Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia


Abstract
1. Molecular and Cellular Aspects of Friedreich’s Ataxia
1.1. Pathophysiology of the Disease
1.2. Current and Prospective Treatments
2. Analysis of Frataxin Function in Drosophila melanogaster
2.1. Methodologies Applied to Generate FRDA Models in Flies
2.2. Fly Models Recapitulate FRDA Features
3. The Contribution of Drosophila to the Analysis and Cure of FRDA
3.1. Understanding the Molecular Physiopathology of the Disease
3.1.1. Biogenesis of Fe–S Clusters: The Beginning of the End
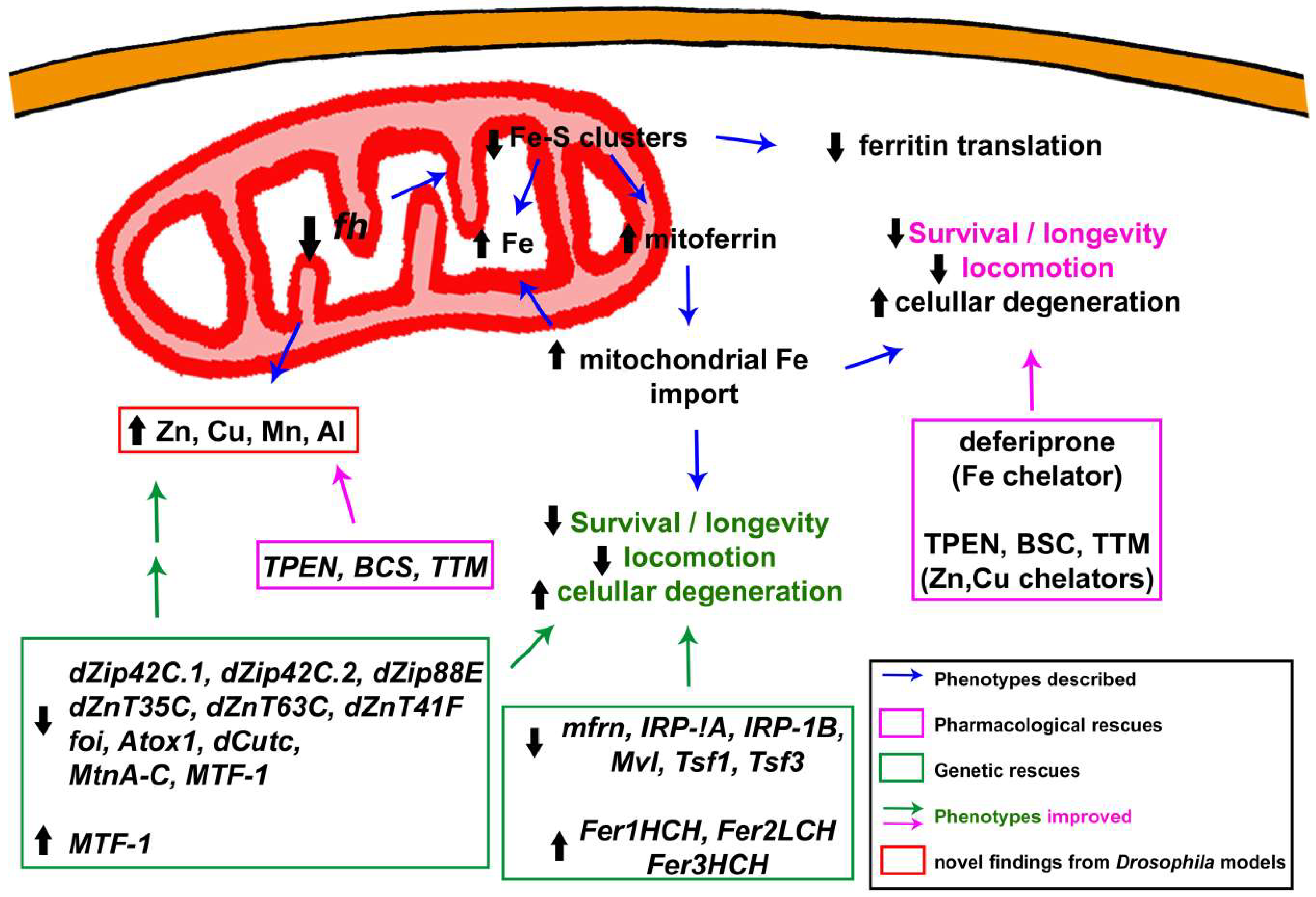
3.1.2. Alterations in Iron Metabolism: The Cornerstone behind FRDA
3.1.3. To Be or Not to Be: The Controversy of Oxidative Stress
3.1.4. Beyond Iron: Importance of Other Metals in the Development and Progression of the Pathology
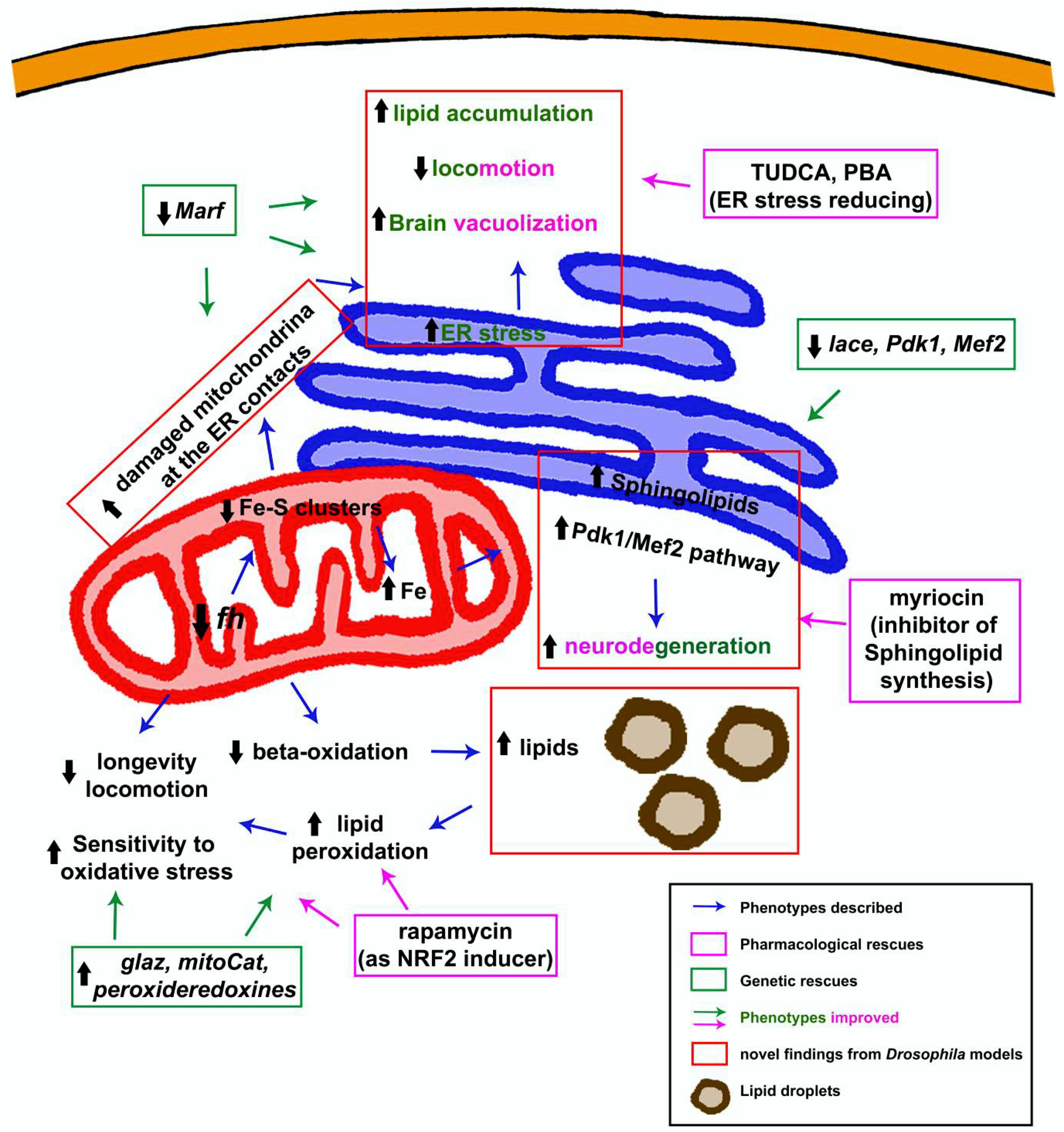
3.1.5. Dyshomeostasis of Lipid Metabolism: Much More Than a Supporting Actor
3.1.6. The Endoplasmic Reticulum Factor: An Alternative Vision to the Pathology
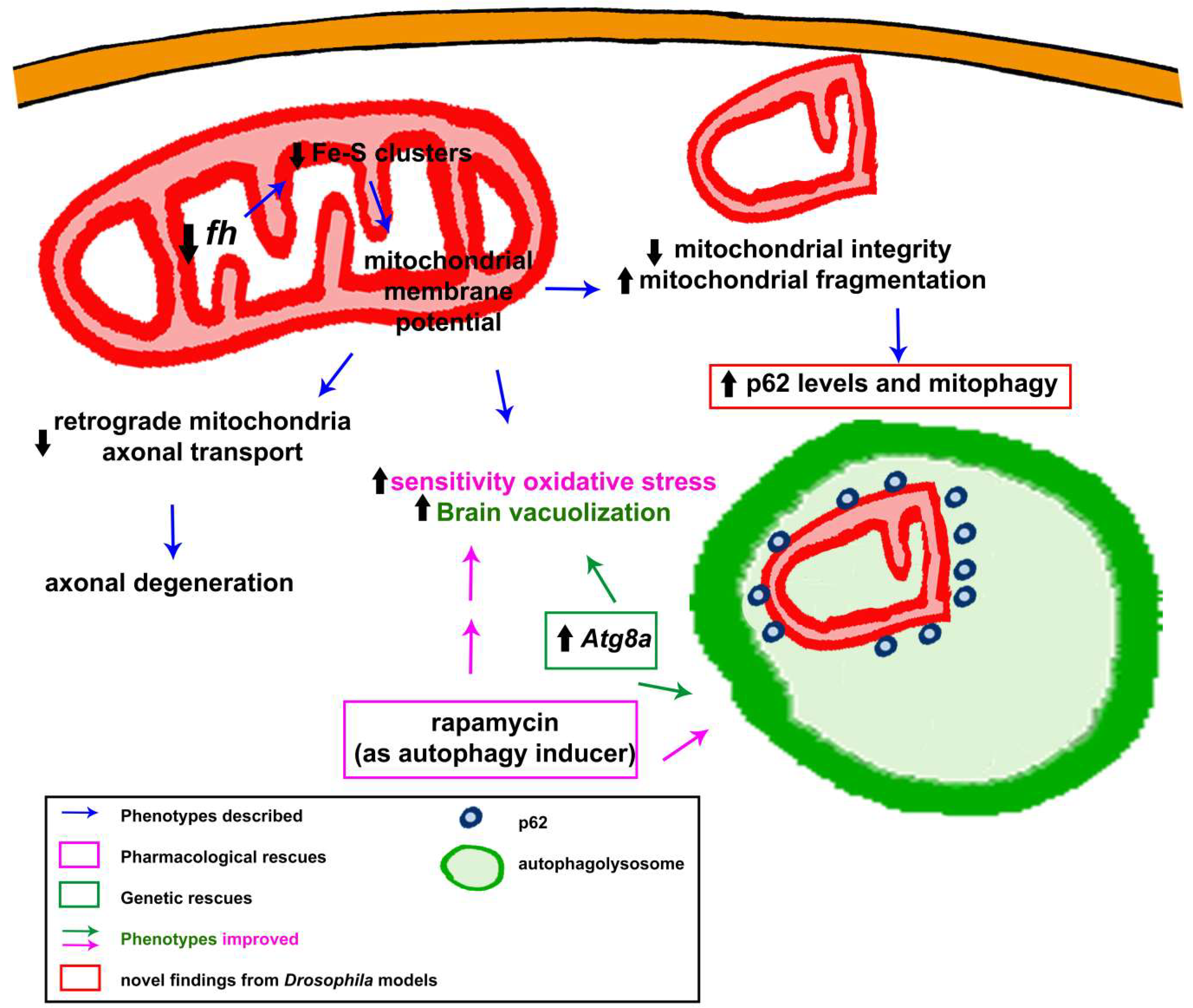
3.1.7. To Be Continued: Additional Pathways That Might Be Participating
3.2. Revealing Possible Therapies in FRDA Fly Models
3.2.1. Analysis of Specific Drugs
3.2.2. Unbiased Drug Screens
4. Future Perspectives
Author Contributions
Conflicts of Interest
Abbreviations
| AAV | adeno-associated virus |
| Al | aluminium |
| AMPK | adenosine monophosphate-dependent protein kinase |
| ATF4 | activating transcription factor |
| BCS | bathocuproine disulphonate |
| BiP | binding immunoglobulin protein |
| Cer | ceramide |
| Cnc | cap-n-collar |
| CNS | central nervous system |
| CRISPR | clustered, regularly interspaced, short palindromic repeat |
| Cu | copper |
| CyaY | Escherichia coli frataxin homolog |
| Da | daughterless |
| DamID | targeted DNA adenine methyltransferase identification |
| DFOB | desferrioxamine B |
| DFP | deferiprone |
| dhCer | dihydroceramide |
| dhSph | dihydrosphingosine |
| DMT1 | divalent metal transporter-1 |
| DN | dentate nucleus |
| DRG | dorsal root ganglia |
| DTT | dithiothreitol |
| eIF2α | α subunit of eukaryotic translation initiation factor 2 |
| EMS | ethyl methanesulfonate |
| ER | endoplasmic reticulum |
| ETC | electron transport chain |
| Fe | iron |
| Fe–S | iron–sulfur |
| FFA | free fatty acid |
| fh | frataxin homolog |
| FLP/FRT | flippase and flippase recognition target |
| FRDA | Friedreich’s ataxia |
| FXN | human frataxin |
| GAA | guanine-adenine-adenine |
| GC/MS | gas chromatography coupled with mass spectrometry |
| Grx1 | glutaredoxine |
| GSH | reduced glutathione |
| GSSG | oxidized glutathione |
| HIFs | hypoxia inducible factors |
| HRE | hypoxia responsive element |
| HSAN1 | hereditary sensory and autonomic neuropathy type 1 |
| IREs | iron responsive elements |
| IRP-1A | iron regulatory protein 1A |
| KO | knock-out |
| lace | palmitoyltransferase |
| MAMs | mitochondria-associated membranes |
| Marf | mitofusin |
| MCK | muscle creatine kinase |
| Mef-2 | myocyte enhancer factor-2 |
| Mn | manganese |
| MPP | mitochondrial processing peptidase |
| Mvl | Malvolio |
| PBA | 4-phenylbutyric acid |
| Pdk1 | protein kinase-1 |
| PNS | peripheral nervous system |
| PPARγ | peroxisome proliferator-activated receptor gamma |
| RNAi | RNA interference |
| ROS | reactive oxygen species |
| SOD1 | cytosolic superoxide dismutase |
| SOD2 | mitochondrial superoxide dismutase |
| Sph | sphingosine |
| SREBP1 | sterol-responsive element-binding protein 1 |
| TPEN | N,N,N′,N′-tetrakis(2-pyridinylmethyl)-1,2-ethanediamine |
| Tsf | transferrins |
| TTM | tetrathiomolybdate |
| TUDCA | tauroursodeoxycholic acid |
| UAS | upstream activating sequence |
| Yfh1 | yeast frataxin homolog 1 |
| Zn | zinc |
References
- Labuda, M.; Labuda, D.; Miranda, C.; Poirier, J.; Soong, B.-W.; Barucha, N.E.; Pandolfo, M. Unique origin and specific ethnic distribution of the Friedreich ataxia GAA expansion. Neurology 2000, 54, 2322–2324. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich ataxia: Neuropathology revised. J. Neuropathol. Exp. Neurol. 2013, 72, 78–90. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.H.; Boesch, S.; Nachbauer, W.; Mariotti, C.; Giunti, P. Clinical features of Friedreich’s ataxia: Classical and atypical phenotypes. J. Neurochem. 2013, 126 (Suppl. 1), 103–117. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Igoillo-Esteve, M.; Rai, M.; Begu, A.; Serroukh, Y.; Depondt, C.; Musuaya, A.E.; Marhfour, I.; Ladrière, L.; Moles Lopez, X.; et al. Central role and mechanisms of β-cell dysfunction and death in friedreich ataxia-associated diabetes. Ann. Neurol. 2012, 72, 971–982. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Campuzano, V.; Foury, F.; Dollé, P.; Cazzalini, O.; Koenig, M. Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat. Genet. 1997, 16, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Cossée, M.; Dürr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschütter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Sandi, C.; Sandi, M.; Anjomani Virmouni, S.; Al-Mahdawi, S.; Pook, M.A. Epigenetic-based therapies for Friedreich ataxia. Front. Genet. 2014, 5, 165. [Google Scholar] [CrossRef] [PubMed]
- Koutnikova, H.; Campuzano, V.; Koenig, M. Maturation of wild-type and mutated frataxin by the mitochondrial processing peptidase. Hum. Mol. Genet. 1998, 7, 1485–1489. [Google Scholar] [CrossRef] [PubMed]
- Condò, I.; Ventura, N.; Malisan, F.; Rufini, A.; Tomassini, B.; Testi, R. In vivo maturation of human frataxin. Hum. Mol. Genet. 2007, 16, 1534–1540. [Google Scholar] [CrossRef] [PubMed]
- Gibson, T.J.; Koonin, E.V.; Musco, G.; Pastore, A.; Bork, P. Friedreich’s ataxia protein: Phylogenetic evidence for mitochondrial dysfunction. Trends Neurosci. 1996, 19, 465–468. [Google Scholar] [CrossRef]
- Adinolfi, S.; Trifuoggi, M.; Politou, A.S.; Martin, S.; Pastore, A. A structural approach to understanding the iron-binding properties of phylogenetically different frataxins. Hum. Mol. Genet. 2002, 11, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Dhe-Paganon, S.; Shigeta, R.; Chi, Y.I.; Ristow, M.; Shoelson, S.E. Crystal structure of human frataxin. J. Biol. Chem. 2000, 275, 30753–30756. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Cho, S.J.; Yang, J.K.; Song, H.K.; Suh, S.W. Crystallization and preliminary X-ray crystallographic analysis of Escherichia coli CyaY, a structural homologue of human frataxin. Acta Crystallogr. Sect. D Biol. Crystallogr. 2000, 56, 920–921. [Google Scholar] [CrossRef]
- He, Y.; Alam, S.L.; Proteasa, S.V.; Zhang, Y.; Lesuisse, E.; Dancis, A.; Stemmler, T.L. Yeast frataxin solution structure, iron binding, and ferrochelatase interaction. Biochemistry 2004, 43, 16254–16262. [Google Scholar] [CrossRef] [PubMed]
- Foury, F.; Pastore, A.; Trincal, M. Acidic residues of yeast frataxin have an essential role in Fe–S cluster assembly. EMBO Rep. 2007, 8, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Adamec, J.; Rusnak, F.; Owen, W.G.; Naylor, S.; Benson, L.M.; Gacy, A.M.; Isaya, G. Iron-dependent self-assembly of recombinant yeast frataxin: Implications for Friedreich ataxia. Am. J. Hum. Genet. 2000, 67, 549–562. [Google Scholar] [CrossRef] [PubMed]
- Gakh, O.; Adamec, J.; Gacy, A.M.; Twesten, R.D.; Owen, W.G.; Isaya, G. Physical evidence that yeast frataxin is an iron storage protein. Biochemistry 2002, 41, 6798–6804. [Google Scholar] [CrossRef] [PubMed]
- Aloria, K.; Schilke, B.; Andrew, A.; Craig, E.A. Iron-induced oligomerization of yeast frataxin homologue Yfh1 is dispensable in vivo. EMBO Rep. 2004, 5, 1096–1101. [Google Scholar] [CrossRef] [PubMed]
- Schmucker, S.; Martelli, A.; Colin, F.; Page, A.; Wattenhofer-Donzé, M.; Reutenauer, L.; Puccio, H. Mammalian frataxin: An essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS ONE 2011, 6, e16199. [Google Scholar] [CrossRef] [PubMed]
- Ahlgren, E.-C.; Fekry, M.; Wiemann, M.; Söderberg, C.A.; Bernfur, K.; Gakh, O.; Rasmussen, M.; Højrup, P.; Emanuelsson, C.; Isaya, G.; et al. Iron-induced oligomerization of human FXN81-210 and bacterial CyaY frataxin and the effect of iron chelators. PLoS ONE 2017, 12, e0188937. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126 (Suppl. 1), 43–52. [Google Scholar] [CrossRef] [PubMed]
- Lill, R. Function and biogenesis of iron-sulphur proteins. Nature 2009, 460, 831–838. [Google Scholar] [CrossRef] [PubMed]
- Rötig, A.; de Lonlay, P.; Chretien, D.; Foury, F.; Koenig, M.; Sidi, D.; Munnich, A.; Rustin, P. Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 1997, 17, 215–217. [Google Scholar] [CrossRef] [PubMed]
- Puccio, H.; Simon, D.; Cossée, M.; Criqui-Filipe, P.; Tiziano, F.; Melki, J.; Hindelang, C.; Matyas, R.; Rustin, P.; Koenig, M. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 2001, 27, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Gerber, J.; Mühlenhoff, U.; Lill, R. An interaction between frataxin and Isu1/Nfs1 that is crucial for Fe/S cluster synthesis on Isu1. EMBO Rep. 2003, 4, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-L.; Barondeau, D.P. Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry 2010, 49, 9132–9139. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Iron-sulfur cluster biosynthesis. Characterization of frataxin as an iron donor for assembly of 2Fe–2S clusters in ISU-type proteins. J. Am. Chem. Soc. 2003, 125, 6078–6084. [Google Scholar] [CrossRef] [PubMed]
- Bridwell-Rabb, J.; Fox, N.G.; Tsai, C.-L.; Winn, A.M.; Barondeau, D.P. Human frataxin activates Fe–S cluster biosynthesis by facilitating sulfur transfer chemistry. Biochemistry 2014, 53, 4904–4913. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, S.; Iannuzzi, C.; Prischi, F.; Pastore, C.; Iametti, S.; Martin, S.R.; Bonomi, F.; Pastore, A. Bacterial frataxin CyaY is the gatekeeper of iron-sulfur cluster formation catalyzed by IscS. Nat. Struct. Mol. Biol. 2009, 16, 390–396. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.-L.; O’Neill, H.A.; Kennedy, M.C.; Ikeda-Saito, M.; Isaya, G.; Szweda, L.I. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science 2004, 305, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [PubMed]
- González-Cabo, P.; Vázquez-Manrique, R.P.; García-Gimeno, M.A.; Sanz, P.; Palau, F. Frataxin interacts functionally with mitochondrial electron transport chain proteins. Hum. Mol. Genet. 2005, 14, 2091–2098. [Google Scholar] [CrossRef] [PubMed]
- Aranca, T.V.; Jones, T.M.; Shaw, J.D.; Staffetti, J.S.; Ashizawa, T.; Kuo, S.-H.; Fogel, B.L.; Wilmot, G.R.; Perlman, S.L.; Onyike, C.U.; et al. Emerging therapies in Friedreich’s ataxia. Neurodegener. Dis. Manag. 2016, 6, 49–65. [Google Scholar] [CrossRef] [PubMed]
- Perdomini, M.; Belbellaa, B.; Monassier, L.; Reutenauer, L.; Messaddeq, N.; Cartier, N.; Crystal, R.G.; Aubourg, P.; Puccio, H. Prevention and reversal of severe mitochondrial cardiomyopathy by gene therapy in a mouse model of Friedreich’s ataxia. Nat. Med. 2014, 20, 542–547. [Google Scholar] [CrossRef] [PubMed]
- Piguet, F.; Montigny, C. de; Vaucamps, N.; Reutenauer, L.; Eisenmann, A.; Puccio, H. Rapid and Complete Reversal of Sensory Ataxia by Gene Therapy in a Novel Model of Friedreich Ataxia. Mol. Ther. J. Am. Soc. Gene Ther. 2018, 26. [Google Scholar] [CrossRef] [PubMed]
- Edenharter, O.; Clement, J.; Schneuwly, S.; Navarro, J.A. Overexpression of Drosophila frataxin triggers cell death in an iron-dependent manner. J. Neurogenet. 2017, 31, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Llorens, J.V.; Soriano, S.; Botella, J.A.; Schneuwly, S.; Martínez-Sebastián, M.J.; Moltó, M.D. Overexpression of human and fly frataxins in Drosophila provokes deleterious effects at biochemical, physiological and developmental levels. PLoS ONE 2011, 6, e21017. [Google Scholar] [CrossRef] [PubMed]
- Vannocci, T.; Notario Manzano, R.; Beccalli, O.; Bettegazzi, B.; Grohovaz, F.; Cinque, G.; de Riso, A.; Quaroni, L.; Codazzi, F.; Pastore, A. Adding a temporal dimension to the study of Friedreich’s ataxia: The effect of frataxin overexpression in a human cell model. Dis. Models Mech. 2018. [Google Scholar] [CrossRef] [PubMed]
- Cañizares, J.; Blanca, J.M.; Navarro, J.A.; Monrós, E.; Palau, F.; Moltó, M.D. dfh is a Drosophila homolog of the Friedreich’s ataxia disease gene. Gene 2000, 256, 35–42. [Google Scholar] [CrossRef]
- Llorens, J.V.; Navarro, J.A.; Martínez-Sebastián, M.J.; Baylies, M.K.; Schneuwly, S.; Botella, J.A.; Moltó, M.D. Causative role of oxidative stress in a Drosophila model of Friedreich ataxia. FASEB J. 2007, 21, 333–344. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, K.C.; Kok, N.M.; Dancis, A.; Stemmler, T.L. Drosophila frataxin: An iron chaperone during cellular Fe–S cluster bioassembly. Biochemistry 2008, 47, 6917–6927. [Google Scholar] [CrossRef] [PubMed]
- Condò, I.; Ventura, N.; Malisan, F.; Tomassini, B.; Testi, R. A pool of extramitochondrial frataxin that promotes cell survival. J. Biol. Chem. 2006, 281, 16750–16756. [Google Scholar] [CrossRef] [PubMed]
- Condò, I.; Malisan, F.; Guccini, I.; Serio, D.; Rufini, A.; Testi, R. Molecular control of the cytosolic aconitase/IRP1 switch by extramitochondrial frataxin. Hum. Mol. Genet. 2010, 19, 1221–1229. [Google Scholar] [CrossRef] [PubMed]
- Long, S.; Jirku, M.; Ayala, F.J.; Lukes, J. Mitochondrial localization of human frataxin is necessary but processing is not for rescuing frataxin deficiency in Trypanosoma brucei. Proc. Natl. Acad. Sci. USA 2008, 105, 13468–13473. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Lin, G.; Haelterman, N.A.; Ho, T.S.-Y.; Li, T.; Li, Z.; Duraine, L.; Graham, B.H.; Jaiswal, M.; Yamamoto, S.; et al. Loss of Frataxin induces iron toxicity, sphingolipid synthesis, and Pdk1/Mef2 activation, leading to neurodegeneration. eLife 2016, 5, e16034. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.H.; Perrimon, N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 1993, 118, 401–415. [Google Scholar] [PubMed]
- Jackson, G.R.; Salecker, I.; Dong, X.; Yao, X.; Arnheim, N.; Faber, P.W.; MacDonald, M.E.; Zipursky, S.L. Polyglutamine-expanded human huntingtin transgenes induce degeneration of Drosophila photoreceptor neurons. Neuron 1998, 21, 633–642. [Google Scholar] [CrossRef]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Kennerdell, J.R.; Carthew, R.W. Heritable gene silencing in Drosophila using double-stranded RNA. Nat. Biotechnol. 2000, 18, 896–898. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Lu, B. Reduction of protein translation and activation of autophagy protect against PINK1 pathogenesis in Drosophila melanogaster. PLoS Genet. 2010, 6, e1001237. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.R.; Kirby, K.; Hilliker, A.J.; Phillips, J.P. RNAi-mediated suppression of the mitochondrial iron chaperone, frataxin, in Drosophila. Hum. Mol. Genet. 2005, 14, 3397–3405. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.R.; Kirby, K.; Orr, W.C.; Hilliker, A.J.; Phillips, J.P. Hydrogen peroxide scavenging rescues frataxin deficiency in a Drosophila model of Friedreich’s ataxia. Proc. Natl. Acad. Sci. USA 2008, 105, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Roman, G.; Endo, K.; Zong, L.; Davis, R.L. PSwitch, a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2001, 98, 12602–12607. [Google Scholar] [CrossRef] [PubMed]
- Osterwalder, T.; Yoon, K.S.; White, B.H.; Keshishian, H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc. Natl. Acad. Sci. USA 2001, 98, 12596–12601. [Google Scholar] [CrossRef] [PubMed]
- Tricoire, H.; Palandri, A.; Bourdais, A.; Camadro, J.-M.; Monnier, V. Methylene blue rescues heart defects in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2014, 23, 968–979. [Google Scholar] [CrossRef] [PubMed]
- Piccin, A.; Salameh, A.; Benna, C.; Sandrelli, F.; Mazzotta, G.; Zordan, M.; Rosato, E.; Kyriacou, C.P.; Costa, R. Efficient and heritable functional knock-out of an adult phenotype in Drosophila using a GAL4-driven hairpin RNA incorporating a heterologous spacer. Nucleic Acids Res. 2001, 29, e55. [Google Scholar] [CrossRef] [PubMed]
- Pianese, L.; Turano, M.; Lo Casale, M.S.; de Biase, I.; Giacchetti, M.; Monticelli, A.; Criscuolo, C.; Filla, A.; Cocozza, S. Real time PCR quantification of frataxin mRNA in the peripheral blood leucocytes of Friedreich ataxia patients and carriers. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1061–1063. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Botella, J.A.; Metzendorf, C.; Lind, M.I.; Schneuwly, S. Mitoferrin modulates iron toxicity in a Drosophila model of Friedreich’s ataxia. Free Radic. Biol. Med. 2015, 85, 71–82. [Google Scholar] [CrossRef] [PubMed]
- Calap-Quintana, P.; Soriano, S.; Llorens, J.V.; Al-Ramahi, I.; Botas, J.; Moltó, M.D.; Martínez-Sebastián, M.J. TORC1 Inhibition by Rapamycin Promotes Antioxidant Defences in a Drosophila Model of Friedreich’s Ataxia. PLoS ONE 2015, 10, e0132376. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Calap-Quintana, P.; Llorens, J.V.; Al-Ramahi, I.; Gutiérrez, L.; Martínez-Sebastián, M.J.; Botas, J.; Moltó, M.D. Metal Homeostasis Regulators Suppress FRDA Phenotypes in a Drosophila Model of the Disease. PLoS ONE 2016, 11, e0159209. [Google Scholar] [CrossRef] [PubMed]
- Edenharter, O.; Schneuwly, S.; Navarro, J.A. Mitofusin-Dependent ER Stress Triggers Glial Dysfunction and Nervous System Degeneration in a Drosophila Model of Friedreich’s Ataxia. Front. Mol. Neurosci. 2018, 11, 38. [Google Scholar] [CrossRef] [PubMed]
- Navarro, J.A.; Ohmann, E.; Sanchez, D.; Botella, J.A.; Liebisch, G.; Moltó, M.D.; Ganfornina, M.D.; Schmitz, G.; Schneuwly, S. Altered lipid metabolism in a Drosophila model of Friedreich’s ataxia. Hum. Mol. Genet. 2010, 19, 2828–2840. [Google Scholar] [CrossRef] [PubMed]
- Shidara, Y.; Hollenbeck, P.J. Defects in mitochondrial axonal transport and membrane potential without increased reactive oxygen species production in a Drosophila model of Friedreich ataxia. J. Neurosci. 2010, 30, 11369–11378. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Jaiswal, M.; Charng, W.-L.; Gambin, T.; Karaca, E.; Mirzaa, G.; Wiszniewski, W.; Sandoval, H.; Haelterman, N.A.; Xiong, B.; et al. A Drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 2014, 159, 200–214. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.-L.; Bridwell-Rabb, J.; Barondeau, D.P. Friedreich’s ataxia variants I154F and W155R diminish frataxin-based activation of the iron-sulfur cluster assembly complex. Biochemistry 2011, 50, 6478–6487. [Google Scholar] [CrossRef] [PubMed]
- Stowers, R.S.; Schwarz, T.L. A genetic method for generating Drosophila eyes composed exclusively of mitotic clones of a single genotype. Genetics 1999, 152, 1631–1639. [Google Scholar] [PubMed]
- Calap-Quintana, P.; Navarro, J.A.; González-Fernández, J.; Martínez-Sebastián, M.J.; Moltó, M.D.; Llorens, J.V. Drosophila melanogaster Models of Friedreich’s Ataxia. BioMed Res. Int. 2018, 2018, 5065190. [Google Scholar] [CrossRef] [PubMed]
- Cossée, M.; Puccio, H.; Gansmuller, A.; Koutnikova, H.; Dierich, A.; LeMeur, M.; Fischbeck, K.; Dollé, P.; Koenig, M. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum. Mol. Genet. 2000, 9, 1219–1226. [Google Scholar] [CrossRef] [PubMed]
- Clark, E.; Butler, J.S.; Isaacs, C.J.; Napierala, M.; Lynch, D.R. Selected missense mutations impair frataxin processing in Friedreich ataxia. Ann. Clin. Transl. Neurol. 2017, 4, 575–584. [Google Scholar] [CrossRef] [PubMed]
- Latouche, M.; Lasbleiz, C.; Martin, E.; Monnier, V.; Debeir, T.; Mouatt-Prigent, A.; Muriel, M.-P.; Morel, L.; Ruberg, M.; Brice, A.; et al. A conditional pan-neuronal Drosophila model of spinocerebellar ataxia 7 with a reversible adult phenotype suitable for identifying modifier genes. J. Neurosci. 2007, 27, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Loría, F.; Díaz-Nido, J. Frataxin knockdown in human astrocytes triggers cell death and the release of factors that cause neuronal toxicity. Neurobiol. Dis. 2015, 76, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.; Genis, L.; Navarro, J.A.; Perez-Domper, P.; Fernandez, A.M.; Schneuwly, S.; Torres Alemán, I. A role for astrocytes in cerebellar deficits in frataxin deficiency: Protection by insulin-like growth factor I. Mol. Cell. Neurosci. 2017, 80, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Simon, D.; Monassier, L.; Criqui-Filipe, P.; Gansmuller, A.; Rustin, P.; Koenig, M.; Puccio, H. Idebenone delays the onset of cardiac functional alteration without correction of Fe–S enzymes deficit in a mouse model for Friedreich ataxia. Hum. Mol. Genet. 2004, 13, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, F.; Rummey, C.; Bijnens, B.; Störk, S.; Jasaityte, R.; Dhooge, J.; Baltabaeva, A.; Sutherland, G.; Schulz, J.B.; Meier, T. The heart in Friedreich ataxia: Definition of cardiomyopathy, disease severity, and correlation with neurological symptoms. Circulation 2012, 125, 1626–1634. [Google Scholar] [CrossRef] [PubMed]
- Chandran, V.; Gao, K.; Swarup, V.; Versano, R.; Dong, H.; Jordan, M.C.; Geschwind, D.H. Inducible and reversible phenotypes in a novel mouse model of Friedreich’s Ataxia. eLife 2017, 6, e30054. [Google Scholar] [CrossRef] [PubMed]
- Beauchamp, M.; Labelle, H.; Duhaime, M.; Joncas, J. Natural history of muscle weakness in Friedreich’s Ataxia and its relation to loss of ambulation. Clin. Orthop. Relat. Res. 1995, 311, 270–275. [Google Scholar]
- Lynch, D.R.; Lech, G.; Farmer, J.M.; Balcer, L.J.; Bank, W.; Chance, B.; Wilson, R.B. Near infrared muscle spectroscopy in patients with Friedreich’s ataxia. Muscle Nerve 2002, 25, 664–673. [Google Scholar] [CrossRef] [PubMed]
- Nachbauer, W.; Boesch, S.; Schneider, R.; Eigentler, A.; Wanschitz, J.; Poewe, W.; Schocke, M. Bioenergetics of the calf muscle in Friedreich ataxia patients measured by 31P-MRS before and after treatment with recombinant human erythropoietin. PLoS ONE 2013, 8, e69229. [Google Scholar] [CrossRef] [PubMed]
- Vorgerd, M.; Schöls, L.; Hardt, C.; Ristow, M.; Epplen, J.T.; Zange, J. Mitochondrial impairment of human muscle in Friedreich ataxia in vivo. Neuromuscul. Disord. NMD 2000, 10, 430–435. [Google Scholar] [CrossRef]
- Palandri, A.; L’hôte, D.; Cohen-Tannoudji, J.; Tricoire, H.; Monnier, V. Frataxin inactivation leads to steroid deficiency in flies and human ovarian cells. Hum. Mol. Genet. 2015, 24, 2615–2626. [Google Scholar] [CrossRef] [PubMed]
- Min, K.T.; Benzer, S. Preventing neurodegeneration in the Drosophila mutant bubblegum. Science 1999, 284, 1985–1988. [Google Scholar] [CrossRef] [PubMed]
- Van der Voet, M.; Harich, B.; Franke, B.; Schenck, A. ADHD-associated dopamine transporter, latrophilin and neurofibromin share a dopamine-related locomotor signature in Drosophila. Mol. Psychiatry 2016, 21, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Lawal, H.O.; Terrell, A.; Lam, H.A.; Djapri, C.; Jang, J.; Hadi, R.; Roberts, L.; Shahi, V.; Chou, M.-T.; Biedermann, T.; et al. Drosophila modifier screens to identify novel neuropsychiatric drugs including aminergic agents for the possible treatment of Parkinson’s disease and depression. Mol. Psychiatry 2014, 19, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Qurashi, A.; Liu, H.; Ray, L.; Nelson, D.L.; Duan, R.; Jin, P. Chemical screen reveals small molecules suppressing fragile X premutation rCGG repeat-mediated neurodegeneration in Drosophila. Hum. Mol. Genet. 2012, 21, 2068–2075. [Google Scholar] [CrossRef] [PubMed]
- Marelja, Z.; Leimkühler, S.; Missirlis, F. Iron Sulfur and Molybdenum Cofactor Enzymes Regulate the Drosophila Life Cycle by Controlling Cell Metabolism. Front. Physiol. 2018, 9, 50. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Casis, G.; Cote, M.; Barbeau, A. Pathology of the heart in Friedreich’s ataxia: Review of the literature and report of one case. Can. J. Neurol. Sci. 1976, 3, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Lamarche, J.B.; Côté, M.; Lemieux, B. The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 1980, 7, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Michael, S.C.; Knutson, M.D.; Haile, D.J.; Qian, J.; Levi, S.; Santambrogio, P.; Garrick, M.D.; Lamarche, J.B. The dentate nucleus in Friedreich’s ataxia: The role of iron-responsive proteins. Acta Neuropathol. 2007, 114, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Morral, J.A.; Davis, A.N.; Qian, J.; Petrocine, S.V.; Knutson, M.D.; Gibson, W.M.; Cusack, M.J.; Li, D. The dorsal root ganglion in Friedreich’s ataxia. Acta Neuropathol. 2009, 118, 763–776. [Google Scholar] [CrossRef] [PubMed]
- Boddaert, N.; Le Quan Sang, K.H.; Rötig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.-C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Harding, I.H.; Raniga, P.; Delatycki, M.B.; Stagnitti, M.R.; Corben, L.A.; Storey, E.; Georgiou-Karistianis, N.; Egan, G.F. Tissue atrophy and elevated iron concentration in the extrapyramidal motor system in Friedreich ataxia: The IMAGE-FRDA study. J. Neurol. Neurosurg. Psychiatry 2016, 87, 1261–1263. [Google Scholar] [CrossRef] [PubMed]
- Soriano, S.; Llorens, J.V.; Blanco-Sobero, L.; Gutiérrez, L.; Calap-Quintana, P.; Morales, M.P.; Moltó, M.D.; Martínez-Sebastián, M.J. Deferiprone and idebenone rescue frataxin depletion phenotypes in a Drosophila model of Friedreich’s ataxia. Gene 2013, 521, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.; Seznec, H.; Gansmuller, A.; Carelle, N.; Weber, P.; Metzger, D.; Rustin, P.; Koenig, M.; Puccio, H. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 2004, 24, 1987–1995. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahdawi, S.; Pinto, R.M.; Varshney, D.; Lawrence, L.; Lowrie, M.B.; Hughes, S.; Webster, Z.; Blake, J.; Cooper, J.M.; King, R.; et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006, 88, 580–590. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Ho, T.S.-Y.; Lin, G.; Tan, K.L.; Rasband, M.N.; Bellen, H.J. Loss of Frataxin activates the iron/sphingolipid/PDK1/Mef2 pathway in mammals. eLife 2016, 5, e20732. [Google Scholar] [CrossRef] [PubMed]
- Wong, A.; Yang, J.; Cavadini, P.; Gellera, C.; Lonnerdal, B.; Taroni, F.; Cortopassi, G. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum. Mol. Genet. 1999, 8, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Chantrel-Groussard, K.; Geromel, V.; Puccio, H.; Koenig, M.; Munnich, A.; Rötig, A.; Rustin, P. Disabled early recruitment of antioxidant defenses in Friedreich’s ataxia. Hum. Mol. Genet. 2001, 10, 2061–2067. [Google Scholar] [CrossRef] [PubMed]
- Whitnall, M.; Suryo Rahmanto, Y.; Huang, M.L.-H.; Saletta, F.; Lok, H.C.; Gutiérrez, L.; Lázaro, F.J.; Fleming, A.J.; St Pierre, T.G.; Mikhael, M.R.; et al. Identification of nonferritin mitochondrial iron deposits in a mouse model of Friedreich ataxia. Proc. Natl. Acad. Sci. USA 2012, 109, 20590–20595. [Google Scholar] [CrossRef] [PubMed]
- Mandilaras, K.; Pathmanathan, T.; Missirlis, F. Iron absorption in Drosophila melanogaster. Nutrients 2013, 5, 1622–1647. [Google Scholar] [CrossRef] [PubMed]
- Calap-Quintana, P.; González-Fernández, J.; Sebastiá-Ortega, N.; Llorens, J.V.; Moltó, M.D. Drosophila melanogaster Models of Metal-Related Human Diseases and Metal Toxicity. Int. J. Mol. Sci. 2017, 18, 1456. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Schmucker, S.; Reutenauer, L.; Mathieu, J.R.R.; Peyssonnaux, C.; Karim, Z.; Puy, H.; Galy, B.; Hentze, M.W.; Puccio, H. Iron regulatory protein 1 sustains mitochondrial iron loading and function in frataxin deficiency. Cell Metab. 2015, 21, 311–323. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Maglioni, S.; Palikaras, K.; Shaik, A.; Strappazzon, F.; Brinkmann, V.; Torgovnick, A.; Castelein, N.; Henau, S. de; Braeckman, B.P.; et al. Iron-Starvation-Induced Mitophagy Mediates Lifespan Extension upon Mitochondrial Stress in C. elegans. Curr. Biol. CB 2015, 25, 1810–1822. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.-H.; Becker, E.M.; Whitnall, M.; Suryo Rahmanto, Y.; Ponka, P.; des Richardson, R. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc. Natl. Acad. Sci. USA 2009, 106, 16381–16386. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, R.L.; Qian, J.; Santambrogio, P.; Levi, S.; Koeppen, A.H. Relation of cytosolic iron excess to cardiomyopathy of Friedreich’s ataxia. Am. J. Cardiol. 2012, 110, 1820–1827. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Becker, A.B.; Bjork, S.T.; Levi, S.; Santambrogio, P.; Parsons, P.J.; Kruger, P.C.; Yang, K.X.; Feustel, P.J.; et al. The pathogenesis of cardiomyopathy in Friedreich ataxia. PLoS ONE 2015, 10, e0116396. [Google Scholar] [CrossRef] [PubMed]
- Missirlis, F.; Hu, J.; Kirby, K.; Hilliker, A.J.; Rouault, T.A.; Phillips, J.P. Compartment-specific protection of iron-sulfur proteins by superoxide dismutase. J. Biol. Chem. 2003, 278, 47365–47369. [Google Scholar] [CrossRef] [PubMed]
- Irazusta, V.; Obis, E.; Moreno-Cermeño, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Yeast frataxin mutants display decreased superoxide dismutase activity crucial to promote protein oxidative damage. Free Radic. Biol. Med. 2010, 48, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Barzilai, A.; Yamamoto, K.-I. DNA damage responses to oxidative stress. DNA Repair 2004, 3, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Berlett, B.S.; Stadtman, E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997, 272, 20313–20316. [Google Scholar] [CrossRef] [PubMed]
- Ayala, A.; Muñoz, M.F.; Argüelles, S. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid. Med. Cell. Longev. 2014, 2014, 360438. [Google Scholar] [CrossRef] [PubMed]
- González-Cabo, P.; Palau, F. Mitochondrial pathophysiology in Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 53–64. [Google Scholar] [CrossRef] [PubMed]
- Vaubel, R.A.; Isaya, G. Iron-sulfur cluster synthesis, iron homeostasis and oxidative stress in Friedreich ataxia. Mol. Cell. Neurosci. 2013, 55, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Isaya, G.; O’Neill, H.A.; Gakh, O.; Park, S.; Mantcheva, R.; Mooney, S.M. Functional studies of frataxin. Acta Paediatr. 2004, 93, 68–71. [Google Scholar] [CrossRef]
- Paupe, V.; Dassa, E.P.; Goncalves, S.; Auchère, F.; Lönn, M.; Holmgren, A.; Rustin, P. Impaired nuclear Nrf2 translocation undermines the oxidative stress response in Friedreich ataxia. PLoS ONE 2009, 4, e4253. [Google Scholar] [CrossRef] [PubMed]
- Floyd, R.A.; Kotake, Y.; Hensley, K.; Nakae, D.; Konishi, Y. Reactive oxygen species in choline deficiency induced carcinogenesis and nitrone inhibition. Mol. Cell. Biochem. 2002, 234–235, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Lupoli, F.; Vannocci, T.; Longo, G.; Niccolai, N.; Pastore, A. The role of oxidative stress in Friedreich’s ataxia. FEBS Lett. 2018, 592, 718–727. [Google Scholar] [CrossRef] [PubMed]
- Irazusta, V.; Moreno-Cermeño, A.; Cabiscol, E.; Ros, J.; Tamarit, J. Major targets of iron-induced protein oxidative damage in frataxin-deficient yeasts are magnesium-binding proteins. Free Radic. Biol. Med. 2008, 44, 1712–1723. [Google Scholar] [CrossRef] [PubMed]
- Babcock, M.; de Silva, D.; Oaks, R.; Davis-Kaplan, S.; Jiralerspong, S.; Montermini, L.; Pandolfo, M.; Kaplan, J. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science 1997, 276, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Auchère, F.; Santos, R.; Planamente, S.; Lesuisse, E.; Camadro, J.-M. Glutathione-dependent redox status of frataxin-deficient cells in a yeast model of Friedreich’s ataxia. Hum. Mol. Genet. 2008, 17, 2790–2802. [Google Scholar] [CrossRef] [PubMed]
- Radisky, D.C.; Babcock, M.C.; Kaplan, J. The yeast frataxin homologue mediates mitochondrial iron efflux. Evidence for a mitochondrial iron cycle. J. Biol. Chem. 1999, 274, 4497–4499. [Google Scholar] [CrossRef] [PubMed]
- Bulteau, A.L.; Planamente, S.; Jornea, L.; Dur, A.; Lesuisse, E.; Camadro, J.M.; Auchère, F. Changes in mitochondrial glutathione levels and protein thiol oxidation in ∆yfh1 yeast cells and the lymphoblasts of patients with Friedreich’s ataxia. Biochim. Biophys. Acta 2012, 1822, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Vázquez-Manrique, R.P.; González-Cabo, P.; Ros, S.; Aziz, H.; Baylis, H.A.; Palau, F. Reduction of Caenorhabditis elegans frataxin increases sensitivity to oxidative stress, reduces lifespan, and causes lethality in a mitochondrial complex II mutant. FASEB J. 2006, 20, 172–174. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Chen, L.S.; Lonnerdal, B.; Gellera, C.; Taroni, F.A.; Cortopassi, G.A. Frataxin expression rescues mitochondrial dysfunctions in FRDA cells. Hum. Mol. Genet. 2001, 10, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Piemonte, F.; Pastore, A.; Tozzi, G.; Tagliacozzi, D.; Santorelli, F.M.; Carrozzo, R.; Casali, C.; Damiano, M.; Federici, G.; Bertini, E. Glutathione in blood of patients with Friedreich’s ataxia. Eur. J. Clin. Investig. 2001, 31, 1007–1011. [Google Scholar] [CrossRef]
- Schulz, J.B.; Dehmer, T.; Schöls, L.; Mende, H.; Hardt, C.; Vorgerd, M.; Bürk, K.; Matson, W.; Dichgans, J.; Beal, M.F.; et al. Oxidative stress in patients with Friedreich ataxia. Neurol. 2000, 55, 1719–1721. [Google Scholar] [CrossRef]
- Haugen, A.C.; Di Prospero, N.A.; Parker, J.S.; Fannin, R.D.; Chou, J.; Meyer, J.N.; Halweg, C.; Collins, J.B.; Durr, A.; Fischbeck, K.; et al. Altered gene expression and DNA damage in peripheral blood cells from Friedreich’s ataxia patients: Cellular model of pathology. PLoS Genet. 2010, 6, e1000812. [Google Scholar] [CrossRef] [PubMed]
- Johnson, W.M.; Wilson-Delfosse, A.L.; Mieyal, J.J. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients 2012, 4, 1399–1440. [Google Scholar] [CrossRef] [PubMed]
- Swarup, V.; Srivastava, A.K.; Padma, M.V.; Rajeswari, M.R. Quantitative profiling and identification of differentially expressed plasma proteins in Friedreich’s ataxia. J. Neurosci. Res. 2013, 91, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Seznec, H.; Simon, D.; Bouton, C.; Reutenauer, L.; Hertzog, A.; Golik, P.; Procaccio, V.; Patel, M.; Drapier, J.-C.; Koenig, M.; et al. Friedreich ataxia: The oxidative stress paradox. Hum. Mol. Genet. 2005, 14, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Macevilly, C.J.; Muller, D.P. Oxidative stress does not appear to be involved in the aetiology of Friedreich’s ataxia. Restor. Neurol. Neurosci. 1997, 11, 131–137. [Google Scholar] [PubMed]
- Lynch, D.R.; Perlman, S.L.; Meier, T. A phase 3, double-blind, placebo-controlled trial of idebenone in friedreich ataxia. Arch. Neurol. 2010, 67, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.H.; Schulz, J.B.; Giunti, P. Co-enzyme Q10 and idebenone use in Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl. 1), 125–141. [Google Scholar] [CrossRef] [PubMed]
- Pryde, K.R.; Taanman, J.W.; Schapira, A.H. A LON-ClpP Proteolytic Axis Degrades Complex I to Extinguish ROS Production in Depolarized Mitochondria. Cell Rep. 2016, 17, 2522–2531. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F.; Boveris, A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem. J. 1980, 191, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, K.; Nakano, M.; Totsune-Nakano, H.; Minakami, H.; Tero-Kubota, S.; Ikegami, Y. Mechanism of O−2 generation in reduction and oxidation cycle of ubiquinones in a model of mitochondrial electron transport systems. Biochim. Biophys. Acta Bioenerget. 1988, 936, 377–385. [Google Scholar] [CrossRef]
- Detienne, G.; de Haes, W.; Mergan, L.; Edwards, S.L.; Temmerman, L.; van Bael, S. Beyond ROS clearance: Peroxiredoxins in stress signaling and aging. Ageing Res. Rev. 2018, 44, 33–48. [Google Scholar] [CrossRef] [PubMed]
- Cotticelli, M.G.; Xia, S.; Kaur, A.; Lin, D.; Wang, Y.; Ruff, E.; Tobias, J.W.; Wilson, R.B. Identification of p38 MAPK as a novel therapeutic target for Friedreich’s ataxia. Sci. Rep. 2018, 8, 5007. [Google Scholar] [CrossRef] [PubMed]
- Hanson, G.T.; Aggeler, R.; Oglesbee, D.; Cannon, M.; Capaldi, R.A.; Tsien, R.Y.; Remington, S.J. Investigating mitochondrial redox potential with redox-sensitive green fluorescent protein indicators. J. Biol. Chem. 2004, 279, 13044–13053. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.C.; Barata, A.G.; Grosshans, J.; Teleman, A.A.; Dick, T.P. In vivo mapping of hydrogen peroxide and oxidized glutathione reveals chemical and regional specificity of redox homeostasis. Cell Metab. 2011, 14, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Kaplowitz, N. The importance and regulation of hepatic glutathione. Yale J. Biol. Med. 1981, 54, 497–502. [Google Scholar] [PubMed]
- Calatrava-Ferreras, L.; Gonzalo-Gobernado, R.; Reimers, D.; Herranz, A.S.; Casarejos, M.J.; Jiménez-Escrig, A.; Regadera, J.; Velasco-Martín, J.; Vallejo-Muñoz, M.; Díaz-Gil, J.J.; et al. Liver Growth Factor (LGF) Upregulates Frataxin Protein Expression and Reduces Oxidative Stress in Friedreich’s Ataxia Transgenic Mice. Int. J. Mol. Sci. 2016, 17, 2066. [Google Scholar] [CrossRef] [PubMed]
- Piermarini, E.; Cartelli, D.; Pastore, A.; Tozzi, G.; Compagnucci, C.; Giorda, E.; D’Amico, J.; Petrini, S.; Bertini, E.; Cappelletti, G.; et al. Frataxin silencing alters microtubule stability in motor neurons: Implications for Friedreich’s ataxia. Hum. Mol. Genet. 2016, 25, 4288–4301. [Google Scholar] [CrossRef] [PubMed]
- Tan, G.; Napoli, E.; Taroni, F.; Cortopassi, G. Decreased expression of genes involved in sulfur amino acid metabolism in frataxin-deficient cells. Hum. Mol. Genet. 2003, 12, 1699–1711. [Google Scholar] [CrossRef] [PubMed]
- Shan, Y.; Schoenfeld, R.A.; Hayashi, G.; Napoli, E.; Akiyama, T.; Iodi Carstens, M.; Carstens, E.E.; Pook, M.A.; Cortopassi, G.A. Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid. Redox Signal. 2013, 19, 1481–1493. [Google Scholar] [CrossRef] [PubMed]
- Runko, A.P.; Griswold, A.J.; Min, K.-T. Overexpression of frataxin in the mitochondria increases resistance to oxidative stress and extends lifespan in Drosophila. FEBS Lett. 2008, 582, 715–719. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Kuntzsch, E.C.; Bjork, S.T.; Ramirez, R.L.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich ataxia: Metal dysmetabolism in dorsal root ganglia. Acta Neuropathol. Commun. 2013, 1, 26. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Ramirez, R.L.; Yu, D.; Collins, S.E.; Qian, J.; Parsons, P.J.; Yang, K.X.; Chen, Z.; Mazurkiewicz, J.E.; Feustel, P.J. Friedreich’s ataxia causes redistribution of iron, copper, and zinc in the dentate nucleus. Cerebellum 2012, 11, 845–860. [Google Scholar] [CrossRef] [PubMed]
- Kruger, P.C.; Yang, K.X.; Parsons, P.J.; Becker, A.B.; Feustel, P.J.; Koeppen, A.H. Abundance and Significance of Iron, Zinc, Copper, and Calcium in the Hearts of Patients with Friedreich Ataxia. Am. J. Cardiol. 2016, 118, 127–131. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Du, Y.; Xue, H.; Wu, Y.; Zhou, B. Aluminum induces neurodegeneration and its toxicity arises from increased iron accumulation and reactive oxygen species (ROS) production. Neurobiol. Aging 2012, 33, 199.e1–199.e12. [Google Scholar] [CrossRef] [PubMed]
- Han, T.H.L.; Camadro, J.M.; Santos, R.; Lesuisse, E.; El Hage Chahine, J.M.; Ha-Duong, N.T. Mechanisms of iron and copper-frataxin interactions. Metall. Integr. Biomet. Sci. 2017, 9, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Sánchez, M.; Palacios, Ò.; Buchensky, C.; Sabio, L.; Gomez-Casati, D.F.; Pagani, M.A.; Capdevila, M.; Atrian, S.; Dominguez-Vera, J.M. Copper redox chemistry of plant frataxins. J. Inorg. Biochem. 2018, 180, 135–140. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Günther, V.; Georgiev, O.; Schaffner, W. Distorted copper homeostasis with decreased sensitivity to cisplatin upon chaperone Atox1 deletion in Drosophila. Biometals 2011, 24, 445–453. [Google Scholar] [CrossRef] [PubMed]
- Xiao, G.; Wan, Z.; Fan, Q.; Tang, X.; Zhou, B. The metal transporter ZIP13 supplies iron into the secretory pathway in Drosophila melanogaster. eLife 2014, 3, e03191. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.-Y.; Jenkitkasemwong, S.; Duarte, S.; Sparkman, B.K.; Shawki, A.; Mackenzie, B.; Knutson, M.D. ZIP8 is an iron and zinc transporter whose cell-surface expression is up-regulated by cellular iron loading. J. Biol. Chem. 2012, 287, 34032–34043. [Google Scholar] [CrossRef] [PubMed]
- Jung, W.-H.; Liu, C.-C.; Yu, Y.-L.; Chang, Y.-C.; Lien, W.-Y.; Chao, H.-C.; Huang, S.-Y.; Kuo, C.-H.; Ho, H.-C.; Chan, C.-C. Lipophagy prevents activity-dependent neurodegeneration due to dihydroceramide accumulation in vivo. EMBO Rep. 2017, 18, 1150–1165. [Google Scholar] [CrossRef] [PubMed]
- Cabirol-Pol, M.-J.; Khalil, B.; Rival, T.; Faivre-Sarrailh, C.; Besson, M.T. Glial lipid droplets and neurodegeneration in a Drosophila model of complex I deficiency. Glia 2018, 66, 874–888. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Uzun, E.; Renganathan, I.; Honda, T.; Pook, M.A.; Giunti, P. Targeting lipid peroxidation and mitochondrial imbalance in Friedreich’s ataxia. Pharmacol. Res. 2015, 99, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Abeti, R.; Parkinson, M.H.; Hargreaves, I.P.; Angelova, P.R.; Sandi, C.; Pook, M.A.; Giunti, P.; Abramov, A.Y. Mitochondrial energy imbalance and lipid peroxidation cause cell death in Friedreich’s ataxia. Cell Death Dis. 2016, 7, e2237. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Burnett, R.; Perlman, S.; Versano, R.; Gao, F.; Plasterer, H.; Rai, M.; Saccá, F.; Filla, A.; Lynch, D.R.; et al. A gene expression phenotype in lymphocytes from Friedreich ataxia patients. Ann. Neurol. 2011, 70, 790–804. [Google Scholar] [CrossRef] [PubMed]
- Coppola, G.; Marmolino, D.; Lu, D.; Wang, Q.; Cnop, M.; Rai, M.; Acquaviva, F.; Cocozza, S.; Pandolfo, M.; Geschwind, D.H. Functional genomic analysis of frataxin deficiency reveals tissue-specific alterations and identifies the PPARgamma pathway as a therapeutic target in Friedreich’s ataxia. Hum. Mol. Genet. 2009, 18, 2452–2461. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Huang, P.; Wu, J.; Song, H. Drosophila protein phosphatase V regulates lipid homeostasis via the AMPK pathway. J. Mol. Cell Biol. 2014, 6, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.L.; Chamberlain, S.; Robinson, N. Lipids and lipoproteins in Friedreich’s ataxia. J. Neurol. Neurosurg. Psychiatry 1980, 43, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Raman, S.V.; Phatak, K.; Hoyle, J.C.; Pennell, M.L.; McCarthy, B.; Tran, T.; Prior, T.W.; Olesik, J.W.; Lutton, A.; Rankin, C.; et al. Impaired myocardial perfusion reserve and fibrosis in Friedreich ataxia: A mitochondrial cardiomyopathy with metabolic syndrome. Eur. Heart J. 2011, 32, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.S.; Nestruck, A.C.; Barbeau, A.; Bouchard, J.P.; Davignon, J. Plasma lipids and lipoproteins in Friedreich’s ataxia and familial spastic ataxia—Evidence for an abnormal composition of high density lipoproteins. Can. J. Neurol. Sci. 1978, 5, 149–156. [Google Scholar] [PubMed]
- Filla, A.; Postiglione, A.; Rubba, P.; Patti, L.; de Michele, G.; Palma, V.; Brescia Morra, V.; Campanella, G. Plasma lipoprotein concentration and erythrocyte membrane lipids in patients with Friedreich’s ataxia. Acta Neurol. 1980, 2, 382–389. [Google Scholar]
- Draper, P.; Huang, Y.S.; Shapcott, D.; Lemieux, B.; Brennan, M.; Barbeau, A.; Davignon, J. Erythrocyte membrane lipids in Friedreich’s ataxia. Can. J. Neurol. Sci. 1979, 6, 291–294. [Google Scholar] [CrossRef] [PubMed]
- Worth, A.J.; Basu, S.S.; Deutsch, E.C.; Hwang, W.-T.; Snyder, N.W.; Lynch, D.R.; Blair, I.A. Stable isotopes and LC-MS for monitoring metabolic disturbances in Friedreich’s ataxia platelets. Bioanalysis 2015, 7, 1843–1855. [Google Scholar] [CrossRef] [PubMed]
- Obis, È.; Irazusta, V.; Sanchís, D.; Ros, J.; Tamarit, J. Frataxin deficiency in neonatal rat ventricular myocytes targets mitochondria and lipid metabolism. Free Radic. Biol. Med. 2014, 73, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Martelli, A.; Friedman, L.S.; Reutenauer, L.; Messaddeq, N.; Perlman, S.L.; Lynch, D.R.; Fedosov, K.; Schulz, J.B.; Pandolfo, M.; Puccio, H. Clinical data and characterization of the liver conditional mouse model exclude neoplasia as a non-neurological manifestation associated with Friedreich’s ataxia. Dis. Models Mech. 2012, 5, 860–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Lin, H.; Xu, Y.; Zhou, F.; Wang, J.; Liu, J.; Zhu, X.; Guo, X.; Tang, Y.; Yao, P. Frataxin-Mediated PINK1-Parkin-Dependent Mitophagy in Hepatic Steatosis: The Protective Effects of Quercetin. Mol. Nutr. Food Res. 2018, e1800164. [Google Scholar] [CrossRef] [PubMed]
- Schiavi, A.; Torgovnick, A.; Kell, A.; Megalou, E.; Castelein, N.; Guccini, I.; Marzocchella, L.; Gelino, S.; Hansen, M.; Malisan, F.; et al. Autophagy induction extends lifespan and reduces lipid content in response to frataxin silencing in C. elegans. Exp. Gerontol. 2013, 48, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Walls, S.M.; Cammarato, A.; Chatfield, D.A.; Ocorr, K.; Harris, G.L.; Bodmer, R. Ceramide-Protein Interactions Modulate Ceramide-Associated Lipotoxic Cardiomyopathy. Cell Rep. 2018, 22, 2702–2715. [Google Scholar] [CrossRef] [PubMed]
- Schulz, J.G.; Laranjeira, A.; van Huffel, L.; Gärtner, A.; Vilain, S.; Bastianen, J.; van Veldhoven, P.P.; Dotti, C.G. Glial β-oxidation regulates Drosophila energy metabolism. Sci. Rep. 2015, 5, 7805. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, K.; Sandoval, H.; Yamamoto, S.; Jaiswal, M.; Sanz, E.; Li, Z.; Hui, J.; Graham, B.H.; Quintana, A.; et al. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell 2015, 160, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; MacKenzie, K.R.; Putluri, N.; Maletić-Savatić, M.; Bellen, H.J. The Glia-Neuron Lactate Shuttle and Elevated ROS Promote Lipid Synthesis in Neurons and Lipid Droplet Accumulation in Glia via APOE/D. Cell Metab. 2017, 26, 719–737.e6. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.-J.; Huang, X.; Kropat, J.; Henras, A.; Merchant, S.S.; Dickson, R.C.; Chanfreau, G.F. Sphingolipid signaling mediates iron toxicity. Cell Metab. 2012, 16, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Hebbar, S.; Sahoo, I.; Matysik, A.; Argudo Garcia, I.; Osborne, K.A.; Papan, C.; Torta, F.; Narayanaswamy, P.; Fun, X.H.; Wenk, M.R.; et al. Ceramides and Stress Signalling Intersect with Autophagic Defects in Neurodegenerative Drosophila blue cheese (bchs) Mutants. Sci. Rep. 2015, 5, 15926. [Google Scholar] [CrossRef] [PubMed]
- Acharya, U.; Patel, S.; Koundakjian, E.; Nagashima, K.; Han, X.; Acharya, J.K. Modulating sphingolipid biosynthetic pathway rescues photoreceptor degeneration. Science 2003, 299, 1740–1743. [Google Scholar] [CrossRef] [PubMed]
- Kraut, R. Roles of sphingolipids in Drosophila development and disease. J. Neurochem. 2011, 116, 764–778. [Google Scholar] [CrossRef] [PubMed]
- West, R.J.H.; Briggs, L.; Perona Fjeldstad, M.; Ribchester, R.R.; Sweeney, S.T. Sphingolipids Regulate Neuromuscular Synapse Structure and function in Drosophila. J. Comp. Neurol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Tamarit, J.; Obis, È.; Ros, J. Oxidative stress and altered lipid metabolism in Friedreich ataxia. Free Radic. Biol. Med. 2016, 100, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Cortopassi, G.; Danielson, S.; Alemi, M.; Zhan, S.S.; Tong, W.; Carelli, V.; Martinuzzi, A.; Marzuki, S.; Majamaa, K.; Wong, A. Mitochondrial disease activates transcripts of the unfolded protein response and cell cycle and inhibits vesicular secretion and oligodendrocyte-specific transcripts. Mitochondrion 2006, 6, 161–175. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Cortopassi, G. Frataxin knockdown causes loss of cytoplasmic iron-sulfur cluster functions, redox alterations and induction of heme transcripts. Arch. Biochem. Biophys. 2007, 457, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.L.-H.; Sivagurunathan, S.; Ting, S.; Jansson, P.J.; Austin, C.J.D.; Kelly, M.; Semsarian, C.; Zhang, D.; Des Richardson, R. Molecular and functional alterations in a mouse cardiac model of Friedreich ataxia: Activation of the integrated stress response, eIF2α phosphorylation, and the induction of downstream targets. Am. J. Pathol. 2013, 183, 745–757. [Google Scholar] [CrossRef] [PubMed]
- Bolinches-Amorós, A.; Mollá, B.; Pla-Martín, D.; Palau, F.; González-Cabo, P. Mitochondrial dysfunction induced by frataxin deficiency is associated with cellular senescence and abnormal calcium metabolism. Front. Cell. Neurosci. 2014, 8, 124. [Google Scholar] [PubMed]
- Zhang, W.; Hietakangas, V.; Wee, S.; Lim, S.C.; Gunaratne, J.; Cohen, S.M. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev. 2013, 27, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Oney, M.; Frizzell, K.; Phadnis, N.; Hollien, J. Drosophila melanogaster activating transcription factor 4 regulates glycolysis during endoplasmic reticulum stress. G3 2015, 5, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.-J.; Vasudevan, D.; Kang, K.; Kim, K.; Park, J.-E.; Zhang, N.; Zeng, X.; Neubert, T.A.; Marr, M.T.; Ryoo, H.D. 4E-BP is a target of the GCN2-ATF4 pathway during Drosophila development and aging. J. Cell Biol. 2017, 216, 115–129. [Google Scholar] [CrossRef] [PubMed]
- Tsuyama, T.; Tsubouchi, A.; Usui, T.; Imamura, H.; Uemura, T. Mitochondrial dysfunction induces dendritic loss via eIF2α phosphorylation. J. Cell Biol. 2017, 216, 815–834. [Google Scholar] [CrossRef] [PubMed]
- Hetz, C.; Mollereau, B. Disturbance of endoplasmic reticulum proteostasis in neurodegenerative diseases. Nat. Rev. Neurosci. 2014, 15, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.Y.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [PubMed]
- Casas-Tinto, S.; Zhang, Y.; Sanchez-Garcia, J.; Gomez-Velazquez, M.; Rincon-Limas, D.E.; Fernandez-Funez, P. The ER stress factor XBP1s prevents amyloid-beta neurotoxicity. Hum. Mol. Genet. 2011, 20, 2144–2160. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Shimoda, M.; Ito, K.; Hanai, S.; Aizawa, H.; Kato, T.; Kawasaki, K.; Yamaguchi, T.; Ryoo, H.D.; Goto-Inoue, N.; et al. Expression of human Gaucher disease gene GBA generates neurodevelopmental defects and ER stress in Drosophila eye. PLoS ONE 2013, 8, e69147. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Martinez, A.; Beavan, M.; Gegg, M.E.; Chau, K.-Y.; Whitworth, A.J.; Schapira, A.H.V. Parkinson disease-linked GBA mutation effects reversed by molecular chaperones in human cell and fly models. Sci. Rep. 2016, 6, 31380. [Google Scholar] [CrossRef] [PubMed]
- Purroy, R.; Britti, E.; Delaspre, F.; Tamarit, J.; Ros, J. Mitochondrial pore opening and loss of Ca2+ exchanger NCLX levels occur after frataxin depletion. Biochim. Biophys. Acta 2018, 1864, 618–631. [Google Scholar] [CrossRef] [PubMed]
- Mincheva-Tasheva, S.; Obis, E.; Tamarit, J.; Ros, J. Apoptotic cell death and altered calcium homeostasis caused by frataxin depletion in dorsal root ganglia neurons can be prevented by BH4 domain of Bcl-xL protein. Hum. Mol. Genet. 2014, 23, 1829–1841. [Google Scholar] [CrossRef] [PubMed]
- Mollá, B.; Muñoz-Lasso, D.C.; Riveiro, F.; Bolinches-Amorós, A.; Pallardó, F.V.; Fernandez-Vilata, A.; La Iglesia-Vaya, M. de; Palau, F.; Gonzalez-Cabo, P. Reversible Axonal Dystrophy by Calcium Modulation in Frataxin-Deficient Sensory Neurons of YG8R Mice. Front. Mol. Neurosci. 2017, 10, 264. [Google Scholar] [CrossRef] [PubMed]
- Crombie, D.E.; Curl, C.L.; Raaijmakers, A.J.; Sivakumaran, P.; Kulkarni, T.; Wong, R.C.; Minami, I.; Evans-Galea, M.V.; Lim, S.Y.; Delbridge, L.; et al. Friedreich’s ataxia induced pluripotent stem cell-derived cardiomyocytes display electrophysiological abnormalities and calcium handling deficiency. Aging 2017, 9, 1440–1452. [Google Scholar] [CrossRef] [PubMed]
- Chorna, T.; Hasan, G. The genetics of calcium signaling in Drosophila melanogaster. Biochim. Biophys. Acta 2012, 1820, 1269–1282. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Quan, X.; Bang, S.; Yoo, H.; Kim, J.; Park, J.; Park, K.-S.; Chung, J. Mitochondrial calcium uniporter in Drosophila transfers calcium between the endoplasmic reticulum and mitochondria in oxidative stress-induced cell death. J. Biol. Chem. 2017, 292, 14473–14485. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Lee, K.-S.; Huh, S.; Liu, S.; Lee, D.-Y.; Hong, S.H.; Yu, K.; Lu, B. Polo Kinase Phosphorylates Miro to Control ER-Mitochondria Contact Sites and Mitochondrial Ca2+ Homeostasis in Neural Stem Cell Development. Dev. Cell 2016, 37, 174–189. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.-C.; Tandarich, L.C.; Hollenbeck, P.J. ROS regulation of axonal mitochondrial transport is mediated by Ca2+ and JNK in Drosophila. PLoS ONE 2017, 12, e0178105. [Google Scholar] [CrossRef] [PubMed]
- Krols, M.; van Isterdael, G.; Asselbergh, B.; Kremer, A.; Lippens, S.; Timmerman, V.; Janssens, S. Mitochondria-associated membranes as hubs for neurodegeneration. Acta Neuropathol. 2016, 131, 505–523. [Google Scholar] [CrossRef] [PubMed]
- Lefevre, S.; Sliwa, D.; Rustin, P.; Camadro, J.-M.; Santos, R. Oxidative stress induces mitochondrial fragmentation in frataxin-deficient cells. Biochem. Biophys. Res. Commun. 2012, 418, 336–341. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, M.J.; Abeliovich, H.; Acevedo Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Lavista-Llanos, S.; Centanin, L.; Irisarri, M.; Russo, D.M.; Gleadle, J.M.; Bocca, S.N.; Muzzopappa, M.; Ratcliffe, P.J.; Wappner, P. Control of the Hypoxic Response in Drosophila melanogaster by the Basic Helix-Loop-Helix PAS Protein Similar. Mol. Cell. Biol. 2002, 22, 6842–6853. [Google Scholar] [CrossRef] [PubMed]
- Oktay, Y.; Dioum, E.; Matsuzaki, S.; Ding, K.; Yan, L.-J.; Haller, R.G.; Szweda, L.I.; Garcia, J.A. Hypoxia-inducible factor 2alpha regulates expression of the mitochondrial aconitase chaperone protein frataxin. J. Biol. Chem. 2007, 282, 11750–11756. [Google Scholar] [CrossRef] [PubMed]
- Strawser, C.; Schadt, K.; Hauser, L.; McCormick, A.; Wells, M.; Larkindale, J.; Lin, H.; Lynch, D.R. Pharmacological therapeutics in Friedreich ataxia: The present state. Expert Rev. Neurother. 2017, 17, 895–907. [Google Scholar] [CrossRef] [PubMed]
- Nanayakkara, G.; Alasmari, A.; Mouli, S.; Eldoumani, H.; Quindry, J.; McGinnis, G.; Fu, X.; Berlin, A.; Peters, B.; Zhong, J.; et al. Cardioprotective HIF-1α-frataxin signaling against ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H867–H879. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Xue, J.; Lai, J.C.K.; Schork, N.J.; White, K.P.; Haddad, G.G. Mechanisms underlying hypoxia tolerance in Drosophila melanogaster: Hairy as a metabolic switch. PLoS Genet. 2008, 4, e1000221. [Google Scholar] [CrossRef] [PubMed]
- Perkins, G.; Hsiao, Y.-h.; Yin, S.; Tjong, J.; Tran, M.T.; Lau, J.; Xue, J.; Liu, S.; Ellisman, M.H.; Zhou, D. Ultrastructural modifications in the mitochondria of hypoxia-adapted Drosophila melanogaster. PLoS ONE 2012, 7, e45344. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.-W.; Tang, L.; Yang, J.; Xu, W.-H. HIF-1 regulates insect lifespan extension by inhibiting c-Myc-TFAM signaling and mitochondrial biogenesis. Biochim. Biophys. Acta 2016, 1863, 2594–2603. [Google Scholar] [CrossRef] [PubMed]
- Hargreaves, I.P. Coenzyme Q10 as a therapy for mitochondrial disease. Int. J. Biochem. Cell Biol. 2014, 49, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.; Orrell, R.W.; Fahey, M.; Brassington, R.; Pandolfo, M. Pharmacological treatments for Friedreich ataxia. Cochrane Database Syst. Rev. 2016, CD007791. [Google Scholar] [CrossRef] [PubMed]
- Khdour, O.M.; Bandyopadhyay, I.; Chowdhury, S.R.; Visavadiya, N.P.; Hecht, S.M. Lipophilic methylene blue analogues enhance mitochondrial function and increase frataxin levels in a cellular model of Friedreich’s ataxia. Bioorg. Med. Chem. 2018, 26, 3359–3369. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, V.; Petrini, S.; Travaglini, L.; Priori, C.; Piermarini, E.; Petrillo, S.; Carletti, B.; Bertini, E.; Piemonte, F. Frataxin deficiency leads to reduced expression and impaired translocation of NF-E2-related factor (Nrf2) in cultured motor neurons. Int. J. Mol. Sci. 2013, 14, 7853–7865. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Piermarini, E.; Pastore, A.; Vasco, G.; Schirinzi, T.; Carrozzo, R.; Bertini, E.; Piemonte, F. Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int. J. Mol. Sci. 2017, 18, 2173. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Vacher, C.; Berger, Z.; Davies, J.E.; Luo, S.; Oroz, L.G.; Scaravilli, F.; Easton, D.F.; Duden, R.; O’Kane, C.J.; et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 2004, 36, 585–595. [Google Scholar] [CrossRef] [PubMed]
- Palomo, G.M.; Cerrato, T.; Gargini, R.; Diaz-Nido, J. Silencing of frataxin gene expression triggers p53-dependent apoptosis in human neuron-like cells. Hum. Mol. Genet. 2011, 20, 2807–2822. [Google Scholar] [CrossRef] [PubMed]
- Marobbio, C.M.T.; Pisano, I.; Porcelli, V.; Lasorsa, F.M.; Palmieri, L. Rapamycin reduces oxidative stress in frataxin-deficient yeast cells. Mitochondrion 2012, 12, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Rusten, T.E.; Katheder, N.; Elvenes, J.; Bruun, J.-A.; Sjøttem, E.; Lamark, T.; Johansen, T. p62/Sequestosome-1, Autophagy-related Gene 8, and Autophagy in Drosophila Are Regulated by Nuclear Factor Erythroid 2-related Factor 2 (NRF2), Independent of Transcription Factor TFEB. J. Biol. Chem. 2015, 290, 14945–14962. [Google Scholar] [CrossRef] [PubMed]
- Seguin, A.; Monnier, V.; Palandri, A.; Bihel, F.; Rera, M.; Schmitt, M.; Camadro, J.-M.; Tricoire, H.; Lesuisse, E. A Yeast/Drosophila Screen to Identify New Compounds Overcoming Frataxin Deficiency. Oxid. Med. Cell. Longev. 2015, 2015, 565140. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Sánchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jiménez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Elincx-Benizri, S.; Glik, A.; Merkel, D.; Arad, M.; Freimark, D.; Kozlova, E.; Cabantchik, I.; Hassin-Baer, S. Clinical Experience With Deferiprone Treatment for Friedreich Ataxia. J. Child Neurol. 2016, 31, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Arpa, J.; Sanz-Gallego, I.; Rodríguez-de-Rivera, F.J.; Domínguez-Melcón, F.J.; Prefasi, D.; Oliva-Navarro, J.; Moreno-Yangüela, M. Triple therapy with deferiprone, idebenone and riboflavin in Friedreich’s ataxia—Open-label trial. Acta Neurol. Scand. 2014, 129, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Le Quan Sang, K.H.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Besse, E.K.; Ha, D.; Kovtunovych, G.; Rouault, T.A. Iron-dependent regulation of frataxin expression: Implications for treatment of Friedreich ataxia. Hum. Mol. Genet. 2008, 17, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Goncalves, S.; Paupe, V.; Dassa, E.P.; Rustin, P. Deferiprone targets aconitase: Implication for Friedreich’s ataxia treatment. BMC Neurol. 2008, 8, 20. [Google Scholar] [CrossRef] [PubMed]
- Palandri, A.; Martin, E.; Russi, M.; Rera, M.; Tricoire, H.; Monnier, V. Identification of cardioprotective drugs by medium-scale in vivo pharmacological screening on a Drosophila cardiac model of Friedreich’s ataxia. Dis. Models Mech. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Creanga, A.; Lum, L.; Beachy, P.A. Prevalence of off-target effects in Drosophila RNA interference screens. Nature 2006, 443, 359–363. [Google Scholar] [CrossRef] [PubMed]
- Seinen, E.; Burgerhof, J.G.M.; Jansen, R.C.; Sibon, O.C.M. RNAi-induced off-target effects in Drosophila melanogaster: Frequencies and solutions. Brief. Funct. Genom. 2011, 10, 206–214. [Google Scholar] [CrossRef] [PubMed]
- Dietzl, G.; Chen, D.; Schnorrer, F.; Su, K.-C.; Barinova, Y.; Fellner, M.; Gasser, B.; Kinsey, K.; Oppel, S.; Scheiblauer, S.; et al. A genome-wide transgenic RNAi library for conditional gene inactivation in Drosophila. Nature 2007, 448, 151–156. [Google Scholar] [CrossRef] [PubMed]
- Ni, J.-Q.; Markstein, M.; Binari, R.; Pfeiffer, B.; Liu, L.-P.; Villalta, C.; Booker, M.; Perkins, L.; Perrimon, N. Vector and parameters for targeted transgenic RNA interference in Drosophila melanogaster. Nat. Methods 2008, 5, 49–51. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Bullock, S.L. Creating Heritable Mutations in Drosophila with CRISPR-Cas9. Methods Mol. Biol. 2016, 1478, 145–160. [Google Scholar] [PubMed]
- Pareek, G.; Thomas, R.E.; Pallanck, L.J. Loss of the Drosophila m-AAA mitochondrial protease paraplegin results in mitochondrial dysfunction, shortened lifespan, and neuronal and muscular degeneration. Cell Death Dis. 2018, 9, 304. [Google Scholar] [CrossRef] [PubMed]
- Port, F.; Bullock, S.L. Augmenting CRISPR applications in Drosophila with tRNA-flanked sgRNAs. Nat. Methods 2016, 13, 852–854. [Google Scholar] [CrossRef] [PubMed]
- Bussmann, J.; Storkebaum, E. Molecular pathogenesis of peripheral neuropathies: Insights from Drosophila models. Curr. Opin. Genet. Dev. 2017, 44, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Oswald, M.C.W.; West, R.J.H.; Lloyd-Evans, E.; Sweeney, S.T. Identification of dietary alanine toxicity and trafficking dysfunction in a Drosophila model of hereditary sensory and autonomic neuropathy type 1. Hum. Mol. Genet. 2015, 24, 6899–6909. [Google Scholar] [PubMed]
- Fernius, J.; Starkenberg, A.; Thor, S. Bar-coding neurodegeneration: Identifying subcellular effects of human neurodegenerative disease proteins using Drosophila leg neurons. Dis. Models Mech. 2017, 10, 1027–1038. [Google Scholar] [CrossRef] [PubMed]
- Dogan, I.; Tinnemann, E.; Romanzetti, S.; Mirzazade, S.; Costa, A.S.; Werner, C.J.; Heim, S.; Fedosov, K.; Schulz, S.; Timmann, D.; et al. Cognition in Friedreich’s ataxia: A behavioral and multimodal imaging study. Ann. Clin. Transl. Neurol. 2016, 3, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Kahsai, L.; Zars, T. Learning and memory in Drosophila: Behavior, genetics, and neural systems. Int. Rev. Neurobiol. 2011, 99, 139–167. [Google Scholar] [PubMed]
- Mandilaras, K.; Missirlis, F. Genes for iron metabolism influence circadian rhythms in Drosophila melanogaster. Metallomics Integr. Biomet. Sci. 2012, 4, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Karaiskos, N.; Wahle, P.; Alles, J.; Boltengagen, A.; Ayoub, S.; Kipar, C.; Kocks, C.; Rajewsky, N.; Zinzen, R.P. The Drosophila embryo at single-cell transcriptome resolution. Science 2017, 358, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Croset, V.; Treiber, C.D.; Waddell, S. Cellular diversity in the Drosophila midbrain revealed by single-cell transcriptomics. eLife 2018, 7, e34550. [Google Scholar] [CrossRef] [PubMed]
- Marshall, O.J.; Southall, T.D.; Cheetham, S.W.; Brand, A.H. Cell-type-specific profiling of protein-DNA interactions without cell isolation using targeted DamID with next-generation sequencing. Nat. Protoc. 2016, 11, 1586–1598. [Google Scholar] [CrossRef] [PubMed]
- Southall, T.D.; Gold, K.S.; Egger, B.; Davidson, C.M.; Caygill, E.E.; Marshall, O.J.; Brand, A.H. Cell-type-specific profiling of gene expression and chromatin binding without cell isolation: Assaying RNA Pol II occupancy in neural stem cells. Dev. Cell 2013, 26, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Alsina, D.; Ros, J.; Tamarit, J. Nitric oxide prevents Aft1 activation and metabolic remodeling in frataxin-deficient yeast. Redox Biol. 2018, 14, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Filadi, R.; Theurey, P.; Pizzo, P. The endoplasmic reticulum-mitochondria coupling in health and disease: Molecules, functions and significance. Cell Calcium 2017, 62, 1–15. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Phenotype | Tissue Affected | Genotype | Fly Phenotype |
|---|---|---|---|
| Reduced longevity | Ubiquitous downregulation Ubiquitous downregulation Panneuronal Panneuronal | Actin-GAL4>UAS-fhIR Da-GAL4>UDIR Elav-GS>UDIR Elav-GAL4; UAS-Dicer2>UDIR | Shortened life span Developmental lethality Shortened life span Shortened life span |
| Ataxia | Ubiquitous downregulation Panneuronal Larval and adult CNS Serotonergic and Dopaminergic neurons | Actin-GAL4>UAS-fhIR Elav-GAL4; UAS-Dicer2>UDIR c698a-GAL4>UAS-fhIR Ddc-GAL4>UAS-fhIR | impaired locomotion impaired locomotion No effect No effect |
| Degeneration and atrophy of DN | mutant photoreceptor neurons | fh1 | Degeneration of neuronal photoreceptors |
| Degeneration of large sensory neurons from the DRG | PNS PNS | C96-GAL4>UDIR Neur-GAL4>UAS-fhIR | Reduced longevity Reduced longevity, impaired locomotion |
| Degeneration of spinocerebellar tracts | Motorneurons | D42-GAL4>UDIR | No effect in life span, reduced mitochondrial transport and axonal degeneration |
| Demyelination of sural nerves and DRG fibres | Panglial | Repo-GAL4>UDIR | Shortened life span, impaired locomotion, brain degeneration, lipid dyshomeostasis |
| Hypertrophic cardiomyopathy | Heart | Hand-GS>UDIR | Heart dilatation an impairment of heart function |
| Abnormal muscle performance and recovery after exercise | Indirect Flight Muscles | Mef2-GAL4>UDIR | Reduced ATP production, shortened life span, impaired locomotion |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monnier, V.; Llorens, J.V.; Navarro, J.A. Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia. Int. J. Mol. Sci. 2018, 19, 1989. https://doi.org/10.3390/ijms19071989
Monnier V, Llorens JV, Navarro JA. Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia. International Journal of Molecular Sciences. 2018; 19(7):1989. https://doi.org/10.3390/ijms19071989
Chicago/Turabian StyleMonnier, Véronique, Jose Vicente Llorens, and Juan Antonio Navarro. 2018. "Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia" International Journal of Molecular Sciences 19, no. 7: 1989. https://doi.org/10.3390/ijms19071989
APA StyleMonnier, V., Llorens, J. V., & Navarro, J. A. (2018). Impact of Drosophila Models in the Study and Treatment of Friedreich’s Ataxia. International Journal of Molecular Sciences, 19(7), 1989. https://doi.org/10.3390/ijms19071989