Na/K-ATPase Signaling and Cardiac Pre/Postconditioning with Cardiotonic Steroids


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cardiac Pre- and Post-Conditioning against Ischemia/Reperfusion Injury
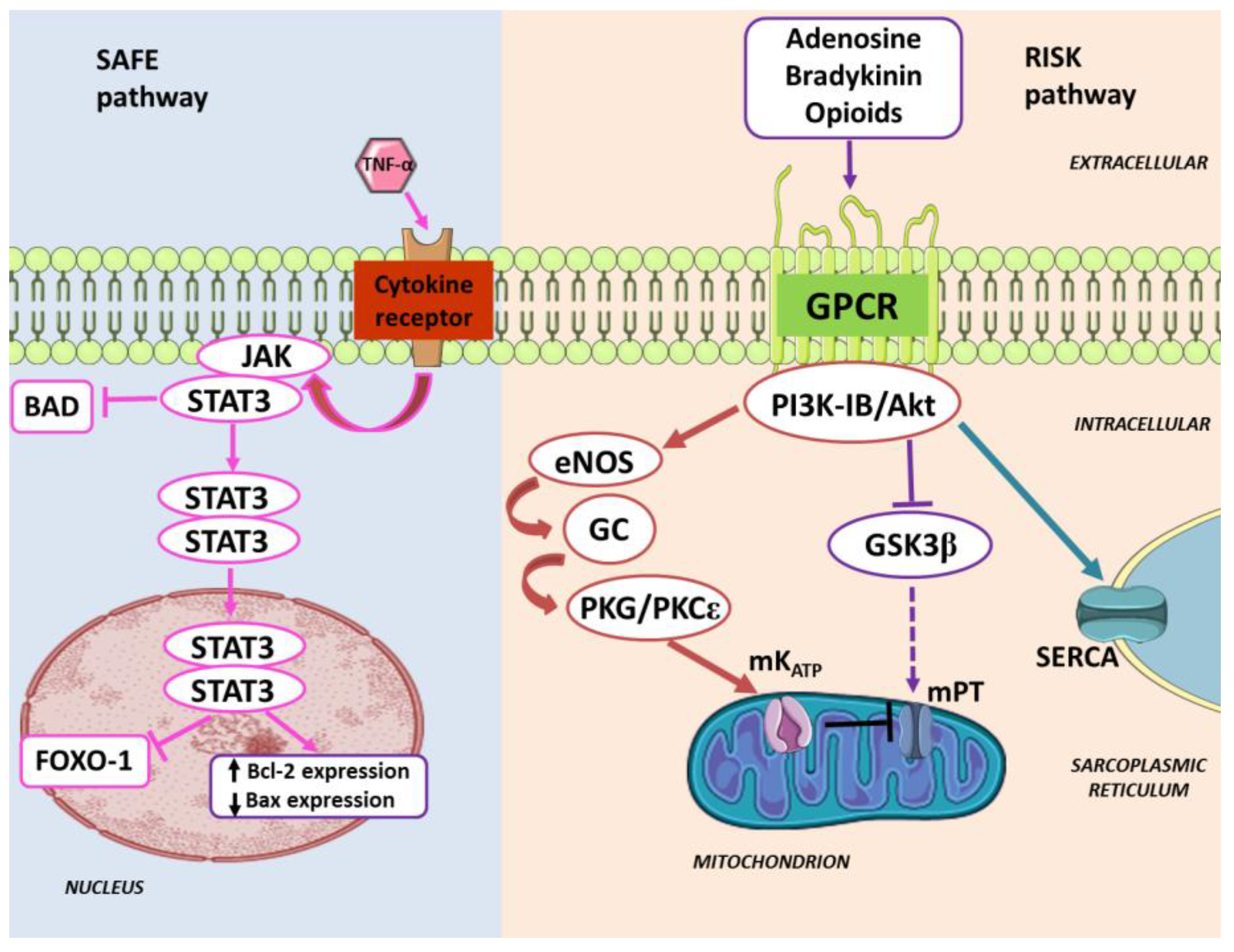
3. Pharmacologically-Induced Cardiac Protection
4. Enduring Challenges and Current Goals for Clinical Application of Conditioning
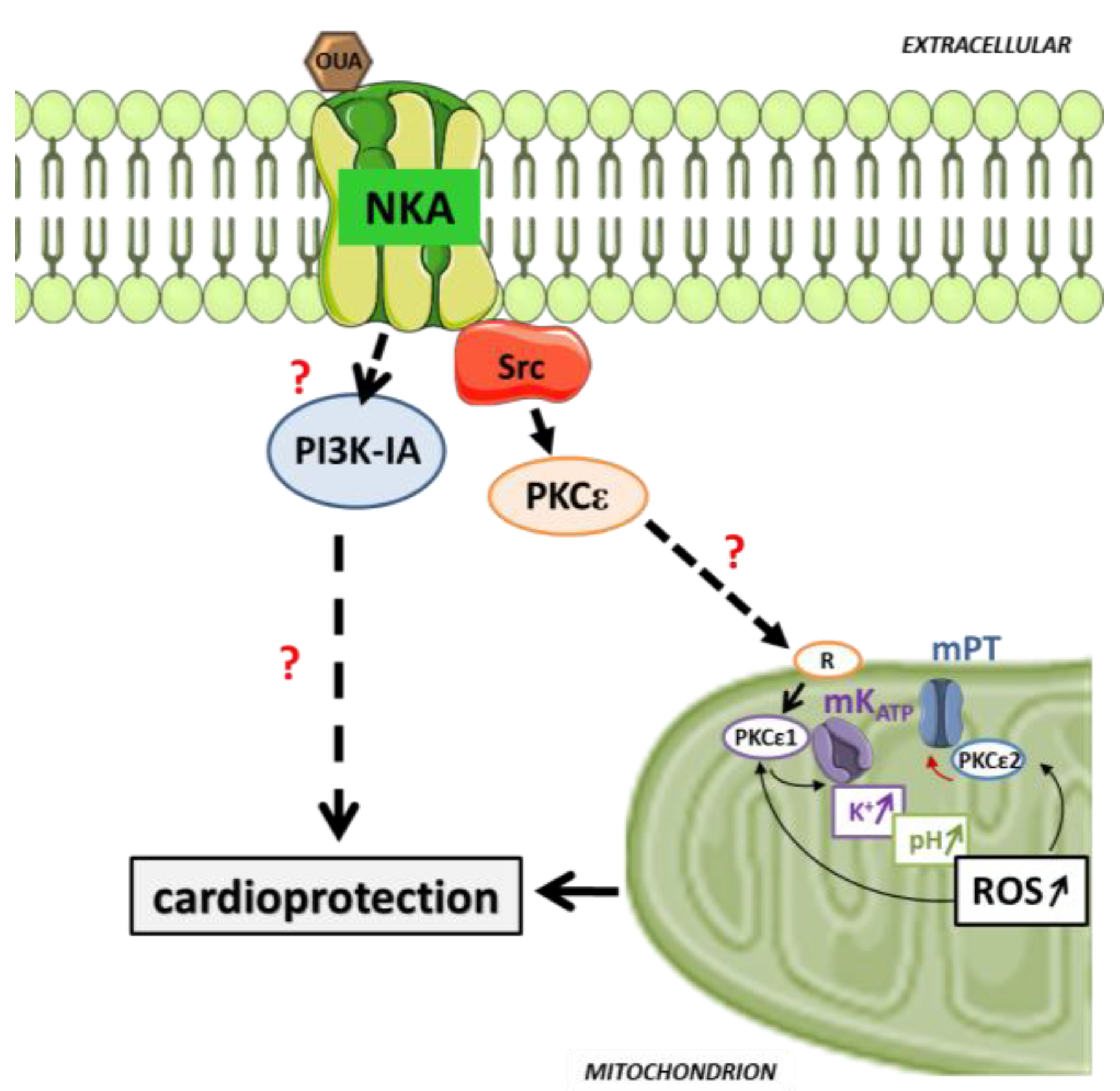
5. Pre/Postconditioning with Cardiotonic Steroid through Cardiac Na/K-ATPase Signaling
6. Prospect and Future Directions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Krumholz, H.M.; Wang, Y.; Chen, J.; Drye, E.E.; Spertus, J.A.; Ross, J.S.; Curtis, J.P.; Nallamothu, B.K.; Lichtman, J.H.; Havranek, E.P.; et al. Reduction in acute myocardial infarction mortality in the United States: Risk-standardized mortality rates from 1995−2006. JAMA 2009, 302, 767–773. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef] [PubMed]
- Fuster, V. Top 10 cardiovascular therapies and interventions for the next decade. Nat. Rev. Cardiol 2014, 11, 671–683. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with ischemia: A delay of lethal cell injury in ischemic myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Garcia-Dorado, D.; Botker, H.E.; Davidson, S.M.; Downey, J.; Engel, F.B.; Jennings, R.; Lecour, S.; Leor, J.; Madonna, R.; et al. Novel targets and future strategies for acute cardioprotection: Position Paper of the European Society of Cardiology Working Group on Cellular Biology of the Heart. Cardiovasc. Res. 2017, 113, 564–585. [Google Scholar] [CrossRef] [PubMed]
- Parratt, J.R. Possibilities for the pharmacological exploitation of ischaemic preconditioning. J. Mol. Cell Cardiol. 1995, 27, 991–1000. [Google Scholar] [CrossRef]
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing myocardial infarct size: Challenges and future opportunities. Heart 2016, 102, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Do Carmo, H.; Arjun, S.; Petrucci, O.; Yellon, D.M.; Davidson, S.M. The Caspase 1 Inhibitor VX-765 Protects the Isolated Rat Heart via the RISK Pathway. Cardiovasc. Drugs Ther. 2018, 32, 165–168. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, J.H.; Werner, J.H.; Moulton, M.J.; Agrawal, D.K. Current Modalities and Mechanisms Underlying Cardioprotection by Ischemic Conditioning. J. Cardiovasc. Transl. Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Heusch, G. Extracellular signalling molecules in the ischaemic/reperfused heart-druggable and translatable for cardioprotection? Br. J. Pharmacol. 2015, 172, 2010–2025. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G. Critical Issues for the Translation of Cardioprotection. Circ. Res. 2017, 120, 1477–1486. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. Ischaemic conditioning and reperfusion injury. Nat. Rev. Cardiol. 2016, 13, 193–209. [Google Scholar] [CrossRef] [PubMed]
- Bessen, H.A. Therapeutic and toxic effects of digitalis: William Withering, 1785. J. Emerg. Med. 1986, 4, 243–248. [Google Scholar] [CrossRef]
- Braunwald, E. Effects of digitalis on the normal and the failing heart. J. Am. Coll. Cardiol. 1985, 5, 51A–59A. [Google Scholar] [CrossRef]
- Skou, J.C. Nobel Lecture. The identification of the sodium pump. Biosci. Rep. 1998, 18, 155–169. [Google Scholar] [CrossRef] [PubMed]
- Skou, J.C. The Identification of the Sodium-Potassium Pump (Nobel Lecture). Angew. Chem. Int. Ed. Engl. 1998, 37, 2320–2328. [Google Scholar] [CrossRef]
- Skou, J.C. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim. Biophys. Acta. 1957, 23, 394–401. [Google Scholar] [CrossRef]
- Lingrel, J.B.; Kuntzweiler, T. Na+,K(+)-ATPase. J. Biol. Chem. 1994, 269, 19659–19662. [Google Scholar] [PubMed]
- Skou, J.C.; Esmann, M. The Na,K-ATPase. J. Bioenerg. Biomembr. 1992, 24, 249–261. [Google Scholar] [PubMed]
- Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 2005, 25, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Shattock, M.J.; Ottolia, M.; Bers, D.M.; Blaustein, M.P.; Boguslavskyi, A.; Bossuyt, J.; Bridge, J.H.; Chen-Izu, Y.; Clancy, C.E.; Edwards, A.; et al. Na+/Ca2+ exchange and Na+/K+-ATPase in the heart. J. Physiol. 2015, 593, 1361–1382. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Morgan, E.E.; Giovannucci, D.R.; Pierre, S.V.; Philipson, K.D.; Askari, A.; Liu, L. Different roles of the cardiac Na+/Ca2+-exchanger in ouabain-induced inotropy, cell signaling, and hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H427–H435. [Google Scholar] [CrossRef] [PubMed]
- Beller, G.A.; Conroy, J.; Smith, T.W. Ischemia-induced alterations in myocardial (Na+ + K+)-ATPase and cardiac glycoside binding. J. Clin. Investig. 1976, 57, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Bersohn, M.M. Sodium pump inhibition in sarcolemma from ischemic hearts. J. Mol. Cell Cardiol. 1995, 27, 1483–1489. [Google Scholar] [CrossRef]
- Murphy, E.; Steenbergen, C. Ion transport and energetics during cell death and protection. Physiology (Bethesda) 2008, 23, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Hilgemann, D.W.; Yaradanakul, A.; Wang, Y.; Fuster, D. Molecular control of cardiac sodium homeostasis in health and disease. J. Cardiovasc. Electrophysiol. 2006, 17, S47–S56. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.J.; Fine, M.; Lu, J.Y.; Hofmann, S.L.; Frazier, G.; Hilgemann, D.W. Massive palmitoylation-dependent endocytosis during reoxygenation of anoxic cardiac muscle. eLife 2013, 2, e01295. [Google Scholar] [CrossRef] [PubMed]
- Fuller, W.; Parmar, V.; Eaton, P.; Bell, J.R.; Shattock, M.J. Cardiac ischemia causes inhibition of the Na/K ATPase by a labile cytosolic compound whose production is linked to oxidant stress. Cardiovasc. Res. 2003, 57, 1044–1051. [Google Scholar] [CrossRef]
- Belliard, A.; Sottejeau, Y.; Duan, Q.; Karabin, J.L.; Pierre, S.V. Modulation of cardiac Na+,K+-ATPase cell surface abundance by simulated ischemia-reperfusion and ouabain preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H94–H103. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Garcia-Dorado, D.; Hernando, V.; Soler-Soler, J. Calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion contributes to cell death after myocardial ischemia. Circ. Res. 2005, 97, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Q.; Moorman, J.R.; Ahlers, B.A.; Carl, L.L.; Lake, D.E.; Song, J.; Mounsey, J.P.; Tucker, A.L.; Chan, Y.M.; Rothblum, L.I.; et al. Phospholemman overexpression inhibits Na+-K+-ATPase in adult rat cardiac myocytes: Relevance to decreased Na+ pump activity in postinfarction myocytes. J. Appl. Physiol. 2006, 100, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Herrick, J.B. Landmark article (JAMA 1912). Clinical features of sudden obstruction of the coronary arteries. JAMA 1983, 250, 1757–1765. [Google Scholar] [CrossRef] [PubMed]
- Dodek, A. Digitalis use in acute myocardial infarction: Current concepts. Can. Med. Assoc. J. 1974, 111, 561, 563–564. [Google Scholar] [PubMed]
- Mohammadi, K.; Kometiani, P.; Xie, Z.; Askari, A. Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J. Biol. Chem. 2001, 276, 42050–42056. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, K.; Liu, L.; Tian, J.; Kometiani, P.; Xie, Z.; Askari, A. Positive inotropic effect of ouabain on isolated heart is accompanied by activation of signal pathways that link Na+/K+-ATPase to ERK1/2. J. Cardiovasc. Pharmacol. 2003, 41, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Tian, J.; Liu, J.; Garlid, K.D.; Shapiro, J.I.; Xie, Z. Involvement of mitogen-activated protein kinases and reactive oxygen species in the inotropic action of ouabain on cardiac myocytes. A potential role for mitochondrial K(ATP) channels. Mol. Cell Biochem. 2003, 242, 181–187. [Google Scholar] [CrossRef] [PubMed]
- Pierre, S.V.; Yang, C.; Yuan, Z.; Seminerio, J.; Mouas, C.; Garlid, K.D.; Dos-Santos, P.; Xie, Z. Ouabain triggers preconditioning through activation of the Na+,K+-ATPase signaling cascade in rat hearts. Cardiovasc. Res. 2007, 73, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Pasdois, P.; Quinlan, C.L.; Rissa, A.; Tariosse, L.; Vinassa, B.; Costa, A.D.; Pierre, S.V.; Dos Santos, P.; Garlid, K.D. Ouabain protects rat hearts against ischemia-reperfusion injury via pathway involving src kinase, mitoKATP, and ROS. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, H1470–H1478. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, G.; Frascarelli, S.; Zucchi, R.; Biver, T.; Montali, U. Cardioprotection by ouabain and digoxin in perfused rat hearts. J. Cardiovasc. Pharmacol. 2008, 52, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.E.; Li, Z.; Stebal, C.; Belliard, A.; Tennyson, G.; Salari, B.; Garlid, K.D.; Pierre, S.V. Preconditioning by subinotropic doses of ouabain in the Langendorff perfused rabbit heart. J. Cardiovasc. Pharmacol. 2010, 55, 234–239. [Google Scholar] [CrossRef] [PubMed]
- Hearse, D.J.; Humphrey, S.M.; Garlick, P.B. Species variation in myocardial anoxic enzyme release, glucose protection and reoxygenation damage. J. Mol. Cell Cardiol. 1976, 8, 329–339. [Google Scholar] [CrossRef]
- Kim, H.D.; Kim, C.H.; Rah, B.J.; Chung, H.I.; Shim, T.S. Quantitative study on the relation between structural and functional properties of the hearts from three different mammals. Anat. Rec. 1994, 238, 199–206. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, H.; Namekata, I.; Nouchi, H.; Shigenobu, K.; Kawanishi, T.; Takahara, A. New aspects for the treatment of cardiac diseases based on the diversity of functional controls on cardiac muscles: Diversity in the excitation-contraction mechanisms of the heart. J. Pharmacol. Sci. 2009, 109, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Ytrehus, K. The ischemic heart-experimental models. Pharmacol. Res. 2000, 42, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Garlid, K.D.; Costa, A.D.; Quinlan, C.L.; Pierre, S.V.; Dos Santos, P. Cardioprotective signaling to mitochondria. J. Mol. Cell Cardiol. 2009, 46, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, C.L.; Costa, A.D.; Costa, C.L.; Pierre, S.V.; Dos Santos, P.; Garlid, K.D. Conditioning the heart induces formation of signalosomes that interact with mitochondria to open mitoKATP channels. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H953–H961. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.; Pierre, S.V.; Cohen, M.V.; Downey, J.M.; Garlid, K.D. cGMP signalling in pre- and post-conditioning: The role of mitochondria. Cardiovasc. Res. 2008, 77, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Belliard, A.; Gulati, G.K.; Duan, Q.; Alves, R.; Brewer, S.; Madan, N.; Sottejeau, Y.; Wang, X.; Kalisz, J.; Pierre, S.V. Ischemia/reperfusion-induced alterations of enzymatic and signaling functions of the rat cardiac Na+/K+-ATPase: Protection by ouabain preconditioning. Physiol. Rep. 2016, 4, e12991. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Garcia-Dorado, D.; Hernando, V.; Barba, I.; Soler-Soler, J. Ischemic preconditioning prevents calpain-mediated impairment of Na+/K+-ATPase activity during early reperfusion. Cardiovasc. Res. 2006, 70, 364–373. [Google Scholar] [CrossRef] [PubMed]
- Inserte, J.; Garcia-Dorado, D. The cGMP/PKG pathway as a common mediator of cardioprotection: Translatability and mechanism. Br. J. Pharmacol. 2015, 172, 1996–2009. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Madan, N.D.; Wu, J.; Kalisz, J.; Doshi, K.Y.; Haldar, S.M.; Liu, L.; Pierre, S.V. Role of phosphoinositide 3-kinase IA (PI3K-IA) activation in cardioprotection induced by ouabain preconditioning. J. Mol. Cell Cardiol. 2015, 80, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Rossello, X.; Riquelme, J.A.; He, Z.; Taferner, S.; Vanhaesebroeck, B.; Davidson, S.M.; Yellon, D.M. The role of PI3Kalpha isoform in cardioprotection. Basic. Res. Cardiol. 2017, 112, 66. [Google Scholar] [CrossRef] [PubMed]
- Duan, Q.; Xu, Y.; Marck, P.V.; Kalisz, J.; Morgan, E.E.; Pierre, S.V. Preconditioning and Postconditioning by Cardiac Glycosides in the Mouse Heart. J. Cardiovasc. Pharmacol. 2018, 71, 95–103. [Google Scholar] [PubMed]
- Kometiani, P.; Li, J.; Gnudi, L.; Kahn, B.B.; Askari, A.; Xie, Z. Multiple signal transduction pathways link Na+/K+-ATPase to growth-related genes in cardiac myocytes. The roles of Ras and mitogen-activated protein kinases. J. Biol. Chem. 1998, 273, 15249–15256. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Kometiani, P.; Liu, J.; Li, J.; Shapiro, J.I.; Askari, A. Intracellular reactive oxygen species mediate the linkage of Na+/K+-ATPase to hypertrophy and its marker genes in cardiac myocytes. J. Biol. Chem. 1999, 274, 19323–19328. [Google Scholar] [CrossRef] [PubMed]
- Kometiani, P.; Tian, J.; Li, J.; Nabih, Z.; Gick, G.; Xie, Z. Regulation of Na/K-ATPase beta1-subunit gene expression by ouabain and other hypertrophic stimuli in neonatal rat cardiac myocytes. Mol. Cell Biochem. 2000, 215, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Li, D.; Du, L.; Baldawi, M.; Gable, M.E.; Askari, A.; Liu, L. Ouabain prevents pathological cardiac hypertrophy and heart failure through activation of phosphoinositide 3-kinase alpha in mouse. Cell Biosci. 2015, 5, 64. [Google Scholar] [CrossRef] [PubMed]
- Sjogren, B.; Parra, S.; Atkins, K.B.; Karaj, B.; Neubig, R.R. Digoxin-Mediated Upregulation of RGS2 Protein Protects against Cardiac Injury. J. Pharmacol. Exp. Ther. 2016, 357, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Lauf, P.K.; Alqahtani, T.; Flues, K.; Meller, J.; Adragna, N.C. Interaction between Na-K-ATPase and Bcl-2 proteins BclXL and Bak. Am. J. Physiol. Cell Physiol. 2015, 308, C51–C60. [Google Scholar] [CrossRef] [PubMed]
- Burlaka, I.; Nilsson, L.M.; Scott, L.; Holtback, U.; Eklof, A.C.; Fogo, A.B.; Brismar, H.; Aperia, A. Prevention of apoptosis averts glomerular tubular disconnection and podocyte loss in proteinuric kidney disease. Kidney Int. 2016, 90, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Kotova, O.; Al-Khalili, L.; Talia, S.; Hooke, C.; Fedorova, O.V.; Bagrov, A.Y.; Chibalin, A.V. Cardiotonic steroids stimulate glycogen synthesis in human skeletal muscle cells via a Src- and ERK1/2-dependent mechanism. J. Biol. Chem. 2006, 281, 20085–20094. [Google Scholar] [CrossRef] [PubMed]
- Hamlyn, J.M.; Manunta, P. Endogenous cardiotonic steroids in kidney failure: A review and an hypothesis. Adv. Chronic. Kidney Dis. 2015, 22, 232–244. [Google Scholar] [CrossRef] [PubMed]
- Bagrov, A.Y.; Fedorova, O.V.; Dmitrieva, R.I.; Howald, W.N.; Hunter, A.P.; Kuznetsova, E.A.; Shpen, V.M. Characterization of a urinary bufodienolide Na+,K+-ATPase inhibitor in patients after acute myocardial infarction. Hypertension 1998, 31, 1097–1103. [Google Scholar] [CrossRef] [PubMed]
- Schoner, W.; Scheiner-Bobis, G. Endogenous and exogenous cardiac glycosides and their mechanisms of action. Am. J. Cardiovasc. Drugs 2007, 7, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, D.J.; Vetteth, S.; Periyasamy, S.M.; Kanj, M.; Fedorova, L.; Khouri, S.; Kahaleh, M.B.; Xie, Z.; Malhotra, D.; Kolodkin, N.I.; et al. Central role for the cardiotonic steroid marinobufagenin in the pathogenesis of experimental uremic cardiomyopathy. Hypertension 2006, 47, 488–495. [Google Scholar] [CrossRef] [PubMed]
- Dvela-Levitt, M.; Cohen-Ben Ami, H.; Rosen, H.; Ornoy, A.; Hochner-Celnikier, D.; Granat, M.; Lichtstein, D. Reduction in maternal circulating ouabain impairs offspring growth and kidney development. J. Am. Soc. Nephrol. 2015, 26, 1103–1114. [Google Scholar] [CrossRef] [PubMed]
- D’Urso, G.; Frascarelli, S.; Balzan, S.; Zucchi, R.; Montali, U. Production of ouabain-like factor in normal and ischemic rat heart. J. Cardiovasc. Pharmacol. 2004, 43, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Khaliulin, I.; Oeltgen, P.R.; Naryzhnaya, N.V.; Pei, J.M.; Brown, S.A.; Lishmanov, Y.B.; Downey, J.M. Prospects for Creation of Cardioprotective and Antiarrhythmic Drugs Based on Opioid Receptor Agonists. Med. Res. Rev. 2016, 36, 871–923. [Google Scholar] [CrossRef] [PubMed]
- Maslov, L.N.; Khaliulin, I.; Zhang, Y.; Krylatov, A.V.; Naryzhnaya, N.V.; Mechoulam, R.; De Petrocellis, L.; Downey, J.M. Prospects for Creation of Cardioprotective Drugs Based on Cannabinoid Receptor Agonists. J. Cardiovasc. Pharmacol. Ther. 2016, 21, 262–272. [Google Scholar] [CrossRef] [PubMed]
- Lasley, R.D. Adenosine Receptor-Mediated Cardioprotection-Current Limitations and Future Directions. Front. Pharmacol. 2018, 9, 310. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Marck, P.V.; Pierre, S.V. Na/K-ATPase Signaling and Cardiac Pre/Postconditioning with Cardiotonic Steroids. Int. J. Mol. Sci. 2018, 19, 2336. https://doi.org/10.3390/ijms19082336
Marck PV, Pierre SV. Na/K-ATPase Signaling and Cardiac Pre/Postconditioning with Cardiotonic Steroids. International Journal of Molecular Sciences. 2018; 19(8):2336. https://doi.org/10.3390/ijms19082336
Chicago/Turabian StyleMarck, Pauline V., and Sandrine V. Pierre. 2018. "Na/K-ATPase Signaling and Cardiac Pre/Postconditioning with Cardiotonic Steroids" International Journal of Molecular Sciences 19, no. 8: 2336. https://doi.org/10.3390/ijms19082336
APA StyleMarck, P. V., & Pierre, S. V. (2018). Na/K-ATPase Signaling and Cardiac Pre/Postconditioning with Cardiotonic Steroids. International Journal of Molecular Sciences, 19(8), 2336. https://doi.org/10.3390/ijms19082336