Beyond the Foam Cell: The Role of LXRs in Preventing Atherogenesis


Abstract
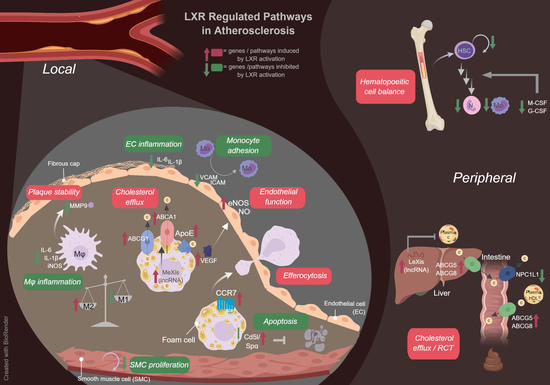
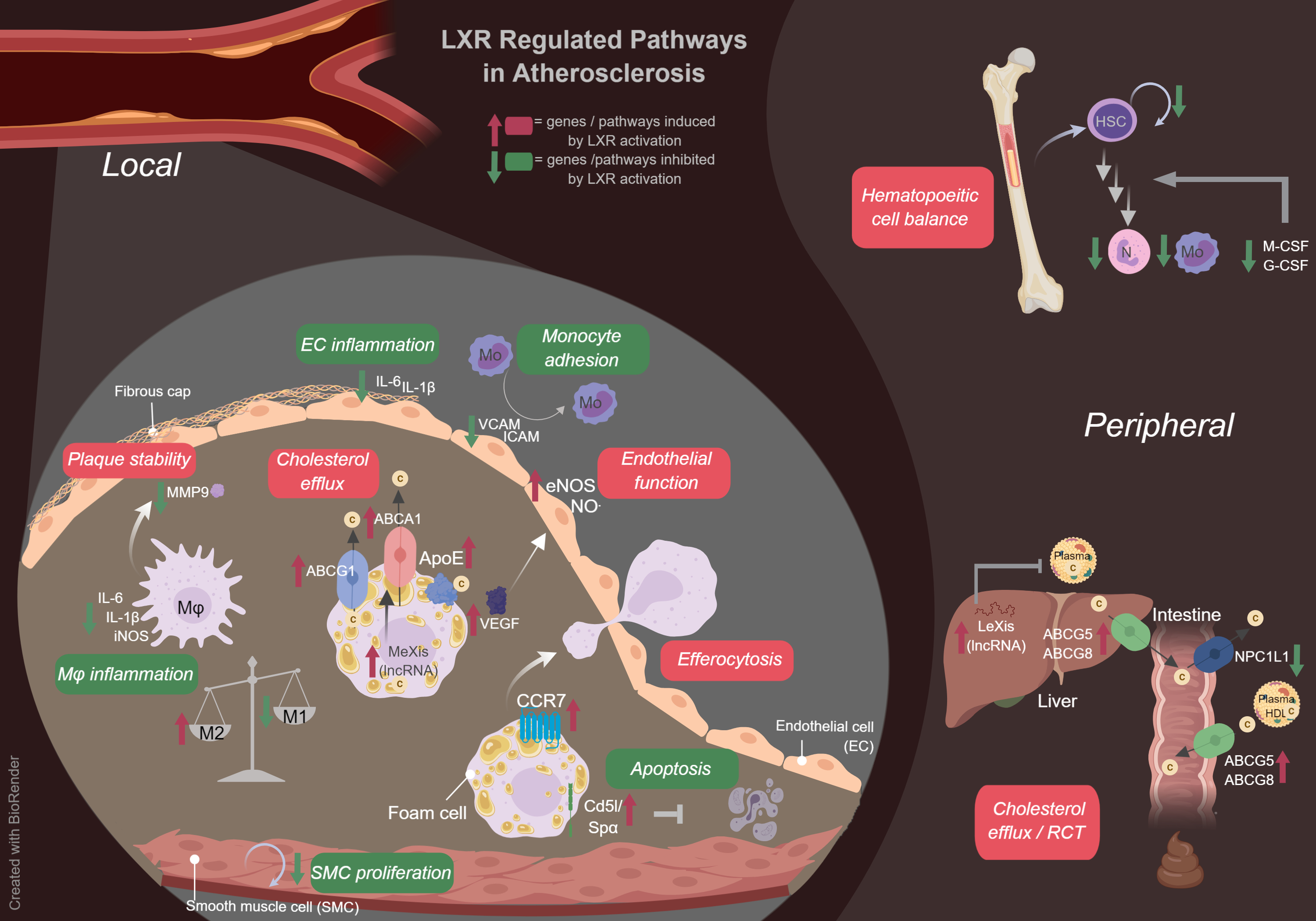
1. Introduction
2. Pathogenesis of Atherosclerosis
2.1. Structure of the Aorta
2.2. Contributions of the Endothelium to Atherosclerosis
2.2.1. Endothelial Dysfunction
2.2.2. Endothelial Activation
2.3. Late Stages of Atherosclerosis
3. Liver X Receptors
3.1. LXRs Preserve Cholesterol Homeostasis
3.2. LXRs Repress Inflammation
4. LXRs and Atherosclerosis: A Macrophage Cholesterol Efflux-Centered Paradigm
5. LXRs and Hematopoietic Cell Types
5.1. Contributions of Hematopoietic Cell Types to Atherosclerosis
5.2. LXRs and Their Target Genes Regulate Hematopoietic Cell Types: Implications for Atherosclerosis
6. LXRs and Vascular Cell Types
6.1. Endothelial Cells
6.2. Smooth Muscle Cells
7. Emerging Mechanisms of LXRs in Atherosclerosis
8. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| CVD | Cardiovascular disease |
| LXR | Liver X receptor |
| SREBP-1c | Sterol response element binding protein-1c |
| ACC | Acetyl-CoA carboxylase |
| FAS | Fatty acid synthase |
| SCD-1 | Stearoyl-CoA desaturase-1 |
| SMC | Smooth muscle cell |
| eNOS | Endothelial nitric oxide synthase |
| LDL | Low-density lipoprotein |
| VLA-4 | Very late antigen-4 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| oxLDL | Oxidized low-density lipoprotein |
| TNFα | Tumor necrosis factor α |
| ICAM-1 | Intercellular adhesion molecule-1 |
| NFκB | Nuclear factor κB |
| M-CSF | Macrophage colony stimulating factor |
| iNOS | Inducible nitric oxide synthase |
| Il | Interleukin |
| MMP | Matrix metalloprotease |
| Apo | Apolipoprotein |
| HDL | High-density lipoprotein |
| Npc1l1 | Niemann-pick C1-like 1 |
| MCP-1 | Monocyte chemoattractant protein-1 |
| LDLR | Low-density lipoprotein receptor |
| CCR7 | C-c chemokine receptor 7 |
| HSC | Hematopoietic stem cell |
| GM-CSF | Granulocyte-monocyte colony stimulating factor |
| Nox2 | NADPH oxidase 2 |
| G-CSF | Granulocyte colony stimulating factor |
| VEGF | Vascular endothelial growth factor |
| CD38 | Cluster of differentiation 38 |
| KLF4 | Krüppel-like factor 4 |
| S1PR2 | Sphingosine-1-phosphate receptor 2 |
| LPS | Lipopolysaccharide |
| LncRNA | Long non-coding RNA |
| DDX17 | DEAD-box helicase 17 |
| TTC39B | Tetratricopeptide repeat domain protein 39B |
| WT | Wildtype |
| WD | Western diet |
| Tg | Transgenic |
| MΦ | Macrophage |
| T09 | T0901317 |
| GW | GW3965 |
| STZ | Streptozotocin |
| BCA | Brachiocephalic artery |
| BM | Bone marrow |
| BMDM | Bone marrow-derived macrophages |
| BMT | Bone marrow transplant |
| OE | Overexpression |
| RCT | Reverse cholesterol transport |
| VLDL | Very low-density lipoprotein |
References
- WHO. Cardiovascular Disease (cvds). Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 22 July 2017).
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [PubMed]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar] [CrossRef] [PubMed]
- AHA. Atherosclerosis. American Heart Association. Available online: https://www.heart.org/en/health-topics/cholesterol/about-cholesterol/atherosclerosis (accessed on 2 August 2017).
- Schultz, J.R.; Tu, H.; Luk, A.; Repa, J.J.; Medina, J.C.; Li, L.; Schwendner, S.; Wang, S.; Thoolen, M.; Mangelsdorf, D.J.; et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000, 14, 2831–2838. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Liang, G.; Ou, J.; Bashmakov, Y.; Lobaccaro, J.M.; Shimomura, I.; Shan, B.; Brown, M.S.; Goldstein, J.L.; Mangelsdorf, D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1C) by oxysterol receptors, LXRα and LXR. Genes Dev. 2000, 14, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Laffitte, B.A.; Patel, P.H.; Watson, M.A.; Matsukuma, K.E.; Walczak, R.; Collins, J.L.; Osborne, T.F.; Tontonoz, P. Direct and indirect mechanisms for regulation of fatty acid synthase gene expression by liver x receptors. J. Biol. Chem. 2002, 277, 11019–11025. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Tontonoz, P. Liver X receptors in lipid metabolism: Opportunities for drug discovery. Nat. Rev. Drug Discov. 2014, 13, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Komati, R.; Spadoni, D.; Zheng, S.; Sridhar, J.; Riley, K.E.; Wang, G. Ligands of therapeutic utility for the liver X receptors. Molecules 2017, 22, 88. [Google Scholar] [CrossRef] [PubMed]
- Rudijanto, A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med. Indones. 2007, 39, 86–93. [Google Scholar] [PubMed]
- Bonetti, P.O.; Lerman, L.O.; Lerman, A. Endothelial dysfunction: A marker of atherosclerotic risk. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Van Hinsbergh, V.W. Endothelium—Role in regulation of coagulation and inflammation. Semin. Immunopathol. 2012, 34, 93–106. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Glagov, S.; Guyton, J.R.; Insull, W., Jr.; Rosenfeld, M.E.; Schaffer, S.A.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Arterioscler. Thromb. 1994, 14, 840–856. [Google Scholar] [CrossRef] [PubMed]
- Karbach, S.; Wenzel, P.; Waisman, A.; Munzel, T.; Daiber, A. Enos uncoupling in cardiovascular diseases—The role of oxidative stress and inflammation. Curr. Pharm. Des. 2014, 20, 3579–3594. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Munzel, T. Endothelial nitric oxide synthase in vascular disease: From marvel to menace. Circulation 2006, 113, 1708–1714. [Google Scholar] [CrossRef] [PubMed]
- Corson, M.A.; James, N.L.; Latta, S.E.; Nerem, R.M.; Berk, B.C.; Harrison, D.G. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ. Res. 1996, 79, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Dimmeler, S.; Assmus, B.; Hermann, C.; Haendeler, J.; Zeiher, A.M. Fluid shear stress stimulates phosphorylation of akt in human endothelial cells: Involvement in suppression of apoptosis. Circ. Res. 1998, 83, 334–341. [Google Scholar] [CrossRef] [PubMed]
- Govers, R.; Rabelink, T.J. Cellular regulation of endothelial nitric oxide synthase. Am. J. Physiol. Renal Physiol. 2001, 280, F193–F206. [Google Scholar] [CrossRef] [PubMed]
- VanderLaan, P.A.; Reardon, C.A.; Getz, G.S. Site specificity of atherosclerosis: Site-selective responses to atherosclerotic modulators. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Grover-Paez, F.; Zavalza-Gomez, A.B. Endothelial dysfunction and cardiovascular risk factors. Diabetes Res. Clin. Pract. 2009, 84, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Davies, P.F.; Remuzzi, A.; Gordon, E.J.; Dewey, C.F., Jr.; Gimbrone, M.A., Jr. Turbulent fluid shear stress induces vascular endothelial cell turnover in vitro. Proc. Natl. Acad. Sci. USA 1986, 83, 2114–2117. [Google Scholar] [CrossRef] [PubMed]
- Warboys, C.M.; de Luca, A.; Amini, N.; Luong, L.; Duckles, H.; Hsiao, S.; White, A.; Biswas, S.; Khamis, R.; Chong, C.K.; et al. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 985–995. [Google Scholar] [CrossRef] [PubMed]
- Schober, A.; Nazari-Jahantigh, M.; Wei, Y.; Bidzhekov, K.; Gremse, F.; Grommes, J.; Megens, R.T.; Heyll, K.; Noels, H.; Hristov, M.; et al. Microrna-126-5p promotes endothelial proliferation and limits atherosclerosis by suppressing dlk1. Nat. Med. 2014, 20, 368–376. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P. Endothelial functions and dysfunctions. In Endothelial Dysfunctions in Vascular Disease; De Caterina, R., Libby, P., Eds.; Blackwell Publishing: Oxford, UK, 2007. [Google Scholar]
- Alon, R.; Kassner, P.D.; Carr, M.W.; Finger, E.B.; Hemler, M.E.; Springer, T.A. The integrin VLA-4 supports tethering and rolling in flow on VCAM-1. J. Cell Biol. 1995, 128, 1243–1253. [Google Scholar] [CrossRef] [PubMed]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.C.; Connelly, P.W.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Cybulsky, M.I.; Gimbrone, M.A., Jr.; Libby, P. An atherogenic diet rapidly induces vcam-1, a cytokine-regulatable mononuclear leukocyte adhesion molecule, in rabbit aortic endothelium. Arterioscler. Thromb. 1993, 13, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Nageh, M.F.; Sandberg, E.T.; Marotti, K.R.; Lin, A.H.; Melchior, E.P.; Bullard, D.C.; Beaudet, A.L. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1517–1520. [Google Scholar] [CrossRef] [PubMed]
- Malek, A.M.; Alper, S.L.; Izumo, S. Hemodynamic shear stress and its role in atherosclerosis. JAMA 1999, 282, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.; Cybulsky, M.I. NF-κB: Pivotal mediator or innocent bystander in atherogenesis? J. Clin. Investig. 2001, 107, 255–264. [Google Scholar] [CrossRef] [PubMed]
- Serbina, N.V.; Pamer, E.G. Monocyte emigration from bone marrow during bacterial infection requires signals mediated by chemokine receptor CCR2. Nat. Immunol. 2006, 7, 311–317. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; Suzuki, M.; Granger, D.N. Nitric oxide: An endogenous modulator of leukocyte adhesion. Proc. Natl. Acad. Sci. USA 1991, 88, 4651–4655. [Google Scholar] [CrossRef] [PubMed]
- Matthews, J.R.; Botting, C.H.; Panico, M.; Morris, H.R.; Hay, R.T. Inhibition of NF-κB DNA binding by nitric oxide. Nucleic Acids Res. 1996, 24, 2236–2242. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.B.; Libby, P.; Liao, J.K. Induction and stabilization of IκBα by nitric oxide mediates inhibition of NF-κB. J. Biol. Chem. 1995, 270, 14214–14219. [Google Scholar] [CrossRef] [PubMed]
- Goss, S.P.; Hogg, N.; Kalyanaraman, B. The effect of nitric oxide release rates on the oxidation of human low density lipoprotein. J. Biol. Chem. 1997, 272, 21647–21653. [Google Scholar] [CrossRef] [PubMed]
- Stary, H.C.; Chandler, A.B.; Dinsmore, R.E.; Fuster, V.; Glagov, S.; Insull, W., Jr.; Rosenfeld, M.E.; Schwartz, C.J.; Wagner, W.D.; Wissler, R.W. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the committee on vascular lesions of the council on arteriosclerosis, american heart association. Circulation 1995, 92, 1355–1374. [Google Scholar] [CrossRef] [PubMed]
- Detmers, P.A.; Hernandez, M.; Mudgett, J.; Hassing, H.; Burton, C.; Mundt, S.; Chun, S.; Fletcher, D.; Card, D.J.; Lisnock, J.; et al. Deficiency in inducible nitric oxide synthase results in reduced atherosclerosis in apolipoprotein e-deficient mice. J. Immunol. 2000, 165, 3430–3435. [Google Scholar] [CrossRef] [PubMed]
- Behr-Roussel, D.; Rupin, A.; Simonet, S.; Bonhomme, E.; Coumailleau, S.; Cordi, A.; Serkiz, B.; Fabiani, J.N.; Verbeuren, T.J. Effect of chronic treatment with the inducible nitric oxide synthase inhibitor n-iminoethyl-l-lysine or with l-arginine on progression of coronary and aortic atherosclerosis in hypercholesterolemic rabbits. Circulation 2000, 102, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Berk, B.C. Vascular smooth muscle growth: Autocrine growth mechanisms. Physiol. Rev. 2001, 81, 999–1030. [Google Scholar] [CrossRef] [PubMed]
- Peiro, C.; Redondo, J.; Rodriguez-Martinez, M.A.; Angulo, J.; Marin, J.; Sanchez-Ferrer, C.F. Influence of endothelium on cultured vascular smooth muscle cell proliferation. Hypertension 1995, 25, 748–751. [Google Scholar] [CrossRef] [PubMed]
- Christodoulides, N.; Durante, W.; Kroll, M.H.; Schafer, A.I. Vascular smooth muscle cell heme oxygenases generate guanylyl cyclase-stimulatory carbon monoxide. Circulation 1995, 91, 2306–2309. [Google Scholar] [CrossRef] [PubMed]
- Keaney, J.F., Jr. Atherosclerosis: From lesion formation to plaque activation and endothelial dysfunction. Mol. Asp. Med. 2000, 21, 99–166. [Google Scholar] [CrossRef]
- Stegemann, J.P.; Hong, H.; Nerem, R.M. Mechanical, biochemical, and extracellular matrix effects on vascular smooth muscle cell phenotype. J. Appl. Physiol. 2005, 98, 2321–2327. [Google Scholar] [CrossRef] [PubMed]
- Rong, J.X.; Shapiro, M.; Trogan, E.; Fisher, E.A. Transdifferentiation of mouse aortic smooth muscle cells to a macrophage-like state after cholesterol loading. Proc. Natl. Acad. Sci. USA 2003, 100, 13531–13536. [Google Scholar] [CrossRef] [PubMed]
- Shankman, L.S.; Gomez, D.; Cherepanova, O.A.; Salmon, M.; Alencar, G.F.; Haskins, R.M.; Swiatlowska, P.; Newman, A.A.; Greene, E.S.; Straub, A.C.; et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat. Med. 2015, 21, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Allahverdian, S.; Chehroudi, A.C.; McManus, B.M.; Abraham, T.; Francis, G.A. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation 2014, 129, 1551–1559. [Google Scholar] [CrossRef] [PubMed]
- Kavurma, M.M.; Rayner, K.J.; Karunakaran, D. The walking dead: Macrophage inflammation and death in atherosclerosis. Curr. Opin. Lipidol. 2017, 28, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Falk, E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006, 47, C7–C12. [Google Scholar] [CrossRef] [PubMed]
- Salvayre, R.; Auge, N.; Benoist, H.; Negre-Salvayre, A. Oxidized low-density lipoprotein-induced apoptosis. Biochim. Biophys. Acta 2002, 1585, 213–221. [Google Scholar] [CrossRef]
- Galis, Z.S.; Sukhova, G.K.; Lark, M.W.; Libby, P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J. Clin. Investig. 1994, 94, 2493–2503. [Google Scholar] [CrossRef] [PubMed]
- Schonbeck, U.; Mach, F.; Sukhova, G.K.; Murphy, C.; Bonnefoy, J.Y.; Fabunmi, R.P.; Libby, P. Regulation of matrix metalloproteinase expression in human vascular smooth muscle cells by t lymphocytes: A role for cd40 signaling in plaque rupture? Circ. Res. 1997, 81, 448–454. [Google Scholar] [CrossRef] [PubMed]
- Glass, C.K.; Witztum, J.L. Atherosclerosis. The road ahead. Cell 2001, 104, 503–516. [Google Scholar] [CrossRef]
- Davies, M.J.; Woolf, N.; Rowles, P.M.; Pepper, J. Morphology of the endothelium over atherosclerotic plaques in human coronary arteries. Br. Heart J. 1988, 60, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Apfel, R.; Benbrook, D.; Lernhardt, E.; Ortiz, M.A.; Salbert, G.; Pfahl, M. A novel orphan receptor specific for a subset of thyroid hormone-responsive elements and its interaction with the retinoid/thyroid hormone receptor subfamily. Mol. Cell. Biol. 1994, 14, 7025–7035. [Google Scholar] [CrossRef] [PubMed]
- Shinar, D.M.; Endo, N.; Rutledge, S.J.; Vogel, R.; Rodan, G.A.; Schmidt, A. Ner, a new member of the gene family encoding the human steroid hormone nuclear receptor. Gene 1994, 147, 273–276. [Google Scholar] [CrossRef]
- Song, C.; Kokontis, J.M.; Hiipakka, R.A.; Liao, S. Ubiquitous receptor: A receptor that modulates gene activation by retinoic acid and thyroid hormone receptors. Proc. Natl. Acad. Sci. USA 1994, 91, 10809–10813. [Google Scholar] [CrossRef] [PubMed]
- Seol, W.; Choi, H.S.; Moore, D.D. Isolation of proteins that interact specifically with the retinoid x receptor: Two novel orphan receptors. Mol. Endocrinol. 1995, 9, 72–85. [Google Scholar] [PubMed]
- Teboul, M.; Enmark, E.; Li, Q.; Wikstrom, A.C.; Pelto-Huikko, M.; Gustafsson, J.A. Or-1, a member of the nuclear receptor superfamily that interacts with the 9-cis-retinoic acid receptor. Proc. Natl. Acad. Sci. USA 1995, 92, 2096–2100. [Google Scholar] [CrossRef] [PubMed]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Hiipakka, R.A.; Kokontis, J.M.; Liao, S. Ubiquitous receptor: Structures, immunocytochemical localization, and modulation of gene activation by receptors for retinoic acids and thyroid hormones. Ann. N. Y. Acad. Sci. 1995, 761, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Bookout, A.L.; Jeong, Y.; Downes, M.; Yu, R.T.; Evans, R.M.; Mangelsdorf, D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell 2006, 126, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Grogan, M.J.; Jones, S.A.; Wisely, G.B.; Kliewer, S.A.; Corey, E.J.; Mangelsdorf, D.J. Structural requirements of ligands for the oxysterol liver X receptors LXRα and LXRβ. Proc. Natl. Acad. Sci. USA 1999, 96, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Janowski, B.A.; Willy, P.J.; Devi, T.R.; Falck, J.R.; Mangelsdorf, D.J. An oxysterol signalling pathway mediated by the nuclear receptor LXRα. Nature 1996, 383, 728–731. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, J.M.; Kliewer, S.A.; Moore, L.B.; Smith-Oliver, T.A.; Oliver, B.B.; Su, J.L.; Sundseth, S.S.; Winegar, D.A.; Blanchard, D.E.; Spencer, T.A.; et al. Activation of the nuclear receptor LXR by oxysterols defines a new hormone response pathway. J. Biol. Chem. 1997, 272, 3137–3140. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Dahlman-Wright, K. Liver X receptor in cholesterol metabolism. J. Endocrinol. 2010, 204, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Peet, D.J.; Turley, S.D.; Ma, W.; Janowski, B.A.; Lobaccaro, J.M.; Hammer, R.E.; Mangelsdorf, D.J. Cholesterol and bile acid metabolism are impaired in mice lacking the nuclear oxysterol receptor LXRα. Cell 1998, 93, 693–704. [Google Scholar] [CrossRef]
- Alberti, S.; Schuster, G.; Parini, P.; Feltkamp, D.; Diczfalusy, U.; Rudling, M.; Angelin, B.; Bjorkhem, I.; Pettersson, S.; Gustafsson, J.A. Hepatic cholesterol metabolism and resistance to dietary cholesterol in LXRβ-deficient mice. J. Clin. Investig. 2001, 107, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Costet, P.; Luo, Y.; Wang, N.; Tall, A.R. Sterol-dependent transactivation of the ABC1 promoter by the liver X receptor/retinoid x receptor. J. Biol. Chem. 2000, 275, 28240–28245. [Google Scholar] [PubMed]
- Klucken, J.; Buchler, C.; Orso, E.; Kaminski, W.E.; Porsch-Ozcurumez, M.; Liebisch, G.; Kapinsky, M.; Diederich, W.; Drobnik, W.; Dean, M.; et al. ABCG1 (ABC8), the human homolog of the drosophila white gene, is a regulator of macrophage cholesterol and phospholipid transport. Proc. Natl. Acad. Sci. USA 2000, 97, 817–822. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Turley, S.D.; Lobaccaro, J.A.; Medina, J.; Li, L.; Lustig, K.; Shan, B.; Heyman, R.A.; Dietschy, J.M.; Mangelsdorf, D.J. Regulation of absorption and Abc1-mediated efflux of cholesterol by RXR heterodimers. Science 2000, 289, 1524–1529. [Google Scholar] [CrossRef] [PubMed]
- Venkateswaran, A.; Repa, J.J.; Lobaccaro, J.M.; Bronson, A.; Mangelsdorf, D.J.; Edwards, P.A. Human white/murine ABC8 mRNA levels are highly induced in lipid-loaded macrophages. A transcriptional role for specific oxysterols. J. Biol. Chem. 2000, 275, 14700–14707. [Google Scholar] [CrossRef] [PubMed]
- Laffitte, B.A.; Repa, J.J.; Joseph, S.B.; Wilpitz, D.C.; Kast, H.R.; Mangelsdorf, D.J.; Tontonoz, P. LXRs control lipid-inducible expression of the apolipoprotein E gene in macrophages and adipocytes. Proc. Natl. Acad. Sci. USA 2001, 98, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Mak, P.A.; Laffitte, B.A.; Desrumaux, C.; Joseph, S.B.; Curtiss, L.K.; Mangelsdorf, D.J.; Tontonoz, P.; Edwards, P.A. Regulated expression of the apolipoprotein E/C-I/C-IV/C-II gene cluster in murine and human macrophages. A critical role for nuclear liver X receptors α and β. J. Biol. Chem. 2002, 277, 31900–31908. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Hammer, R.E.; Li-Hawkins, J.; Von Bergmann, K.; Lutjohann, D.; Cohen, J.C.; Hobbs, H.H. Disruption of Abcg5 and Abcg8 in mice reveals their crucial role in biliary cholesterol secretion. Proc. Natl. Acad. Sci. USA 2002, 99, 16237–16242. [Google Scholar] [CrossRef] [PubMed]
- Berge, K.E.; Tian, H.; Graf, G.A.; Yu, L.; Grishin, N.V.; Schultz, J.; Kwiterovich, P.; Shan, B.; Barnes, R.; Hobbs, H.H. Accumulation of dietary cholesterol in sitosterolemia caused by mutations in adjacent Abc transporters. Science 2000, 290, 1771–1775. [Google Scholar] [CrossRef] [PubMed]
- Repa, J.J.; Berge, K.E.; Pomajzl, C.; Richardson, J.A.; Hobbs, H.; Mangelsdorf, D.J. Regulation of ATP-binding cassette sterol transporters Abcg5 and Abcg8 by the liver X receptors α and β. J. Biol. Chem. 2002, 277, 18793–18800. [Google Scholar] [CrossRef] [PubMed]
- Duval, C.; Touche, V.; Tailleux, A.; Fruchart, J.C.; Fievet, C.; Clavey, V.; Staels, B.; Lestavel, S. Niemann-pick C1 like 1 gene expression is down-regulated by LXR activators in the intestine. Biochem. Biophys. Res. Commun. 2006, 340, 1259–1263. [Google Scholar] [CrossRef] [PubMed]
- Kruit, J.K.; Plosch, T.; Havinga, R.; Boverhof, R.; Groot, P.H.; Groen, A.K.; Kuipers, F. Increased fecal neutral sterol loss upon liver X receptor activation is independent of biliary sterol secretion in mice. Gastroenterology 2005, 128, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.U.; Wang, X.; Da Silva, J.S.; Jaye, M.; Macphee, C.H.; Reilly, M.P.; Billheimer, J.T.; Rothblat, G.H.; Rader, D.J. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation 2006, 113, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Rogers, P.M.; Su, C.; Varga, G.; Stayrook, K.R.; Burris, T.P. Regulation of cholesterologenesis by the oxysterol receptor, LXRα. J. Biol. Chem. 2008, 283, 26332–26339. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; Castrillo, A.; Laffitte, B.A.; Mangelsdorf, D.J.; Tontonoz, P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003, 9, 213–219. [Google Scholar] [CrossRef] [PubMed]
- Myhre, A.E.; Agren, J.; Dahle, M.K.; Tamburstuen, M.V.; Lyngstadaas, S.P.; Collins, A.J.; Foster, S.J.; Thiemermann, C.; Aasen, A.O.; Wang, J.E. Liver X receptor is a key regulator of cytokine release in human monocytes. Shock 2008, 29, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Steffensen, K.R.; Jakobsson, T.; Gustafsson, J.A. Targeting liver X receptors in inflammation. Expert Opin. Ther. Targets 2013, 17, 977–990. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel sumoylation-dependent pathways mediate gene- and signal-specific transrepression by LXRs and PPARγ. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Valledor, A.F.; Hsu, L.C.; Ogawa, S.; Sawka-Verhelle, D.; Karin, M.; Glass, C.K. Activation of liver X receptors and retinoid x receptors prevents bacterial-induced macrophage apoptosis. Proc. Natl. Acad. Sci. USA 2004, 101, 17813–17818. [Google Scholar] [CrossRef] [PubMed]
- Fowler, A.J.; Sheu, M.Y.; Schmuth, M.; Kao, J.; Fluhr, J.W.; Rhein, L.; Collins, J.L.; Willson, T.M.; Mangelsdorf, D.J.; Elias, P.M.; et al. Liver X receptor activators display anti-inflammatory activity in irritant and allergic contact dermatitis models: Liver-x-receptor-specific inhibition of inflammation and primary cytokine production. J. Investig. Dermatol. 2003, 120, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Nunomura, S.; Okayama, Y.; Matsumoto, K.; Hashimoto, N.; Endo-Umeda, K.; Terui, T.; Makishima, M.; Ra, C. Activation of LXRs using the synthetic agonist GW3965 represses the production of pro-inflammatory cytokines by murine mast cells. Allergol. Int. 2015, 64, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Joseph, S.B.; McKilligin, E.; Pei, L.; Watson, M.A.; Collins, A.R.; Laffitte, B.A.; Chen, M.; Noh, G.; Goodman, J.; Hagger, G.N.; et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 7604–7609. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.N.; Hong, C.; Chen, M.; Joseph, S.B.; Wilpitz, D.C.; Wang, X.; Lusis, A.J.; Collins, A.; Hseuh, W.A.; Collins, J.L.; et al. Ligand activation of LXRβ reverses atherosclerosis and cellular cholesterol overload in mice lacking LXRα and apoE. J. Clin. Investig. 2007, 117, 2337–2346. [Google Scholar] [CrossRef] [PubMed]
- Bischoff, E.D.; Daige, C.L.; Petrowski, M.; Dedman, H.; Pattison, J.; Juliano, J.; Li, A.C.; Schulman, I.G. Non-redundant roles for LXRα and LXRβ in atherosclerosis susceptibility in low density lipoprotein receptor knockout mice. J. Lipid Res. 2010, 51, 900–906. [Google Scholar] [CrossRef] [PubMed]
- Tangirala, R.K.; Bischoff, E.D.; Joseph, S.B.; Wagner, B.L.; Walczak, R.; Laffitte, B.A.; Daige, C.L.; Thomas, D.; Heyman, R.A.; Mangelsdorf, D.J.; et al. Identification of macrophage liver X receptors as inhibitors of atherosclerosis. Proc. Natl. Acad. Sci. USA 2002, 99, 11896–11901. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Ranalletta, M.; Wang, N.; Han, S.; Terasaka, N.; Li, R.; Welch, C.; Tall, A.R. Combined deficiency of abca1 and abcg1 promotes foam cell accumulation and accelerates atherosclerosis in mice. J. Clin. Investig. 2007, 117, 3900–3908. [Google Scholar] [CrossRef] [PubMed]
- Levin, N.; Bischoff, E.D.; Daige, C.L.; Thomas, D.; Vu, C.T.; Heyman, R.A.; Tangirala, R.K.; Schulman, I.G. Macrophage liver X receptor is required for antiatherogenic activity of LXR agonists. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Teupser, D.; Kretzschmar, D.; Tennert, C.; Burkhardt, R.; Wilfert, W.; Fengler, D.; Naumann, R.; Sippel, A.E.; Thiery, J. Effect of macrophage overexpression of murine liver X receptor-α (LXR-α) on atherosclerosis in LDL-receptor deficient mice. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 2009–2015. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Breevoort, S.R.; Angdisen, J.; Fu, M.; Schmidt, D.R.; Holmstrom, S.R.; Kliewer, S.A.; Mangelsdorf, D.J.; Schulman, I.G. Liver LXRα expression is crucial for whole body cholesterol homeostasis and reverse cholesterol transport in mice. J. Clin. Investig. 2012, 122, 1688–1699. [Google Scholar] [CrossRef] [PubMed]
- Lo Sasso, G.; Murzilli, S.; Salvatore, L.; D’Errico, I.; Petruzzelli, M.; Conca, P.; Jiang, Z.Y.; Calabresi, L.; Parini, P.; Moschetta, A. Intestinal specific LXR activation stimulates reverse cholesterol transport and protects from atherosclerosis. Cell Metab. 2010, 12, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Murphy, A.J.; Wang, M.; Pagler, T.A.; Vengrenyuk, Y.; Kappus, M.S.; Gorman, D.J.; Nagareddy, P.R.; Zhu, X.; Abramowicz, S.; et al. Deficiency of ATP-binding cassette transporters a1 and G1 in macrophages increases inflammation and accelerates atherosclerosis in mice. Circ. Res. 2013, 112, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Kappus, M.S.; Murphy, A.J.; Abramowicz, S.; Ntonga, V.; Welch, C.L.; Tall, A.R.; Westerterp, M. Activation of liver X receptor decreases atherosclerosis in Ldlr−/− mice in the absence of ATP-binding cassette transporters A1 and G1 in myeloid cells. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Trogan, E.; Feig, J.E.; Dogan, S.; Rothblat, G.H.; Angeli, V.; Tacke, F.; Randolph, G.J.; Fisher, E.A. Gene expression changes in foam cells and the role of chemokine receptor CCR7 during atherosclerosis regression in ApoE-deficient mice. Proc. Natl. Acad. Sci. USA 2006, 103, 3781–3786. [Google Scholar] [CrossRef] [PubMed]
- Verschuren, L.; de Vries-van der Weij, J.; Zadelaar, S.; Kleemann, R.; Kooistra, T. LXR agonist suppresses atherosclerotic lesion growth and promotes lesion regression in apoE* 3Leiden mice: Time course and mechanisms. J. Lipid Res. 2009, 50, 301–311. [Google Scholar] [CrossRef] [PubMed]
- van der Stoep, M.; Li, Z.; Calpe-Berdiel, L.; van der Sluis, R.J.; Saleh, P.; McKinnon, H.J.; Smit, M.J.; Korporaal, S.J.; Van Berkel, T.J.; Van Eck, M.; et al. Elimination of macrophages drives LXR-induced regression both in initial and advanced stages of atherosclerotic lesion development. Biochem. Pharmacol. 2013, 86, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Feig, J.E.; Pineda-Torra, I.; Sanson, M.; Bradley, M.N.; Vengrenyuk, Y.; Bogunovic, D.; Gautier, E.L.; Rubinstein, D.; Hong, C.; Liu, J.; et al. LXR promotes the maximal egress of monocyte-derived cells from mouse aortic plaques during atherosclerosis regression. J. Clin. Investig. 2010, 120, 4415–4424. [Google Scholar] [CrossRef] [PubMed]
- Seita, J.; Weissman, I.L. Hematopoietic stem cell: Self-renewal versus differentiation. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010, 2, 640–653. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.M.; Verfaillie, C.M. Regulation of hematopoiesis. In Blood and Bone Marrow Pathology, 2nd ed.; Elsevier Ltd.: Toronto, ON, Canada, 2011; pp. 63–76. [Google Scholar]
- Soehnlein, O.; Swirski, F.K. Hypercholesterolemia links hematopoiesis with atherosclerosis. Trends Endocrinol. Metab. 2013, 24, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Wantha, S.; Alard, J.E.; Megens, R.T.; van der Does, A.M.; Doring, Y.; Drechsler, M.; Pham, C.T.; Wang, M.W.; Wang, J.M.; Gallo, R.L.; et al. Neutrophil-derived cathelicidin promotes adhesion of classical monocytes. Circ. Res. 2013, 112, 792–801. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, N.; Nakano, K.; Aratani, Y.; Koyama, H.; Kodama, T.; Niki, E. Role of myeloperoxidase in the neutrophil-induced oxidation of low density lipoprotein as studied by myeloperoxidase-knockout mouse. J. Biochem. 2000, 127, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.; Tabas, I. Dendritic cells in atherosclerosis. Semin. Immunopathol. 2014, 36, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.; Gautier, E.L.; Avagyan, S.; Siry, R.L.; Han, S.; Welch, C.L.; Wang, N.; Randolph, G.J.; Snoeck, H.W.; et al. ATP-binding cassette transporters and hdl suppress hematopoietic stem cell proliferation. Science 2010, 328, 1689–1693. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.J.; Akhtari, M.; Tolani, S.; Pagler, T.; Bijl, N.; Kuo, C.L.; Wang, M.; Sanson, M.; Abramowicz, S.; Welch, C.; et al. ApoE regulates hematopoietic stem cell proliferation, monocytosis, and monocyte accumulation in atherosclerotic lesions in mice. J. Clin. Investig. 2011, 121, 4138–4149. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Schouteden, S.; Geenens, R.; Van Duppen, V.; Herijgers, P.; Holvoet, P.; Van Veldhoven, P.P.; Verfaillie, C.M. Hematopoietic stem/progenitor cell proliferation and differentiation is differentially regulated by high-density and low-density lipoproteins in mice. PLoS ONE 2012, 7, e47286. [Google Scholar] [CrossRef] [PubMed]
- Tolani, S.; Pagler, T.A.; Murphy, A.J.; Bochem, A.E.; Abramowicz, S.; Welch, C.; Nagareddy, P.R.; Holleran, S.; Hovingh, G.K.; Kuivenhoven, J.A.; et al. Hypercholesterolemia and reduced hdl-c promote hematopoietic stem cell proliferation and monocytosis: Studies in mice and fh children. Atherosclerosis 2013, 229, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, A.; Tsai, R.; Cummins, C.L. Loss of the liver X receptors disrupts the balance of hematopoietic populations, with detrimental effects on endothelial progenitor cells. J. Am. Heart Assoc. 2018, 7, e007787. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Pagler, T.A.; Seimon, T.A.; Thorp, E.; Welch, C.L.; Witztum, J.L.; Tabas, I.; Tall, A.R. ABCA1 and ABCG1 protect against oxidative stress-induced macrophage apoptosis during efferocytosis. Circ. Res. 2010, 106, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Castrillo, A.; Joseph, S.B.; Marathe, C.; Mangelsdorf, D.J.; Tontonoz, P. Liver X receptor-dependent repression of matrix metalloproteinase-9 expression in macrophages. J. Biol. Chem. 2003, 278, 10443–10449. [Google Scholar] [CrossRef] [PubMed]
- Arai, S.; Shelton, J.M.; Chen, M.; Bradley, M.N.; Castrillo, A.; Bookout, A.L.; Mak, P.A.; Edwards, P.A.; Mangelsdorf, D.J.; Tontonoz, P.; et al. A role for the apoptosis inhibitory factor AIM/SPΑ/API6 in atherosclerosis development. Cell Metab. 2005, 1, 201–213. [Google Scholar] [CrossRef] [PubMed]
- Walczak, R.; Joseph, S.B.; Laffitte, B.A.; Castrillo, A.; Pei, L.; Tontonoz, P. Transcription of the vascular endothelial growth factor gene in macrophages is regulated by liver X receptors. J. Biol. Chem. 2004, 279, 9905–9911. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Lim, E.J.; Yoon, Y.S.; Ahn, Y.H.; Park, E.M.; Kim, H.S.; Kang, J.L. Liver X receptor and STAT1 cooperate downstream of Gas6/Mer to induce anti-inflammatory arginase 2 expression in macrophages. Sci. Rep. 2016, 6, 29673. [Google Scholar] [CrossRef] [PubMed]
- Marathe, C.; Bradley, M.N.; Hong, C.; Lopez, F.; Ruiz de Galarreta, C.M.; Tontonoz, P.; Castrillo, A. The arginase ii gene is an anti-inflammatory target of liver X receptor in macrophages. J. Biol. Chem. 2006, 281, 32197–32206. [Google Scholar] [CrossRef] [PubMed]
- Pourcet, B.; Feig, J.E.; Vengrenyuk, Y.; Hobbs, A.J.; Kepka-Lenhart, D.; Garabedian, M.J.; Morris, S.M., Jr.; Fisher, E.A.; Pineda-Torra, I. LXRα regulates macrophage arginase 1 through PU.1 and interferon regulatory factor 8. Circ. Res. 2011, 109, 492–501. [Google Scholar] [CrossRef] [PubMed]
- Baitsch, D.; Bock, H.H.; Engel, T.; Telgmann, R.; Muller-Tidow, C.; Varga, G.; Bot, M.; Herz, J.; Robenek, H.; von Eckardstein, A.; et al. Apolipoprotein E induces antiinflammatory phenotype in macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Zhu, X.; Gao, C.; Shewale, S.; Cao, Q.; Liu, M.; Boudyguina, E.; Gebre, A.K.; Wilson, M.D.; Brown, A.L.; et al. Myeloid cell-specific atp-binding cassette transporter A1 deletion has minimal impact on atherogenesis in atherogenic diet-fed low-density lipoprotein receptor knockout mice. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1888–1899. [Google Scholar] [CrossRef] [PubMed]
- Hong, C.; Kidani, Y.; Noelia, A.G.; Phung, T.; Ito, A.; Rong, X.; Ericson, K.; Mikkola, H.; Beaven, S.W.; Miller, L.S.; et al. Coordinate regulation of neutrophil homeostasis by liver X receptors in mice. J. Clin. Investig. 2012, 122, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Acebes, M.; Pitaval, C.; Weiss, L.A.; Nombela-Arrieta, C.; Chevre, R.; Noelia, A.-G.; Kunisaki, Y.; Zhang, D.; van Rooijen, N.; Silberstein, L.E.; et al. Rhythmic modulation of the hematopoietic niche through neutrophil clearance. Cell 2013, 153, 1025–1035. [Google Scholar] [CrossRef] [PubMed]
- Beceiro, S.; Pap, A.; Czimmerer, Z.; Sallam, T.; Guillen, J.A.; Gallardo, G.; Hong, C.; Noelia, A.-G.; Tabraue, C.; Diaz, M.; et al. LXR nuclear receptors are transcriptional regulators of dendritic cell chemotaxis. Mol. Cell Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; Fu, Y.; Hou, Y.; Wang, N.; Guan, Y.; Tang, C.; Shyy, J.Y.; Zhu, Y. Laminar shear stress regulates liver X receptor in vascular endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Martin, M.; Zhang, J.; Huang, H.Y.; Bai, L.; Zhang, J.; Kang, J.; He, M.; Li, J.; Maurya, M.R.; et al. Kruppel-like factor 4 regulation of cholesterol-25-hydroxylase and liver X receptor mitigates atherosclerosis susceptibility. Circulation 2017, 136, 1315–1330. [Google Scholar] [CrossRef] [PubMed]
- Peter, A.; Weigert, C.; Staiger, H.; Rittig, K.; Cegan, A.; Lutz, P.; Machicao, F.; Haring, H.U.; Schleicher, E. Induction of stearoyl-coa desaturase protects human arterial endothelial cells against lipotoxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E339–E349. [Google Scholar] [CrossRef] [PubMed]
- Morello, F.; Saglio, E.; Noghero, A.; Schiavone, D.; Williams, T.A.; Verhovez, A.; Bussolino, F.; Veglio, F.; Mulatero, P. LXR-activating oxysterols induce the expression of inflammatory markers in endothelial cells through LXR-independent mechanisms. Atherosclerosis 2009, 207, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Spillmann, F.; Van Linthout, S.; Miteva, K.; Lorenz, M.; Stangl, V.; Schultheiss, H.P.; Tschope, C. LXR agonism improves tnf-α-induced endothelial dysfunction in the absence of its cholesterol-modulating effects. Atherosclerosis 2014, 232, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Yuhanna, I.S.; Umetani, J.; Lee, W.R.; Korach, K.S.; Shaul, P.W.; Umetani, M. LXRβ/estrogen receptor-α signaling in lipid rafts preserves endothelial integrity. J. Clin. Investig. 2013, 123, 3488–3497. [Google Scholar] [CrossRef] [PubMed]
- Fan, A.; Wang, Q.; Yuan, Y.; Cheng, J.; Chen, L.; Guo, X.; Li, Q.; Chen, B.; Huang, X.; Huang, Q. Liver X receptor-α and miR-130a-3p regulate expression of sphingosine 1-phosphate receptor 2 in human umbilical vein endothelial cells. Am. J. Physiol. Cell Physiol. 2016, 310, C216–C226. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Song, J.; Gao, J.; Zhao, J.; Wang, M.; Scipione, C.A.; Koschinsky, M.L.; Wang, Z.V.; Xu, S.; Fu, G. Activation of liver X receptor attenuates lysophosphatidylcholine-induced il-8 expression in endothelial cells via the NF-κB pathway and sumoylation. J. Cell. Mol. Med. 2016, 20, 2249–2258. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Kotani, H.; Yamaguchi, T.; Taguchi, K.; Iida, M.; Ina, K.; Maeda, M.; Kuzuya, M.; Hattori, Y.; Ignarro, L.J. Endothelial cellular senescence is inhibited by liver X receptor activation with an additional mechanism for its atheroprotection in diabetes. Proc. Natl. Acad. Sci. USA 2014, 111, 1168–1173. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Zeng, Y.; Guan, Y.; Hu, Z.; Zhong, D.; Shen, X.; Zhang, L.; Xu, Z.; Gong, W.; Zhang, Y.; et al. Activation of liver X receptor attenuates endothelin-1 expression in vascular endothelial cells. Int. J. Biochem. Cell Biol. 2012, 44, 2299–2307. [Google Scholar] [CrossRef] [PubMed]
- Whetzel, A.M.; Sturek, J.M.; Nagelin, M.H.; Bolick, D.T.; Gebre, A.K.; Parks, J.S.; Bruce, A.C.; Skaflen, M.D.; Hedrick, C.C. ABCG1 deficiency in mice promotes endothelial activation and monocyte-endothelial interactions. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Yu, S.; Yvan-Charvet, L.; Wang, N.; Mzhavia, N.; Langlois, R.; Pagler, T.; Li, R.; Welch, C.L.; Goldberg, I.J.; et al. ABCG1 and HDl protect against endothelial dysfunction in mice fed a high-cholesterol diet. J. Clin. Investig. 2008, 118, 3701–3713. [Google Scholar] [CrossRef] [PubMed]
- Terasaka, N.; Westerterp, M.; Koetsveld, J.; Fernandez-Hernando, C.; Yvan-Charvet, L.; Wang, N.; Sessa, W.C.; Tall, A.R. ATP-binding cassette transporter G1 and high-density lipoprotein promote endothelial no synthesis through a decrease in the interaction of caveolin-1 and endothelial no synthase. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2219–2225. [Google Scholar] [CrossRef] [PubMed]
- Westerterp, M.; Tsuchiya, K.; Tattersall, I.W.; Fotakis, P.; Bochem, A.E.; Molusky, M.M.; Ntonga, V.; Abramowicz, S.; Parks, J.S.; Welch, C.L.; et al. Deficiency of ATP-binding cassette transporters A1 and G1 in endothelial cells accelerates atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Delvecchio, C.J.; Bilan, P.; Radford, K.; Stephen, J.; Trigatti, B.L.; Cox, G.; Parameswaran, K.; Capone, J.P. Liver X receptor stimulates cholesterol efflux and inhibits expression of proinflammatory mediators in human airway smooth muscle cells. Mol. Endocrinol. 2007, 21, 1324–1334. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, F.; Leppanen, O.; Takata, Y.; Caglayan, E.; Liu, J.; Fishbein, M.C.; Kappert, K.; Nakayama, K.I.; Collins, A.R.; Fleck, E.; et al. Liver X receptor agonists suppress vascular smooth muscle cell proliferation and inhibit neointima formation in balloon-injured rat carotid arteries. Circ. Res. 2004, 95, e110–e123. [Google Scholar] [CrossRef] [PubMed]
- Leik, C.E.; Carson, N.L.; Hennan, J.K.; Basso, M.D.; Liu, Q.Y.; Crandall, D.L.; Nambi, P. GW3965, a synthetic liver X receptor (LXR) agonist, reduces angiotensin ii-mediated pressor responses in sprague-dawley rats. Br. J. Pharmacol. 2007, 151, 450–456. [Google Scholar] [CrossRef] [PubMed]
- Sallam, T.; Jones, M.C.; Gilliland, T.; Zhang, L.; Wu, X.; Eskin, A.; Sandhu, J.; Casero, D.; Vallim, T.Q.; Hong, C.; et al. Feedback modulation of cholesterol metabolism by the lipid-responsive non-coding RNA lexis. Nature 2016, 534, 124–128. [Google Scholar] [CrossRef] [PubMed]
- Sallam, T.; Jones, M.; Thomas, B.J.; Wu, X.; Gilliland, T.; Qian, K.; Eskin, A.; Casero, D.; Zhang, Z.; Sandhu, J.; et al. Transcriptional regulation of macrophage cholesterol efflux and atherogenesis by a long noncoding RNA. Nat. Med. 2018, 24, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, J.; Koseki, M.; Molusky, M.M.; Yakushiji, E.; Ichi, I.; Westerterp, M.; Iqbal, J.; Chan, R.B.; Abramowicz, S.; Tascau, L.; et al. TTC39B deficiency stabilizes LXR reducing both atherosclerosis and steatohepatitis. Nature 2016, 535, 303–307. [Google Scholar] [CrossRef] [PubMed]
- Schuster, G.U.; Parini, P.; Wang, L.; Alberti, S.; Steffensen, K.R.; Hansson, G.K.; Angelin, B.; Gustafsson, J.A. Accumulation of foam cells in liver X receptor-deficient mice. Circulation 2002, 106, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Lammers, B.; Zhao, Y.; Hoekstra, M.; Hildebrand, R.B.; Ye, D.; Meurs, I.; Van Berkel, T.J.; Van Eck, M. Augmented atherogenesis in LDL receptor deficient mice lacking both macrophage ABCA1 and ApoE. PLoS ONE 2011, 6, e26095. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
| Reference | Description of Study 2 | Major Findings | Conclusions | |
|---|---|---|---|---|
| Whole-body gain and loss-of-function studies | ||||
| [148] | Chow-fed WT vs. Lxrαβ−/− at 18 months | ↑ lipid in aortic root of Lxrαβ−/− | LXRs regulate atherosclerotic development | |
| [89] | WD-fed Apoe−/−, Ldlr−/− ± GW (12 weeks) | ↓ aortic root lesion area with GW | ||
| [94] | WD-fed Ldlr−/− ± T09 (oral gavage, 6 weeks) | T09: ↓ en face and aortic root lesion area, ↓ MΦ content, ↑ collagen content | ||
| [101] | WD-fed Apoe*3Leiden Tg (18 weeks) + cholesterol-depleted diet ± T09 (8 weeks) | T09: ↓ aortic root lesion area, ↓ MΦ content | LXR activation promotes plaque regression | |
| [102] | WD-fed Ldlr−/− (6 weeks) + chow ± T09 (3 weeks) | T09: ↓ aortic root lesion area | ||
| [90] | WD-fed Apoe−/−, Lxrα−/−Apoe−/− ± GW (11 weeks) | ↑ aortic root lesion area of Lxrα−/−Apoe−/− compared to Apoe−/− | Basal LXRβ does not compensate for loss of LXRα w.r.t. lesion development; however, activation of LXRβ ↓atherosclerotic plaques in the absence of LXRα | |
| GW ↓ aortic root lesion area in Apoe−/− and Lxrα−/−Apoe−/− | ||||
| [91] | WD-fed Ldlr−/−, Lxrα−/−Ldlr−/−, Lxrβ−/− Ldlr−/− ± T09 (12 weeks) | ↑ aortic root lesion area in Lxrα−/−Ldlr−/− vs. Ldlr−/− | ||
| T09: ↓ aortic root lesion area in Ldlr−/−, Lxrα−/−Ldlr−/−, and Lxrβ−/− Ldlr−/− | ||||
| [147] | WD-fed Ttc39b−/− vs. WT | Ttc39b−/−: ↓ steatohepatitis, cholesterol absorption, LXRα ubiquitination, ↑ HDL cholesterol | Increasing LXRα stability in the liver promotes its anti-atherogenic effects, while preventing negative effects associated with LXR activation | |
| WD-fed Ttc39b−/− Ldlr−/− vs. Ldlr−/− | ↓ en face lesion area in Ttc39b−/−Ldlr−/− vs. Ldlr−/− | |||
| [136] | Sprague–Dawley rats + STZ ± T09 | T09: ↓ en face lesion area, aortic intimal senescence | LXR decreases aortic endothelial cell senescence, decreasing atherosclerosis | |
| Bone marrow transplant studies 3 | ||||
| [92] | 1) Apoe−/− 2) WT 3) Lxrαβ−/− 4) Ldlr−/− 5) WT 6) Lxrαβ−/− | ➔ Apoe−/− ➔ Apoe−/− ➔ Apoe−/− ➔ Ldlr−/− ➔ Ldlr−/− ➔ Ldlr−/− | ↑ en face lesion area in Apoe−/− and Ldlr−/− mice receiving Lxrαβ−/− BM (Apoe−/−: 3 > 1 > 2; Ldlr−/−: 6 > 4,5) | Cholesterol efflux in macrophages is responsible for the LXR-mediated effects on reducing atherosclerotic lesions |
| [94] | 1) Ldlr−/− 2) WT 3) Lxrαβ−/− | ➔ Ldlr−/− ± T09 ➔ Ldlr−/− ± T09 ➔ Ldlr−/− ± T09 | T09: ↓ en face lesion area from WT and Ldlr−/− donors | |
| [102] | 1) WT ➔ Apoe−/− (3 days WD-early lesion) 2) WT ➔ Apoe−/− (3 weeks WD-advanced lesion) After BMT, recipients switched to chow ± T09 | T09: ↓ early and late aortic root lesion area, ↓ MΦ content | LXR activation promotes plaque regression | |
| [103] | WD-fed Apoe−/− ± T09 | T09: ↓ aortic arch MΦ content | ||
| Aortic Arch transplants to WT mice after the following BMTs and 16 weeks WD: | ↑ Aortic plaque lesion area and monocyte area of mice from Lxrα−/− Apoe−/− or Lxrβ−/− Apoe−/− donors (2, 3 > 1) | |||
| 1) Apoe−/− 2) Lxrα−/−Apoe−/− 3) Lxrβ−/−Apoe−/− | ➔ Apoe−/− ➔ Apoe−/− ➔ Apoe−/− | |||
| [91] | 1) Ldlr−/− 2) Lxrα−/−Ldlr−/− 3) Ldlr−/− 4) Lxrα−/−Ldlr−/− | ➔ Ldlr−/− ➔ Ldlr−/− ➔ Lxrα−/−Ldlr−/− ➔ Lxrα−/−Ldlr−/− | ↑ en face lesion area in 2 vs. 1 ↑ en face lesion area in 4 vs. 2 ↑ en face lesion area in 4 vs. 3 ↑ en face lesion area in 3 vs. 1 | LXRα also has anti-atherogenic effects in non-hematopoietic cells (3 vs. 1) |
| [99] | 1) WT 2) Abca1/g1−/− 3) Abca1/g1−/− 4) Abca1/g1−/−(myeloid) | ➔ Ldlr−/− ± T09 ➔ Ldlr−/− ± T09 ➔ Ldlr−/− ± GW ➔ Ldlr−/− ± GW | T09: ↓ aortic root lesion area in 2 but not 1; ↓ inflammatory cell infiltration in 2 GW: ↓ aortic root lesion area 3 and 4; (greater ↓ in 3 vs. 4) | LXRs can mediate anti-atherogenic effects via BM cells independent of cholesterol efflux from myeloid cells Loss of LXR target genes in BM cells ↑ atherosclerosis Under chow feeding, Abca1/g1−/− from whole BM ↑ lesion area relative to Abca1/g1−/− only in myeloid cells |
| [98] | 1) WT and fl/fl 2) Abca1/g1−/− 3) Abca1/g1−/−(myeloid) | ➔ Ldlr−/− (chow) ➔ Ldlr−/− (chow) ➔ Ldlr−/− (chow) | Aortic root lesion area: 2 > 3 > 1 | |
| [149] | 1) WT 2) Abca1−/− 3) Apoe−/− 4) Abca1−/−Apoe−/− | ➔ Ldlr−/− ➔ Ldlr−/− ➔ Ldlr−/− ➔ Ldlr−/− | En face and aortic root lesion area: 4 > 3 = 2 > 1 | |
| [124] | 1) Ldlr−/− 2) Abca1−/−(myeloid) Ldlr−/− | ➔ Ldlr−/− ➔ Ldlr−/− | Aortic root lesion area: 2 > 1 (chow-fed); 2 = 1 (WD-fed) | |
| Tissue-specific gain and loss-of-function studies | ||||
| [95] | WD-fed Lxrα-Tg(macrophage) Ldlr−/− vs. Ldlr−/− | Lxrα-Tg(macrophage)Ldlr−/−: ↓ BCA lesion area, ↑ BMDM cholesterol efflux, ↓ BMDM nitric oxide production | Macrophage OE of LXRs ↓ lesion area | |
| [96] | Lxrα−/−(liver) vs. WT ± T09 | Lxrα−/−(liver): ↓ T09-induced increases in circulating triglycerides & cholesterol excretion Lxrα−/−(liver): ↓ T09-induced decrease in intestinal cholesterol absorption | Hepatic LXRα is required for agonist-mediated RCT but not ↓ atherosclerosis, whereas intestinal LXRα OE does facilitate RCT and ↓ atherosclerosis | |
| [96] | WD-fed Lxrα−/−(liver) Ldlr−/− vs. Ldlr−/− ± T09 | T09: ↓ en face lesion area in both genotypes | ||
| [97] | WD-fed Lxrα-Tg(intestine) vs. WT | Lxrα-Tg(intestine): ↓ hepatic cholesterol & triglycerides, ↑ circulating VLDL triglycerides, ↑ HDL cholesterol | ||
| [97] | WD-fed Lxrα-Tg(intestine) Ldlr−/− vs. Ldlr−/− | Lxrα-Tg (intestine)Ldlr−/−: ↓ en face & aortic sinus lesion area | ||
| [143] | Carotid artery injury in Sprague–Dawley rats ± T09 | T09: ↓ neointimal formation | LXR target genes can also affect non-hematopoietic cells (i.e., endothelial, smooth muscle) to ↓ atherogenesis | |
| [140] | 1) WD-fed WT 2) WD-fed Abca1−/− 3) WD-fed Abcg1−/− 4) WD-fed Abca1−/−Abcg1−/− | Aortic eNOS-caveolin interaction: 4 > 3 > 2 > 1 | ||
| [138] | Abcg1−/− vs. WT | Abcg1−/−: ↑ monocyte adherence to aortic endothelium | ||
| [141] | Abca1/g1−/−(endothelial)Ldlr−/− vs. Ldlr−/− | Abca1/g1−/−(endothelial): ↑ en face and aortic root lesion area, ↑ MΦ content, ↑ monocyte recruitment, ↑ LPS-induced endothelial expression of pro-inflammatory and adhesion molecules | ||
| LncRNAs | ||||
| [145] | Chow-fed Ad-LeXis vs. Ad-GFP | Ad-LeXis: ↓ plasma cholesterol, hepatic cholesterol biosynthetic gene expression | LncRNA targets of LXRs work to enhance cholesterol efflux and repress cholesterol synthesis, together enhancing the anti-atherogenic effects of LXRs | |
| WD-fed LeXis−/− vs. WT | LeXis−/−: ↑ hepatic cholesterol | |||
| [146] | BMT3: 1) WT ➔ Ldlr−/− 2) MeXis−/− ➔ Ldlr−/− | MeXis−/−: ↑ en face lesion area, ↓ aortic MΦ Abca1 | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rasheed, A.; Cummins, C.L. Beyond the Foam Cell: The Role of LXRs in Preventing Atherogenesis. Int. J. Mol. Sci. 2018, 19, 2307. https://doi.org/10.3390/ijms19082307
Rasheed A, Cummins CL. Beyond the Foam Cell: The Role of LXRs in Preventing Atherogenesis. International Journal of Molecular Sciences. 2018; 19(8):2307. https://doi.org/10.3390/ijms19082307
Chicago/Turabian StyleRasheed, Adil, and Carolyn L. Cummins. 2018. "Beyond the Foam Cell: The Role of LXRs in Preventing Atherogenesis" International Journal of Molecular Sciences 19, no. 8: 2307. https://doi.org/10.3390/ijms19082307
APA StyleRasheed, A., & Cummins, C. L. (2018). Beyond the Foam Cell: The Role of LXRs in Preventing Atherogenesis. International Journal of Molecular Sciences, 19(8), 2307. https://doi.org/10.3390/ijms19082307