Epithelial Cell Cycle Behaviour in the Injured Kidney



Abstract
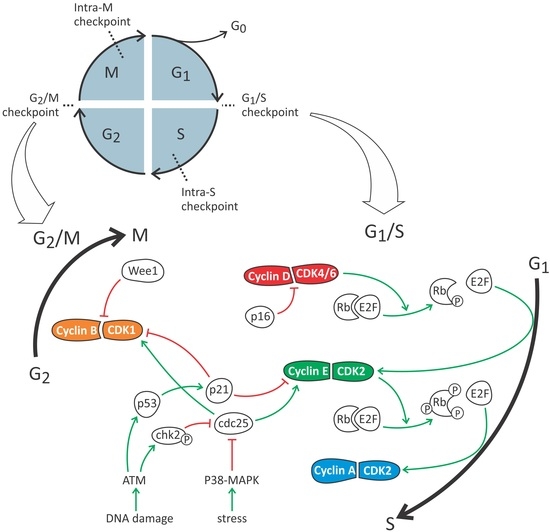
{kind=link}
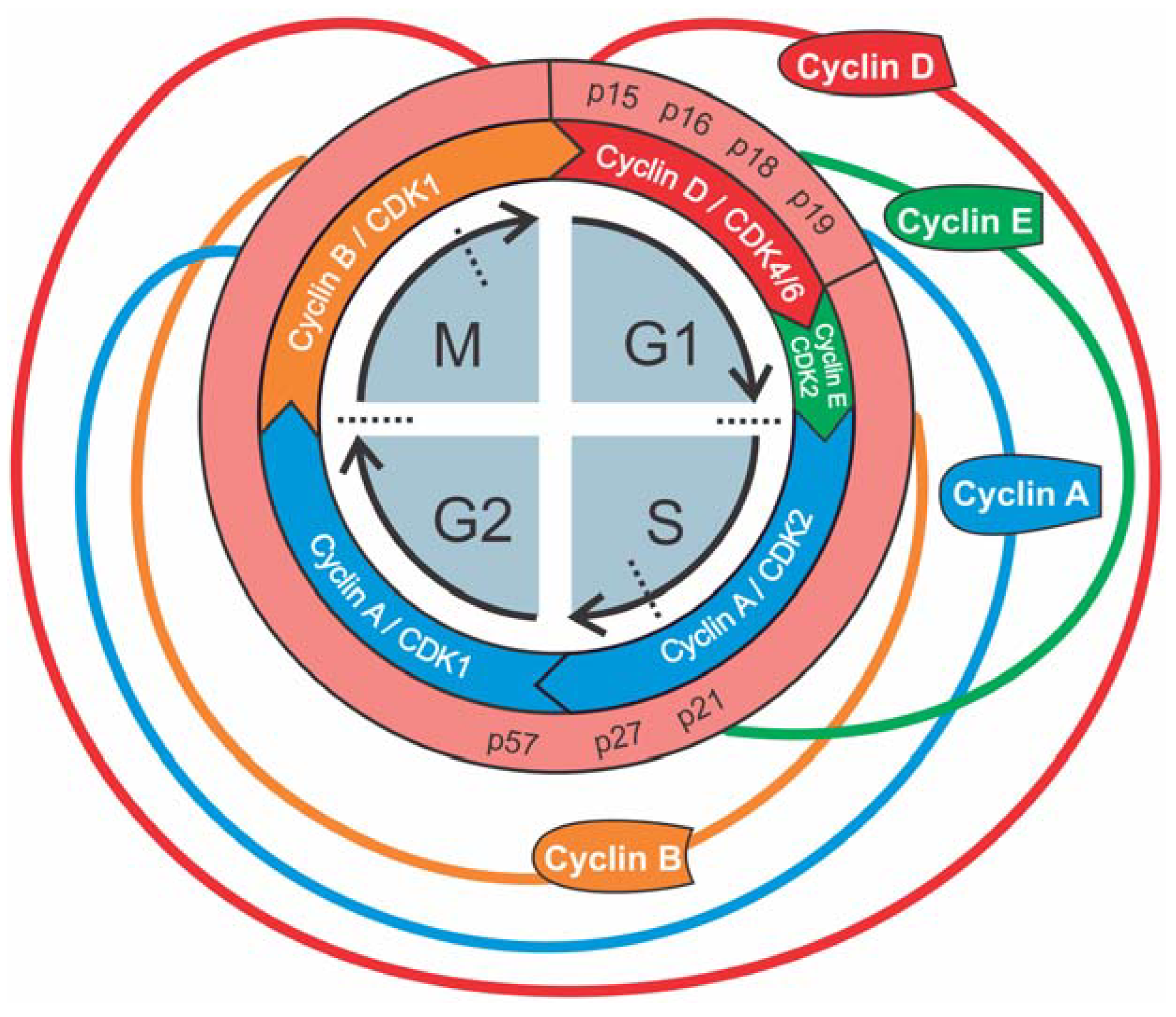{kind=link}
{kind=link}
1. Introduction
2. The Cell Cycle: A General Overview
3. Cell Cycle Phases of Proximal Tubular Epithelial Cells in Health and Renal Injury
3.1. G0 Phase
3.2. G1/S Checkpoint
3.3. Intra-S Checkpoint
3.4. G2/M Checkpoint
3.5. Intra-M Checkpoint
4. Cell Cycle Intervention as Potential Innovative Strategy in the Treatment of Renal Injury
4.1. G1/S
4.2. G2/M
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AAN | Aristolochic acid toxic nephropathy |
| AKI | Acute kidney injury |
| APC | Anaphase-promoting complex |
| ATM | Ataxia telangiectasia mutated protein |
| ATR | Ataxia telangiectasia and Rad3-related protein |
| CCN2 | Connective tissue growth factor 2 |
| cdc25 | Cell division cycle 25 |
| CDK | Cyclin-dependent kinase |
| CHK | Checkpoint kinase |
| CIP | CDK2 interacting protein |
| CKD | Chronic kidney disease |
| CKI | Cyclin-dependent kinase inhibitor |
| Col I | Collagen 1 |
| CTGF | Connective tissue growth factor 2 |
| CVD | Cardiovascular disease |
| CXCR2 | C-X-C motif chemokine receptor |
| DDR | DNA-damage response |
| E2F | E2 transcription factor |
| ECM | Extracellular matrix |
| EGFR | Epidermal growth factor receptor |
| FAN1 | Fanconi anemia-associated nuclease 1 |
| FDA | Food and drug administration |
| GFR | Glomerular filtration rate |
| IL-8 | Interleukin-8 |
| INK4 | Inhibitors of CDK4 |
| IRI | Ischemia-reperfusion injury |
| JNK | c-Jun NH2-terminal kinase |
| KDIGO | Kidney disease: Improving global outcomes |
| KIP | Kinase inhibiting protein |
| MAPK | Mitogen-activated protein kinase |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| NF-κβ | Nuclear factor kappa beta |
| PTBA | 4-phenylthiol-butanoic acid |
| PTC | Proximal tubular epithelium cell |
| Rb | Retinoblastoma protein |
| ROS | Reactive oxygen species |
| SASP | Senescent associated secretory phenotype |
| TGFβ | Transforming growth factor beta |
| UUO | Unilateral ureteric obstruction |
References
- KDIGO Group. KDIGO clinical practice guideline for acute kidney injury. Kidney Int. Suppl. 2012, 2, 1–138. [Google Scholar]
- Basile, D.P.; Anderson, M.D.; Sutton, T.A. Pathophysiology of acute kidney injury. Compr. Physiol. 2012, 2, 1303–1353. [Google Scholar] [PubMed]
- Levey, A.S.; Eckardt, K.U.; Tsukamoto, Y.; Levin, A.; Coresh, J.; Rossert, J.; De, Z.D.; Hostetter, T.H.; Lameire, N.; Eknoyan, G. Definition and classification of chronic kidney disease: A position statement from kidney disease: Improving global outcomes (kdigo). Kidney Int. 2005, 67, 2089–2100. [Google Scholar] [CrossRef] [PubMed]
- Hill, N.R.; Fatoba, S.T.; Oke, J.L.; Hirst, J.A.; O’Callaghan, C.A.; Lasserson, D.S.; Hobbs, F.D. Global prevalence of chronic kidney disease—A systematic review and meta-analysis. PLoS ONE 2016, 11, e0158765. [Google Scholar] [CrossRef] [PubMed]
- De Broe, M.E.; Gharbi, M.B.; Zamd, M.; Elseviers, M. Why overestimate or underestimate chronic kidney disease when correct estimation is possible? Nephrol. Dial. Transplant. 2017, 32, ii136–ii141. [Google Scholar] [CrossRef] [PubMed]
- Lu, K.C.; Wu, C.C.; Yen, J.F.; Liu, W.C. Vascular calcification and renal bone disorders. Sci. World J. 2014, 2014, 637065. [Google Scholar] [CrossRef] [PubMed]
- Palevsky, P.M.; Liu, K.D.; Brophy, P.D.; Chawla, L.S.; Parikh, C.R.; Thakar, C.V.; Tolwani, A.J.; Waikar, S.S.; Weisbord, S.D. Kdoqi us commentary on the 2012 kdigo clinical practice guideline for acute kidney injury. Am. J. Kidney Dis. 2013, 61, 649–672. [Google Scholar] [CrossRef] [PubMed]
- Ioannidis, I. Diabetes treatment in patients with renal disease: Is the landscape clear enough? World J. Diabetes 2014, 5, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Polichnowski, A.J.; Griffin, K.A.; Picken, M.M.; Licea-Vargas, H.; Long, J.; Williamson, G.A.; Bidani, A.K. Hemodynamic basis for the limited renal injury in rats with angiotensin ii-induced hypertension. Am. J. Physiol. Ren. Physiol. 2015, 308, F252–F260. [Google Scholar] [CrossRef] [PubMed]
- Henegar, J.R.; Zhang, Y.; De Rama, R.; Hata, C.; Hall, M.E.; Hall, J.E. Catheter-based radiorefrequency renal denervation lowers blood pressure in obese hypertensive dogs. Am. J. Hypertens. 2014, 27, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Kazancioglu, R. Risk factors for chronic kidney disease: An update. Kidney Int. Suppl. 2013, 3, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Weiner, D.E. Causes and consequences of chronic kidney disease: Implications for managed health care. J. Manag. Care Pharm. 2007, 13, S1–S9. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S. Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 2014, 124, 2299–2306. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury. J. Clin. Investig. 2011, 121, 4210–4221. [Google Scholar] [CrossRef] [PubMed]
- Granger, D.N.; Kvietys, P.R. Reperfusion injury and reactive oxygen species: The evolution of a concept. Redox Biol. 2015, 6, 524–551. [Google Scholar] [CrossRef] [PubMed]
- Hosohata, K. Role of oxidative stress in drug-induced kidney injury. Int. J. Mol. Sci. 2016, 17. [Google Scholar] [CrossRef] [PubMed]
- Canaud, G.; Bonventre, J.V. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol. Dial. Transplant. 2015, 30, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Grgic, I.; Campanholle, G.; Bijol, V.; Wang, C.; Sabbisetti, V.S.; Ichimura, T.; Humphreys, B.D.; Bonventre, J.V. Targeted proximal tubule injury triggers interstitial fibrosis and glomerulosclerosis. Kidney Int. 2012, 82, 172–183. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Maladaptive proximal tubule repair: Cell cycle arrest. Nephron Clin. Pract. 2014, 127, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Czerniak, S.; DiRocco, D.P.; Hasnain, W.; Cheema, R.; Bonventre, J.V. Repair of injured proximal tubule does not involve specialized progenitors. Proc. Natl. Acad. Sci. USA 2011, 108, 9226–9231. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Dedifferentiation and proliferation of surviving epithelial cells in acute renal failure. J. Am. Soc. Nephrol. 2003, 14, S55–S61. [Google Scholar] [CrossRef] [PubMed]
- Thadhani, R.; Pascual, M.; Bonventre, J.V. Acute renal failure. N. Engl. J. Med. 1996, 334, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Venkatachalam, M.A.; Jones, D.B.; Rennke, H.G.; Sandstrom, D.; Patel, Y. Mechanism of proximal tubule brush border loss and regeneration following mild renal ischemia. Lab. Investig. 1981, 45, 355–365. [Google Scholar] [PubMed]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, R.; Cantley, L.G. The impact of aging on kidney repair. Am. J. Physiol. Ren. Physiol. 2008, 294, F1265–F1272. [Google Scholar] [CrossRef] [PubMed]
- Prescott, L.F. The normal urinary excretion rates of renal tubular cells, leucocytes and red blood cells. Clin. Sci. 1966, 31, 425–435. [Google Scholar] [PubMed]
- Vogetseder, A.; Picard, N.; Gaspert, A.; Walch, M.; Kaissling, B.; Le Hir, M. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. Am. J. Physiol. Cell Physiol. 2008, 294, C22–C28. [Google Scholar] [CrossRef] [PubMed]
- Kamura, T.; Hara, T.; Matsumoto, M.; Ishida, N.; Okumura, F.; Hatakeyama, S.; Yoshida, M.; Nakayama, K.; Nakayama, K.I. Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27kip1 at G1 phase. Nat. Cell Biol. 2004, 6, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Sutterluty, H.; Chatelain, E.; Marti, A.; Wirbelauer, C.; Senften, M.; Muller, U.; Krek, W. P45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat. Cell Biol. 1999, 1, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Iwakura, T.; Fujigaki, Y.; Fujikura, T.; Ohashi, N.; Kato, A.; Yasuda, H. A high ratio of G1 to G0 phase cells and an accumulation of G1 phase cells before s phase progression after injurious stimuli in the proximal tubule. Physiol. Rep. 2014, 2. [Google Scholar] [CrossRef] [PubMed]
- Witzgall, R.; Brown, D.; Schwarz, C.; Bonventre, J.V. Localization of proliferating cell nuclear antigen, vimentin, c-Fos, and clusterin in the postischemic kidney. Evidence for a heterogenous genetic response among nephron segments, and a large pool of mitotically active and dedifferentiated cells. J. Clin. Investig. 1994, 93, 2175–2188. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. The cell cycle and acute kidney injury. Kidney Int. 2009, 76, 604–613. [Google Scholar] [CrossRef] [PubMed]
- Kellum, J.A.; Chawla, L.S. Cell-cycle arrest and acute kidney injury: The light and the dark sides. Nephrol. Dial. Transplant. 2016, 31, 16–22. [Google Scholar] [CrossRef] [PubMed]
- van Willigenburg, H.; de Keizer, P.L.J.; de Bruin, R.W.F. Cellular senescence as a therapeutic target to improve renal transplantation outcome. Pharmacol. Res. 2018, 130, 322–330. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Serrano, M. Cellular senescence: From physiology to pathology. Nat. Rev. Mol. Cell Biol. 2014, 15, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Nasrallah, R.; Touyz, R.M.; Hebert, R.L. NADPH oxidases, reactive oxygen species, and the kidney: Friend and foe. J. Am. Soc. Nephrol. 2013, 24, 1512–1518. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Lee, H.B. Reactive oxygen species as glucose signaling molecules in mesangial cells cultured under high glucose. Kidney Int. Suppl. 2000, 77, S19–S25. [Google Scholar] [CrossRef] [PubMed]
- Ohye, H.; Sugawara, M. Dual oxidase, hydrogen peroxide and thyroid diseases. Exp. Biol. Med. 2010, 235, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, B.J.; Mahadev, K.; Wu, X.; Zhu, L.; Motoshima, H. Role of insulin-induced reactive oxygen species in the insulin signaling pathway. Antioxid. Redox Signal. 2005, 7, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Kaelin, W.G., Jr. Ros: Really involved in oxygen sensing. Cell Metab. 2005, 1, 357–358. [Google Scholar] [CrossRef] [PubMed]
- Sedeek, M.; Callera, G.; Montezano, A.; Gutsol, A.; Heitz, F.; Szyndralewiez, C.; Page, P.; Kennedy, C.R.; Burns, K.D.; Touyz, R.M.; et al. Critical role of Nox4-based NADPH oxidase in glucose-induced oxidative stress in the kidney: Implications in type 2 diabetic nephropathy. Am. J. Physiol. Ren. Physiol. 2010, 299, F1348–F1358. [Google Scholar] [CrossRef] [PubMed]
- Mouche, S.; Mkaddem, S.B.; Wang, W.; Katic, M.; Tseng, Y.H.; Carnesecchi, S.; Steger, K.; Foti, M.; Meier, C.A.; Muzzin, P.; et al. Reduced expression of the NADPH oxidase Nox4 is a hallmark of adipocyte differentiation. Biochim. Biophys. Acta 2007, 1773, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The Nox family of ROS-generating nadph oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Buetler, T.M.; Krauskopf, A.; Ruegg, U.T. Role of superoxide as a signaling molecule. News Physiol. Sci. 2004, 19, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, I.; Clarke, P.R.; Marcote, M.J.; Karsenti, E.; Draetta, G. Phosphorylation and activation of human cdc25-C by cdc2--cyclin B and its involvement in the self-amplification of MPF at mitosis. EMBO J. 1993, 12, 53–63. [Google Scholar] [PubMed]
- Brisson, M.; Nguyen, T.; Wipf, P.; Joo, B.; Day, B.W.; Skoko, J.S.; Schreiber, E.M.; Foster, C.; Bansal, P.; Lazo, J.S. Redox regulation of cdc25B by cell-active quinolinediones. Mol. Pharmacol. 2005, 68, 1810–1820. [Google Scholar] [CrossRef] [PubMed]
- Yamaura, M.; Mitsushita, J.; Furuta, S.; Kiniwa, Y.; Ashida, A.; Goto, Y.; Shang, W.H.; Kubodera, M.; Kato, M.; Takata, M.; et al. NADPH oxidase 4 contributes to transformation phenotype of melanoma cells by regulating G2-M cell cycle progression. Cancer Res. 2009, 69, 2647–2654. [Google Scholar] [CrossRef] [PubMed]
- Verbon, E.H.; Post, J.A.; Boonstra, J. The influence of reactive oxygen species on cell cycle progression in mammalian cells. Gene 2012, 511, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Seok, Y.M.; Jung, K.J.; Park, K.M. Reactive oxygen species/oxidative stress contributes to progression of kidney fibrosis following transient ischemic injury in mice. Am. J. Physiol. Ren. Physiol. 2009, 297, F461–F470. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.R.; Rhind, N. The intra-S checkpoint responses to DNA damage. Genes 2017, 8, 74. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.; Tang, C.; Ma, Z.; Huang, S.; Dong, Z. DNA damage response in nephrotoxic and ischemic kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Astuti, P.; Pike, T.; Widberg, C.; Payne, E.; Harding, A.; Hancock, J.; Gabrielli, B. MAPK pathway activation delays G2/M progression by destabilizing cdc25B. J. Biol. Chem. 2009, 284, 33781–33788. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.; Raatz, Y.; Wolkenhauer, O.; Kottek, T.; Bhattacharya, A.; Simon, J.C.; Kunz, M. Chk1 and wee1 control genotoxic-stress induced G2-M arrest in melanoma cells. Cell Signal. 2015, 27, 951–960. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Bhatt, K.; Dong, Z. Checkpoint kinase 1 (Chk1)-short is a splice variant and endogenous inhibitor of chk1 that regulates cell cycle and DNA damage checkpoints. Proc. Natl. Acad. Sci. USA 2012, 109, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Abraham, R.T. Cell cycle checkpoint signaling through the atm and atr kinases. Genes Dev. 2001, 15, 2177–2196. [Google Scholar] [CrossRef] [PubMed]
- Goodarzi, A.A.; Block, W.D.; Lees-Miller, S.P. The role of ATM and ATR in DNA damage-induced cell cycle control. Prog. Cell Cycle Res. 2003, 5, 393–411. [Google Scholar] [PubMed]
- Zhou, W.; Otto, E.A.; Cluckey, A.; Airik, R.; Hurd, T.W.; Chaki, M.; Diaz, K.; Lach, F.P.; Bennett, G.R.; Gee, H.Y.; et al. FAN1 mutations cause karyomegalic interstitial nephritis, linking chronic kidney failure to defective DNA damage repair. Nat. Genet. 2012, 44, 910–915. [Google Scholar] [CrossRef] [PubMed]
- Bencokova, Z.; Kaufmann, M.R.; Pires, I.M.; Lecane, P.S.; Giaccia, A.J.; Hammond, E.M. Atm activation and signaling under hypoxic conditions. Mol. Cell. Biol. 2009, 29, 526–537. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; He, G.; Nelman-Gonzalez, M.; Ashorn, C.L.; Gallick, G.E.; Stukenberg, P.T.; Kirschner, M.W.; Kuang, J. Regulation of Cdc25C by ERK-MAP kinases during the G2/M transition. Cell 2007, 128, 1119–1132. [Google Scholar] [CrossRef] [PubMed]
- Harvey, S.L.; Charlet, A.; Haas, W.; Gygi, S.P.; Kellogg, D.R. Cdk1-dependent regulation of the mitotic inhibitor wee1. Cell 2005, 122, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Matheson, C.J.; Backos, D.S.; Reigan, P. Targeting wee1 kinase in cancer. Trends Pharmacol. Sci. 2016, 37, 872–881. [Google Scholar] [CrossRef] [PubMed]
- Le Clef, N.; Verhulst, A.; D’Haese, P.C.; Vervaet, B.A. Unilateral renal ischemia-reperfusion as a robust model for acute to chronic kidney injury in mice. PLoS ONE 2016, 11, e0152153. [Google Scholar] [CrossRef] [PubMed]
- Alexakis, C.; Maxwell, P.; Bou-Gharios, G. Organ-specific collagen expression: Implications for renal disease. Nephron Exp. Nephrol. 2006, 102, e71–e75. [Google Scholar] [CrossRef] [PubMed]
- Ko, Y.A.; Mohtat, D.; Suzuki, M.; Park, A.S.D.; Izquierdo, M.C.; Han, S.Y.; Kang, H.M.; Si, H.; Hostetter, T.; Pullman, J.M.; et al. Cytosine methylation changes in enhancer regions of core pro-fibrotic genes characterize kidney fibrosis development. Genome Biol. 2013, 14, R108. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Inoue, T.; Kikuta, T.; Kato, N.; Kanno, Y.; Hirosawa, N.; Sakamoto, Y.; Sugaya, T.; Suzuki, H. Poly(ADP-ribose) polymerase-1 enhances transcription of the profibrotic CCN2 gene. J. Am. Soc. Nephrol. 2008, 19, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, W.; McGoohan, S.; Zeisberg, E.M.; Muller, G.A.; Kalbacher, H.; Salant, D.J.; Muller, C.A.; Kalluri, R.; Zeisberg, M. Methylation determines fibroblast activation and fibrogenesis in the kidney. Nat. Med. 2010, 16, 544–550. [Google Scholar] [CrossRef] [PubMed]
- Leask, A.; Abraham, D.J. TGF-β signaling and the fibrotic response. FASEB J. 2004, 18, 816–827. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, M.; Yang, C.; Martino, M.; Duncan, M.B.; Rieder, F.; Tanjore, H.; Kalluri, R. Fibroblasts derive from hepatocytes in liver fibrosis via epithelial to mesenchymal transition. J. Biol. Chem. 2007, 282, 23337–23347. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo Cosentino, C.; Skrypnyk, N.I.; Brilli, L.L.; Chiba, T.; Novitskaya, T.; Woods, C.; West, J.; Korotchenko, V.N.; McDermott, L.; Day, B.W.; et al. Histone deacetylase inhibitor enhances recovery after AKI. J. Am. Soc. Nephrol. 2013, 24, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Liu, N.; Tolbert, E.; Ponnusamy, M.; Ma, L.; Gong, R.; Bayliss, G.; Yan, H.; Zhuang, S. Sustained activation of EGFR triggers renal fibrogenesis after acute kidney injury. Am. J. Pathol. 2013, 183, 160–172. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Safirstein, R.; Price, P.M. Induction of p21 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J. Am. Soc. Nephrol. 1997, 8, A2823. [Google Scholar]
- Megyesi, J.; Price, P.M.; Tamayo, E.; Safirstein, R.L. The lack of a functional p21WAF1/CIP1 gene ameliorates progression to chronic renal failure. Proc. Natl. Acad. Sci. USA 1999, 96, 10830–10835. [Google Scholar] [CrossRef] [PubMed]
- Megyesi, J.; Andrade, L.; Vieira, J.M.; Safirstein, R.L.; Price, P.M. Positive effect of the induction of p21wa1/cip1 on the course of ischemic acute renal failure. Kidney Int. 2001, 60, 2164–2172. [Google Scholar] [CrossRef] [PubMed]
- Wei, Q.; Dong, G.; Yang, T.; Megyesi, J.; Price, P.M.; Dong, Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am. J. Physiol. Ren. Physiol. 2007, 293, F1282–F1291. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Fu, P.; Huang, X.R.; Liu, F.; Lai, K.N.; Lan, H.Y. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J. Am. Soc. Nephrol. 2010, 21, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Khalid, U.; Bowen, T.; Fraser, D.J.; Jenkins, R.H. Acute kidney injury: A paradigm for mirna regulation of the cell cycle. Biochem. Soc. Trans. 2014, 42, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- De Borst, M.H.; Prakash, J.; Sandovici, M.; Klok, P.A.; Hamming, I.; Kok, R.J.; Navis, G.; van Goor, H. C-Jun NH2-terminal kinase is crucially involved in renal tubulo-interstitial inflammation. J. Pharmacol. Exp. Ther. 2009, 331, 896–905. [Google Scholar] [CrossRef] [PubMed]
- Acosta, J.C.; O’Loghlen, A.; Banito, A.; Guijarro, M.V.; Augert, A.; Raguz, S.; Fumagalli, M.; Da Costa, M.; Brown, C.; Popov, N.; et al. Chemokine signaling via the CXCR2 receptor reinforces senescence. Cell 2008, 133, 1006–1018. [Google Scholar] [CrossRef] [PubMed]
- Thorn, T.; Gniadecki, R.; Petersen, A.B.; Vicanova, J.; Wulf, H.C. Differences in activation of G2/M checkpoint in keratinocytes after genotoxic stress induced by hydrogen peroxide and ultraviolet a radiation. Free Radic. Res. 2001, 35, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Chung, Y.W.; Jeong, D.W.; Won, J.Y.; Choi, E.J.; Choi, Y.H.; Kim, I.Y. H2O2-induced AP-1 activation and its effect on p21WAF1/CIP1-mediated G2/M arrest in a p53-deficient human lung cancer cell. Biochem. Biophys. Res. Commun. 2002, 293, 1248–1253. [Google Scholar] [CrossRef]
- May, K.M.; Hardwick, K.G. The spindle checkpoint. J. Cell Sci. 2006, 119, 4139–4142. [Google Scholar] [CrossRef] [PubMed]
- Price, P.M.; Safirstein, R.L.; Megyesi, J. Protection of renal cells from cisplatin toxicity by cell cycle inhibitors. Am. J. Physiol. Ren. Physiol. 2004, 286, F378–F384. [Google Scholar] [CrossRef] [PubMed]
- Shapiro, G.I. Cyclin-dependent kinase pathways as targets for cancer treatment. J. Clin. Oncol. 2006, 24, 1770–1783. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.M.; Torrice, C.D.; Bell, J.F.; Monahan, K.B.; Jiang, Q.; Wang, Y.; Ramsey, M.R.; Jin, J.; Wong, K.K.; Su, L.; et al. Mitigation of hematologic radiation toxicity in mice through pharmacological quiescence induced by CDK4/6 inhibition. J. Clin. Investig. 2010, 120, 2528–2536. [Google Scholar] [CrossRef] [PubMed]
- Pabla, N.; Gibson, A.A.; Buege, M.; Ong, S.S.; Li, L.; Hu, S.; Du, G.; Sprowl, J.A.; Vasilyeva, A.; Janke, L.J.; et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc. Natl. Acad. Sci. USA 2015, 112, 5231–5236. [Google Scholar] [CrossRef] [PubMed]
- DiRocco, D.P.; Bisi, J.; Roberts, P.; Strum, J.; Wong, K.K.; Sharpless, N.; Humphreys, B.D. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am. J. Physiol. Ren. Physiol. 2014, 306, F379–F388. [Google Scholar] [CrossRef] [PubMed]
- Roberts, P.J.; Bisi, J.E.; Strum, J.C.; Combest, A.J.; Darr, D.B.; Usary, J.E.; Zamboni, W.C.; Wong, K.K.; Perou, C.M.; Sharpless, N.E. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J. Natl. Cancer Inst. 2012, 104, 476–487. [Google Scholar] [CrossRef] [PubMed]
- Preisig, P.A.; Franch, H.A. Renal epithelial cell hyperplasia and hypertrophy. Semin. Nephrol. 1995, 15, 327–340. [Google Scholar] [PubMed]
- Derynck, R.; Akhurst, R.J.; Balmain, A. TGF-β signaling in tumor suppression and cancer progression. Nat. Genet. 2001, 29, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Fu, S.; Tang, Y.; Huang, X.R.; Feng, M.; Xu, A.P.; Lan, H.Y. Smad7 protects against acute kidney injury by rescuing tubular epithelial cells from the g1 cell cycle arrest. Clin. Sci. (Lond.) 2017, 131, 1955–1969. [Google Scholar] [CrossRef] [PubMed]
- Finn, W.F. Enhanced recovery from postischemic acute renal failure. Micropuncture studies in the rat. Circ. Res. 1980, 46, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Finn, W.F.; Fernandez-Repollet, E.; Goldfarb, D.; Iaina, A.; Eliahou, H.E. Attenuation of injury due to unilateral renal ischemia: Delayed effects of contralateral nephrectomy. J. Lab. Clin. Med. 1984, 103, 193–203. [Google Scholar] [PubMed]
- Christophorou, M.A.; Ringshausen, I.; Finch, A.J.; Swigart, L.B.; Evan, G.I. The pathological response to DNA damage does not contribute to p53-mediated tumour suppression. Nature 2006, 443, 214–217. [Google Scholar] [CrossRef] [PubMed]
- Donehower, L.A.; Lozano, G. 20 years studying p53 functions in genetically engineered mice. Nat. Rev. Cancer 2009, 9, 831–841. [Google Scholar] [CrossRef] [PubMed]
- Thomasova, D.; Anders, H.J. Cell cycle control in the kidney. Nephrol. Dial. Transplant. 2015, 30, 1622–1630. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Finn, W.F. Renal counterbalance. J. Lab. Clin. Med. 1985, 105, 523–530. [Google Scholar] [PubMed]
- Hinman, F. Experimental hdronephrosis—Repair following ureterocystoneostomy in white rats with complete ureteral obstruction. J. Urol. 1919, 3, 147–174. [Google Scholar] [CrossRef]
- Hickson, I.; Zhao, Y.; Richardson, C.J.; Green, S.J.; Martin, N.M.; Orr, A.I.; Reaper, P.M.; Jackson, S.P.; Curtin, N.J.; Smith, G.C. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase atm. Cancer Res. 2004, 64, 9152–9159. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J. Four faces of cellular senescence. J. Cell Biol. 2011, 192, 547–556. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; van de Sluis, B.; Kirkland, J.L.; van Deursen, J.M. Clearance of p16ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Bonventre, J.V. Primary proximal tubule injury leads to epithelial cell cycle arrest, fibrosis, vascular rarefaction, and glomerulosclerosis. Kidney Int. Suppl. (2011) 2014, 4, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Coca, S.G.; Singanamala, S.; Parikh, C.R. Chronic kidney disease after acute kidney injury: A systematic review and meta-analysis. Kidney Int. 2012, 81, 442–448. [Google Scholar] [CrossRef] [PubMed]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moonen, L.; D’Haese, P.C.; Vervaet, B.A. Epithelial Cell Cycle Behaviour in the Injured Kidney. Int. J. Mol. Sci. 2018, 19, 2038. https://doi.org/10.3390/ijms19072038
Moonen L, D’Haese PC, Vervaet BA. Epithelial Cell Cycle Behaviour in the Injured Kidney. International Journal of Molecular Sciences. 2018; 19(7):2038. https://doi.org/10.3390/ijms19072038
Chicago/Turabian StyleMoonen, Lies, Patrick C. D’Haese, and Benjamin A. Vervaet. 2018. "Epithelial Cell Cycle Behaviour in the Injured Kidney" International Journal of Molecular Sciences 19, no. 7: 2038. https://doi.org/10.3390/ijms19072038
APA StyleMoonen, L., D’Haese, P. C., & Vervaet, B. A. (2018). Epithelial Cell Cycle Behaviour in the Injured Kidney. International Journal of Molecular Sciences, 19(7), 2038. https://doi.org/10.3390/ijms19072038