain of RXR agonists has little effect on permissive RXR-heterodimer activation. Bioorg. Med. Chem. Lett. 2010, 20, 5139–5142. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, M.; Yamada, S.; Ohsawa, F.; Ohta, Y.; Kawata, K.; Makishima, M.; Kakuta, H. Discovery of a Potent Retinoid X Receptor Antagonist Structurally Closely Related to RXR Agonist NEt-3IB. ACS Med. Chem. Lett. 2011, 2, 896–900. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Liu, G.; James, S.Y.; Hobbs, P.D.; Peterson, V.J.; Bhattacharya, A.A.; Kolluri, S.K.; Zhang, X.K.; Leid, M.; Abagyan, R.; et al. Determinants of Retinoid X Receptor Transcriptional Antagonism. J. Med. Chem. 2004, 47, 4360–4372. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Dawson, M.I.; Hu, Q.Y.; Xia, Z.; Dambacher, J.D.; Ye, M.; Zhang, X.K.; Li, E. The effect of antagonists on the conformational exchange of the retinoid X receptor alpha ligand-binding domain. Magn. Reson. Chem. 2009, 47, 1071–1080. [Google Scholar] [CrossRef] [PubMed]
- Vuligonda, V.; Thacher, S.M.; Chandraratna, R.A. Enantioselective Syntheses of Potent Retinoid X Receptor Ligands: Differential Biological Activities of Individual Antipodes. J. Med. Chem. 2001, 44, 2298–2303. [Google Scholar] [CrossRef] [PubMed]
- Nahoum, V.; Pérez, E.; Germain, P.; Rodríguez-Barrios, F.; Manzo, F.; Kammerer, S.; Lemaire, G.; Hirsch, O.; Royer, C.A.; Gronemeyer, H.; et al. Modulators of the structural dynamics of the retinoid X receptor to reveal receptor function. Proc. Natl. Acad. Sci. USA 2007, 104, 17323–17328. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Janesick, A.; Wu, L.; Hu, L.; Tang, W.; Blumberg, B.; Shi, H. The unexpected teratogenicity of RXR antagonist UVI3003 via activation of PPARγ in Xenopus tropicalis. Toxicol. Appl. Pharmacol. 2017, 314, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Ebisawa, M.; Umemiya, H.; Ohta, K.; Fukasawa, H.; Kawachi, E.; Christoffel, G.; Gronemeyer, H.; Tsuji, M.; Hashimoto, Y.; Shudo, K.; et al. Retinoid X receptor-antagonistic diazepinylbenzoic acids. Chem. Pharm. Bull. 1999, 47, 1778–1786. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, J.; Konishi, K.; Kishida, M.; Gunji, H.; Kanazawa, T.; Uchiyama, H.; Fukaya, H.; Mitani, H.; Kimura, M. Synthesis and structure-activity relationship of RXR antagonists based on the diazepinylbenzoic acid structure. Bioorg. Med. Chem. Lett. 2007, 17, 4808–4811. [Google Scholar] [CrossRef] [PubMed]
- Sakaki, J.; Kishida, M.; Konishi, K.; Gunji, H.; Toyao, A.; Matsumoto, Y.; Kanazawa, T.; Uchiyama, H.; Fukaya, H.; Mitani, H.; et al. Synthesis and structure–activity relationship of novel RXR antagonists: Orally active anti-diabetic and anti-obesity agents. Bioorg. Med. Chem. Lett. 2007, 17, 4804–4807. [Google Scholar] [CrossRef] [PubMed]
- Ohta, K.; Iijima, T.; Kawachi, E.; Kagechika, H.; Endo, Y. Novel retinoid X receptor (RXR) antagonists having a dicarba-closo-dodecaborane as a hydrophobic moiety. Bioorg. Med. Chem. Lett. 2004, 14, 5913–5918. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhou, R.; Li, L.; Chen, J.; Chen, L.; Li, C.; Ding, H.; Yu, L.; Hu, L.; Jiang, H.; et al. Danthron Functions as a Retinoic X Receptor Antagonist by Stabilizing Tetramers of the Receptor. J. Biol. Chem. 2011, 286, 1868–1875. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chen, L.; Chen, J.; Jiang, H.; Shen, X. Structural Basis for Retinoic X Receptor Repression on the Tetramer. J. Biol. Chem. 2011, 286, 24593–24598. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Narayanasamy, S.; Curley, R.W., Jr.; Harrison, E.H. β-Apo-13-carotenone regulates retinoid X receptor transcriptional activity through tetramerization of the receptor. J. Biol. Chem. 2014, 289, 33118–33124. [Google Scholar] [CrossRef] [PubMed]
- Kolluri, S.K.; Corr, M.; James, S.Y.; Bernasconi, M.; Lu, D.; Liu, W.; Cottam, H.B.; Leoni, L.M.; Carson, D.A.; Zhang, X.K. The R-enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid X receptor and induces tumor-selective apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Liu, W.; Su, Y.; Wei, Z.; Liu, J.; Kolluri, S.K.; Wu, H.; Cao, Y.; Chen, J.; Wu, Y.; et al. NSAID Sulindac and Its Analogs Bind RXRα and Inhibit RXRα-dependent AKT Signaling. Cancer Cell 2010, 17, 560–573. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.G.; Chen, L.; Chen, J.; Zheng, J.F.; Gao, W.; Zeng, Z.; Zhou, H.; Zhang, X.K.; Huang, P.Q.; Su, Y. Synthesis and SAR study of modulators inhibiting tRXRα-dependent AKT activation. Eur. J. Med. Chem. 2013, 62, 632–648. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, Z.G.; Aleshin, A.E.; Chen, F.; Chen, J.; Jiang, F.; Alitongbieke, G.; Zeng, Z.; Ma, Y.; Huang, M.; et al. Sulindac-derived RXRα modulators inhibit cancer cell growth by binding to a novel site of RXRα. Chem. Biol. 2014, 21, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Liu, J.; Liu, J.; Zhang, C.; Jiang, F.; Wu, H.; Chen, L.; Zeng, W.; Cao, X.; Yan, T.; et al. Antagonist effect of triptolide on AKT activation by truncated retinoid X receptor-alpha. PLoS ONE 2012, 7, e35722. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.Y.; Zeng, W.J.; Liu, J.; Wu, Y.L.; Ma, Y.; Zeng, Z.; Pang, J.Y.; Zhang, X.K.; Yan, X.; Wong, A.S.T.; et al. TRC4, an improved triptolide derivative, specifically targets to truncated form of retinoid X receptor-alpha in cancer cells. Biochem. Pharmacol. 2017, 124, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Chen, J.; Lin, J.; Cheltsov, A.V.; Xu, L.; Chen, Y.; Zeng, Z.; Chen, L.; Huang, M.; Hu, M.; et al. NSC-640358 acts as RXRα ligand to promote TNFα-mediated apoptosis of cancer cell. Protein Cell 2015, 6, 654–666. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Sun, Z.; Huang, M.; Zhang, W.; Liu, J.; Chen, L.; Chen, F.; Zhou, Y.; Lin, J.; Huang, F.; et al. Nitrostyrene Derivatives Act as RXRα Ligands to Inhibit TNFα Activation of NF-κB. Cancer Res. 2015, 75, 2049–2060. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Liu, J.; Huang, M.; Hu, M.; Su, Y.; Zhang, X.K. Identification of a New RXRα Antagonist Targeting the Coregulator-Binding Site. ACS Med. Chem. Lett. 2014, 5, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Xu, D.; Cai, L.; Guo, S.; Xie, L.; Yin, M.; Chen, Z.; Zhou, H.; Su, Y.; Zeng, Z.; Zhang, X. Virtual screening and experimental validation identify novel modulators of nuclear receptor RXRα from Drugbank database. Bioorg. Med. Chem. Lett. 2017, 27, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Boehm, M.F.; Zhang, L.; Badea, B.A.; White, S.K.; Mais, D.E.; Berger, E.; Suto, C.M.; Goldman, M.E.; Heyman, R.A. Synthesis and Structure-Activity Relationships of Novel Retinoid XReceptor-Selective Retinoids. J. Med. Chem. 1994, 37, 2930–2941. [Google Scholar] [CrossRef] [PubMed]
- Schulman, I.G.; Li, C.; Schwabe, J.W.; Evans, R.M. The phantom ligand effect: Allosteric control of transcription by the retinoid X receptor. Genes Dev. 1997, 11, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Lala, D.S.; Mukherjee, R.; Schulman, I.G.; Koch, S.S.; Dardashti, L.J.; Nadzan, A.M.; Croston, G.E.; Evans, R.M.; Heyman, R.A. Activation of specific RXR heterodimers by an antagonist of RXR homodimers. Nature 1996, 383, 450–453. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, R.; Jow, L.; Croston, G.E.; Paterniti, J.R., Jr. Identification, characterization, and tissue distribution of human peroxisome proliferator-activated receptor (PPAR) isoforms PPARgamma2 versus PPARgamma1 and activation with retinoid X receptor agonists and antagonists. J. Biol. Chem. 1997, 272, 8071–8076. [Google Scholar] [CrossRef] [PubMed]
- Cesario, R.M.; Klausing, K.; Razzaghi, H.; Crombie, D.; Rungta, D.; Heyman, R.A.; Lala, D.S. The Rexinoid LG100754 Is a Novel RXR:PPARγ Agonist and Decreases Glucose Levels in Vivo. Mol. Endocrinol. 2001, 15, 1360–1369. [Google Scholar] [PubMed]
- Sato, Y.; Ramalanjaona, N.; Huet, T.; Potier, N.; Osz, J.; Antony, P.; Peluso-Iltis, C.; Poussin-Courmontagne, P.; Ennifar, E.; Mély, Y.; et al. The “Phantom Effect” of the Rexinoid LG100754: Structural and functional insights. PLoS ONE 2010, 5, e15119. [Google Scholar] [CrossRef] [PubMed]
- Dawson, M.I.; Hobbs, P.D.; Jong, L.; Xiao, D.; Chao, W.R.; Pan, C.; Zhang, X.K. sp2-Bridged Diaryl Retinoids: Efects of Bridge-Region Substitution on Retinoid X Receptor (RXR) Selectivity. Bioorg. Med. Chem. Lett. 2000, 10, 1307–1310. [Google Scholar] [CrossRef]
- Pérez Santín, E.; Germain, P.; Quillard, F.; Khanwalkar, H.; Rodríguez-Barrios, F.; Gronemeyer, H.; de Lera, A.R.; Bourguet, W. Modulating Retinoid X Receptor with a Series of (E)-3-[4-Hydroxy-3-(3-alkoxy-5,5,8,8-tetramethyl-5,6,7,8-tetrahydronaphthalen-2-yl)phenyl]acrylic Acids and Their 4-Alkoxy Isomers. J. Med. Chem. 2009, 52, 3150–3158. [Google Scholar] [CrossRef] [PubMed]
- Yotsumoto, T.; Naitoh, T.; Kanaki, T.; Tsuruzoe, N. A retinoid X receptor antagonist, HX531, improves leptin resistance without increasing plasma leptin level in KK-Ay mice under normal dietary conditions. Metabolism 2005, 54, 573–578. [Google Scholar] [CrossRef] [PubMed]
- Kawata, K.; Morishita, K.; Nakayama, M.; Yamada, S.; Kobayashi, T.; Furusawa, Y.; Arimoto-Kobayashi, S.; Oohashi, T.; Makishima, M.; Naitou, H.; et al. RXR Partial Agonist Produced by Side Chain Repositioning of Alkoxy RXR Full Agonist Retains Antitype 2 Diabetes Activity without the Adverse Effects. J. Med. Chem. 2015, 58, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Lazareno, S.; Birdsall, N.J. Estimation of competitive antagonist affinity from functional inhibition curves using the Gaddum, Schild and Cheng-Prusoff equations. Br. J. Pharmacol. 1993, 109, 1110–1119. [Google Scholar] [CrossRef] [PubMed]
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Ayala-Peña, V.B.; Pilotti, F.; Volonté, Y.; Rotstein, N.P.; Politi, L.E.; German, O.L. Protective effects of retinoid x receptors on retina pigment epithelium cells. Biochim. Biophys. Acta 2016, 1863, 1134–1145. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Carpentier, A.; Cheng, X.; Block, P.D.; Zhao, Y.; Zhang, Z.; Protzer, U.; Liang, T.J. Human stem cell-derived hepatocytes as a model for hepatitis B virus infection, spreading and virus-host interactions. J. Hepatol. 2017, 66, 494–503. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Xu, T.; Li, D.; Zhou, J. A representative retinoid X receptor antagonist UVI3003 induced teratogenesis in zebrafish embryos. J. Appl. Toxicol. 2015, 35, 280–286. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Yu, J.; Shi, H.; Xia, L.; Xin, Q.; Zhang, Q.; Zhao, H.; Luo, J.; Jin, W.; Li, D.; Zhou, J. Quantitative toxicoproteomic analysis of zebrafish embryos exposed to a retinoid X receptor antagonist UVI3003. J. Appl. Toxicol. 2015, 35, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Shi, H.; Zhu, P.; Hu, L.; Wu, L.; Yang, Y.; Rotchell, J.M. Effects of antagonist of retinoid X receptor (UVI3003) on morphology and gene profile of Xenopus tropicalis embryos. Environ. Toxicol. Pharmacol. 2014, 38, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Mengeling, B.J.; Goodson, M.L.; Furlow, J.D. RXR Ligands Modulate Thyroid Hormone Signaling Competence in Young Xenopus laevis Tadpoles. Endocrinology 2018, 159, 2576–2595. [Google Scholar] [CrossRef] [PubMed]
- Mihály, J.; Gericke, J.; Lucas, R.; de Lera, A.R.; Alvarez, S.; Törőcsik, D.; Rühl, R. TSLP expression in the skin is mediated via RARγ-RXR pathways. Immunobiology 2016, 221, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Shan, P.R.; Xu, W.W.; Huang, Z.Q.; Pu, J.; Huang, W.J. Protective role of retinoid X receptor in H9c2 cardiomyocytes from hypoxia/reoxygenation injury in rats. World J. Emerg. Med. 2014, 5, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Natrajan, M.S.; de la Fuente, A.G.; Crawford, A.H.; Linehan, E.; Nuñez, V.; Johnson, K.R.; Wu, T.; Fitzgerald, D.C.; Ricote, M.; Bielekova, B.; et al. Retinoid X receptor activation reverses age-related deficiencies in myelin debris phagocytosis and remyelination. Brain 2015, 138, 3581–3597. [Google Scholar] [CrossRef] [PubMed]
- Litwa, E.; Rzemieniec, J.; Wnuk, A.; Lason, W.; Krzeptowski, W.; Kajta, M. Apoptotic and neurotoxic actions of 4-para-nonylphenol are accompanied by activation of retinoid X receptor and impairment of classical estrogen receptor signaling. J. Steroid Biochem. Mol. Biol. 2014, 144, 334–347. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.J.; Hopwood, T.; Gallagher, A.L.; Partridge, F.A.; Burgis, T.; Sattelle, D.B.; Else, K.J. An antagonist of the retinoid X receptor reduces the viability of Trichuris muris in vitro. BMC Infect. Dis. 2014, 14, 520. [Google Scholar] [CrossRef] [PubMed]
- Unsworth, A.J.; Flora, G.D.; Sasikumar, P.; Bye, A.P.; Sage, T.; Kriek, N.; Crescente, M.; Gibbins, J.M. RXR Ligands Negatively Regulate Thrombosis and Hemostasis. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 812–822. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, A.; Rzemieniec, J.; Litwa, E.; Lasoń, W.; Krzeptowski, W.; Wójtowicz, A.K.; Kajta, M. The Crucial Involvement of Retinoid X Receptors in DDE Neurotoxicity. Neurotox. Res. 2016, 29, 155–172. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.H.; Watt, J.; Huang, C.K.; Gerstenfeld, L.C.; Schlezinger, J.J. Tributyltin engages multiple nuclear receptor pathways and suppresses osteogenesis in bone marrow multipotent stromal cells. Chem. Res. Toxicol. 2015, 28, 1156–1166. [Google Scholar] [CrossRef] [PubMed]
- Urushitani, H.; Katsu, Y.; Kagechika, H.; Sousa, A.C.A.; Barroso, C.M.; Ohta, Y.; Shiraishi, H.; Iguchi, T.; Horiguchi, T. Characterization and comparison of transcriptional activities of the retinoid X receptors by various organotin compounds in three prosobranch gastropods; Thais clavigera, Nucella lapillus and Babylonia japonica. Aquat. Toxicol. 2018, 199, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.H.; Jiang, F.Q.; Duan, Y.H.; Zeng, Z.P.; Chen, F.; Dai, Y.; Chen, J.B.; Liu, J.X.; Liu, J.; Zhou, H.; et al. Targeting Truncated Retinoid X Receptor-α by CF31 Induces TNF-α-Dependent Apoptosis. Cancer Res. 2013, 73, 307–318. [Google Scholar] [CrossRef] [PubMed]
- Gao, W.; Liu, J.; Hu, M.; Huang, M.; Cai, S.; Zeng, Z.; Lin, B.; Cao, X.; Chen, J.; Zeng, J.Z.; et al. Regulation of proteolytic cleavage of retinoid X receptor-α by GSK-3β. Carcinogenesis 2013, 34, 1208–1215. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.K.; Su, Y.; Chen, L.; Chen, F.; Liu, J.; Zhou, H. Regulation of the nongenomic actions of retinoid X receptor-α by targeting the coregulator-binding sites. Acta Pharmacol. Sin. 2015, 36, 102–112. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ma, F.; Hu, X.; Jin, T.; Xiong, C.; Teng, X. Elevated COX2 expression and PGE2 production by downregulation of RXRα in senescent macrophages. Biochem. Biophys. Res. Commun. 2013, 440, 157–162. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Term | Definition/Description/Examples |
|---|---|
| AF-1 | Activation function-1. AF-1 consists of the N-terminal region (domains A/B), which can operate autonomously. This region can interact with cofactors such as co-activators or other transcription factors. The activation is independent of ligand binding. The activity of AF-1 is regulated by growth factors acting through the MAP kinase pathway. |
| AF-2 | Activation function-2. AF-2 is the C-terminal helix 12, located in domain E, which mediates ligand-dependent transactivation. |
| DBD | DNA-binding domain. Domain C of nuclear receptors. This region binds to a specific DNA sequence, called the hormone response element (HRE). |
| LBD | Ligand-binding domain. Domain E of nuclear receptors. The LBD contains (1) a dimerization surface, which mediates interaction with partner LBDs; (2) the LBP; (3) a co-regulator binding surface, and 4) an activation function helix, termed AF-2. |
| LBP | Ligand-binding pocket (LBP), which interacts with small molecules. The LBP is generally located behind helix 3 and in front of helices 7 and 10, and is lined with mostly hydrophobic amino acids. |
| Ligands for NRs | Compounds that bind reversibly to NRs at the C-terminal LBP. |
| Agonists | Ligands that induce an active conformation of the receptor |
| Inverse agonists | Ligands that can promote co-repressor recruitment. |
| Antagonists | Ligands that produce a conformation and an action of the receptor distinct from that produced by an agonist. |
| Partial agonists | Agonists that in a given tissue, under specific conditions, cannot elicit as large an effect (even when applied at high concentration, so that all the receptors should be occupied), as can another agonist acting through the same receptors in the same tissue |
| NR modulators | Compounds that bind to NRs, which include ligands, SNuRMs, and SNuRDs [26]. |
| Orthosteric modulators | Compounds that bind to the same site of endogenous ligands. |
| Allosteric modulators | Compounds that bind to the different site of endogenous ligands. The term “allo-” means “other”. |
| Positive allosteric modulators (PAMs) | Allosteric modulators that induce an amplification of the effect of the primary ligand. |
| Negative allosteric modulators (NAMs) | Allosteric modulators that reduce the effect of the primary ligand. |
| Silent allosteric modulators (SAMs) | Allosteric modulators that occupy the allosteric binding site and behave functionally neutral; also called neutral or null modulators. |
| SNuRMs | Selective nuclear receptor modulators. Selective ligands with partial function-, cell-, and/or promoter-specific action. |
| SNuRDs | Selective nuclear receptor Down-regulators. Compounds that cause NR to be degraded and thus down-regulated. A subclass of antagonists. Fluvestrant is a selective estrogen receptor down-regulator (SERD) [31]. |
| Selective agonists and antagonists | Ligands with an affinity difference (preferably greater than 100-fold) between their primary target and other receptors. |
| Name | Subtypes | Nomenclature | Sequence | References |
|---|---|---|---|---|
| TR | α | NR1A1 | DR4 | [33] |
| β | NR1A2 | [33] | ||
| RAR | α | NR1B1 | DR2, DR5 | [33,34,35] |
| β | NR1B2 | [33] | ||
| γ | NR1B3 | [33,36] | ||
| PPAR | α | NR1C1 | DR1 | [33] |
| β/δ | NR1C2 | [33,34,35] | ||
| γ | NR1C3 | [33,34,35,36] | ||
| LXR | α | NR1H1 | DR4 | [33,34,35,36] |
| β | NR1H2 | [33,34,35,36] | ||
| VDR | NR1I1 | DR3 | [33,34,35,36] | |
| PXR | NR1I2 | DR3–5 | [37] | |
| LXR | α | NR1H1 | DR4 | [33,34,35,36] |
| FXR | NR1H4 | IR1 * | [38] | |
| RXR | α | NR2B1 | DR1 | [33,34,35,36] |
| β | NR2B2 | [33,34,35,36] | ||
| γ | NR2B3 | [33,34,35,36] | ||
| Nur77 | NR4A1 | DR5 | [36,39,40,41] | |
| Nurr1 | NR4A2 | DR5 | [36,40] |
| Compounds | Structures | Binding | Transactivity (RXRα) | Ref. |
|---|---|---|---|---|
| LG100754 (3) |  | Ki = 3 nM (RXRα, [3H]1) Ki = 8 nM (RXRα, [3H]2) | IC50 = 16 nM (vs. 32 nM 2, CV-1 cells) | [42] |
| AGN195393 (4) |  | N.D. | N.D. | [43] |
| Ro26-5405 (5) |  | Ki = 0.9 nM (RXRα, [3H]2) | N.D. | [43,44] |
| LG101506 (6) |  | Ki = 3 nM (RXRα, [3H]1) Ki = 3 nM (RXRα, [3H]2) | IC50 = 8 nM (CV-1 cells) | [43,45] |
| 7 |  | Ki = 9.9 nM (RXRα, [3H]1) | IC50 = 10.3 nM (CV-1 cells) | [46] |
| 8 |  | Ki = 3 nM (RXRα, [3H]1) | IC50 = 8 nM (CV-1 cells) | [47] |
| PA451 (9a) R = n-Pen |  | N.D. | N.D. | [48] |
| PA452 (9b) R = n-Hex | N.D. | pA2 = 7.11 (vs. NEt-TMN: EC50 = 5.28 nM [49], COS-1 cell) | [48,50] | |
| Bl-1003 (10a) X = O, Y = CH |  | Kd = 26 nM (RXRα-LBD, fluorescence titration) IC50 = 46 nM (RXRα-LBD, [3H]1) | IC50 = 1100 nM (vs. 1 @ 0.1 μM, CV-1 cells) | [51,52] |
| Bl-1005 (10b) X = O, Y = N | Kd = 329 nM (RXRα-LBD, fluorescence titration) IC50 = 1200 nM (RXRα-LBD, [3H]1) | IC50 ≥ 10,000 nM (vs. 1 @ 0.1 μM, CV-1 cells) | [51,52] | |
| SR11179 (10c) C = CH2, Y = CH | Kd = 15 nM (RXRα-LBD, fluorescence titration) IC50 = 450 nM (RXRα-LBD, [3H]1) | IC50 = 67 nM (vs. 1 @ 0.1 μM, CV-1 cells) | [51,52] | |
| UVI3003 (11) |  | N.D. | IC50 = 0.24 μM (vs. IRX4204: EC50 = 0.2 nM [53] @ 10 nM, COS-7 cells) | [54,55] |
| Compounds | Structures | Binding | Transactivity (RXRα) | Ref. |
|---|---|---|---|---|
| HX531 (12) |  | N.D. | IC50 = 1.0 μM (vs. IRX4204: EC50 = 0.2 nM [53] @ 10 nM, COS-7 cells) | [55,56] |
| 13a R1 = Et, R2 = NHSO2-(3-CF3)Ph, X = H |  | N.D. | IC50 = 0.095 μM (vs. 1 @ 20 nM, HEK-293 cells) | [57] |
| 13b R1 = n-Pr, R2 = NHSO2-(3-CF3)Ph, X = H | N.D. | IC50 = 0.076 μM (vs. 1 @ 20 nM, HEK-293 cells) | [57] | |
| 13c R1 = Et, R2 = CN, X = F | N.D. | IC50 = 0.50 μM (vs. 1, HEK-293 cells) | [58] | |
| 14 |  | N.D. | N.D. | [59] |
| 15a X = Cl |  | N.D. | IC50 = 4.1 μM (vs. 2 @ 10 nM, COS-1 cells) | [29] |
| 15b X = CF3 | N.D. | IC50 = 3.2 μM (vs. 2 @ 10 nM, COS-1 cells) | [29] | |
| 16 |  | N.D. | pA2 = 8.23 (vs. NEt-TMN: EC50 = 5.28 nM [49], COS-1 cells) | [50] |
| Compounds | Structures | Binding | Transactivity (RXRα) | Ref. |
|---|---|---|---|---|
| Danthron (17a) R = H |  | Kd = 6.2 μM (RXRα-LBD, SPR) Kd = 7.5 μM (RXRα-LBD, ITC) | IC50 = 0.11 μM (vs. 1 @ 0.1 μM, HEK-293T cells) | [60] |
| Rhein (17b) R = CO2H | N.D. * | IC50 = 0.75 μM (vs. 1 @ 0.1 μM, HEK-293T cells) | [61] | |
| β-Apo-13-carotenone (18) |  | N.D. | IC50 value is not described (vs. 1 @ 0.01~1000 nM, COS-7 cells) | [62] |
| R-Etodolac (19) |  | IC50 ≈ 200 μM (RXRα-LBD, [3H]1) | N.D. | [63] |
| Sulindac sulfide (20) |  | IC50 = 80 μM (RXRα-LBD, [3H]1) | N.D. | [64] |
| K-80003 (21a) X = F, R = CO2H |  | IC50 = 2.4 μM (RXRα-LBD, [3H]1) | N.D. | [64,65] |
| K-8008 (21b) X = H R =  | IC50 = 16.8 μM TR-FRET, GST-RXRα-LBD, 1 @ 10 nM) | IC50 = 13.2 μM (vs. 1 @ 100 nM, HCT-116 cells) | [65,66] | |
| Triptolide (22a) R = H |  | N.D. | N.D. | [67] |
| TRC4 (22b) R =  | N.D. | N.D. | [68] | |
| NSC-640358 (23) |  | Ki = 15.7 μM (RXRα-LBD, [3H]1) | N.D. | [69] |
| 24 |  | Ki = 0.28 μM (RXRα-LBD, [3H]1) | N.D. | [70] |
| 25 |  | Ki = 0.81 μM (RXRα-LBD, [3H]1) | N.D. | [70] |
| 26 |  | N.D. | IC50 = 2 μM (vs. 1 @ 0.1 μM, HEK-293T cells) | [71] |
| 27 |  | Kd = 488 nM (RXRα-LBD, SPR) | IC50 = 2.45 μM (vs. 1 @ 0.1 μM, HEK-293T cells) | [71] |
| Fluvastatin (28) |  | Kd = 11.04 μM (RXRα-LBD, SPR) | IC50 value is not described. (vs. 1 @ 100 nM, MCF-7 cells) | [72] |
| Pitavastatin (29) |  | Kd = 13.30 μM (RXRα-LBD, SPR) | IC50 value is not described. (vs. 1 @ 10 nM, MCF-7 cells) | [72] |
| 30 |  | Kd = 5.12 μM (RXRα-LBD, SPR) | IC50 value is not described. (vs. 1 @ 100 nM, MCF-7 cells) | [72] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Watanabe, M.; Kakuta, H. Retinoid X Receptor Antagonists. Int. J. Mol. Sci. 2018, 19, 2354. https://doi.org/10.3390/ijms19082354
Watanabe M, Kakuta H. Retinoid X Receptor Antagonists. International Journal of Molecular Sciences. 2018; 19(8):2354. https://doi.org/10.3390/ijms19082354
Chicago/Turabian StyleWatanabe, Masaki, and Hiroki Kakuta. 2018. "Retinoid X Receptor Antagonists" International Journal of Molecular Sciences 19, no. 8: 2354. https://doi.org/10.3390/ijms19082354
APA StyleWatanabe, M., & Kakuta, H. (2018). Retinoid X Receptor Antagonists. International Journal of Molecular Sciences, 19(8), 2354. https://doi.org/10.3390/ijms19082354