Recent Insights on Alzheimer’s Disease Originating from Yeast Models

 , , and
, , and
Abstract
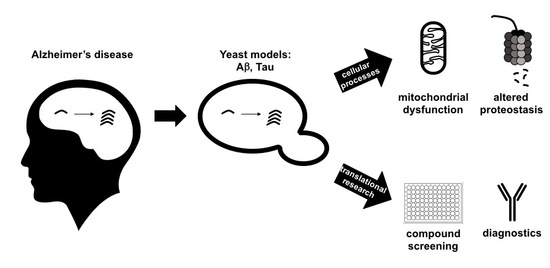
1. Introduction
1.1. Alzheimer’s Disease (AD)
1.2. Yeast as a Model Organism to Study AD: Advantages
1.3. Yeast as a Model Organism to Study AD: Limitations
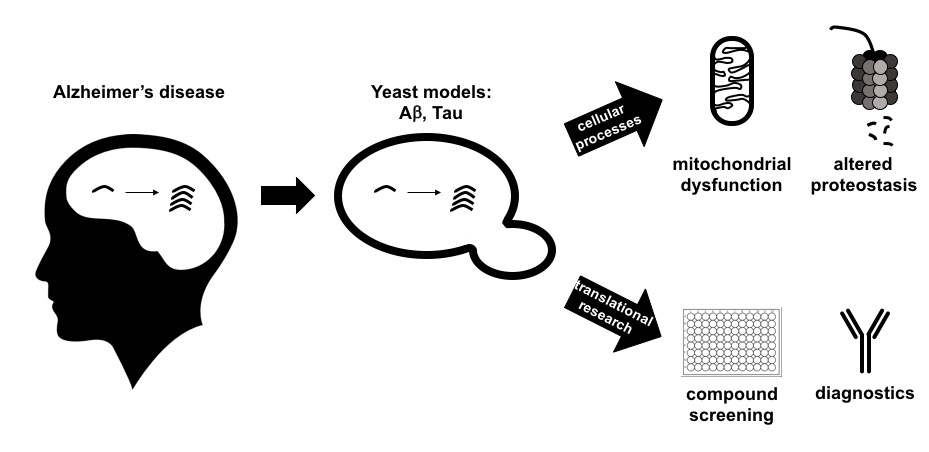
2. Humanized Yeast Models to Study Tau Biology
2.1. Protein Tau: Structure, Functions and Modifications
2.2. From Complementary Disease Models to AD Diagnostics
2.3. Future Perspectives
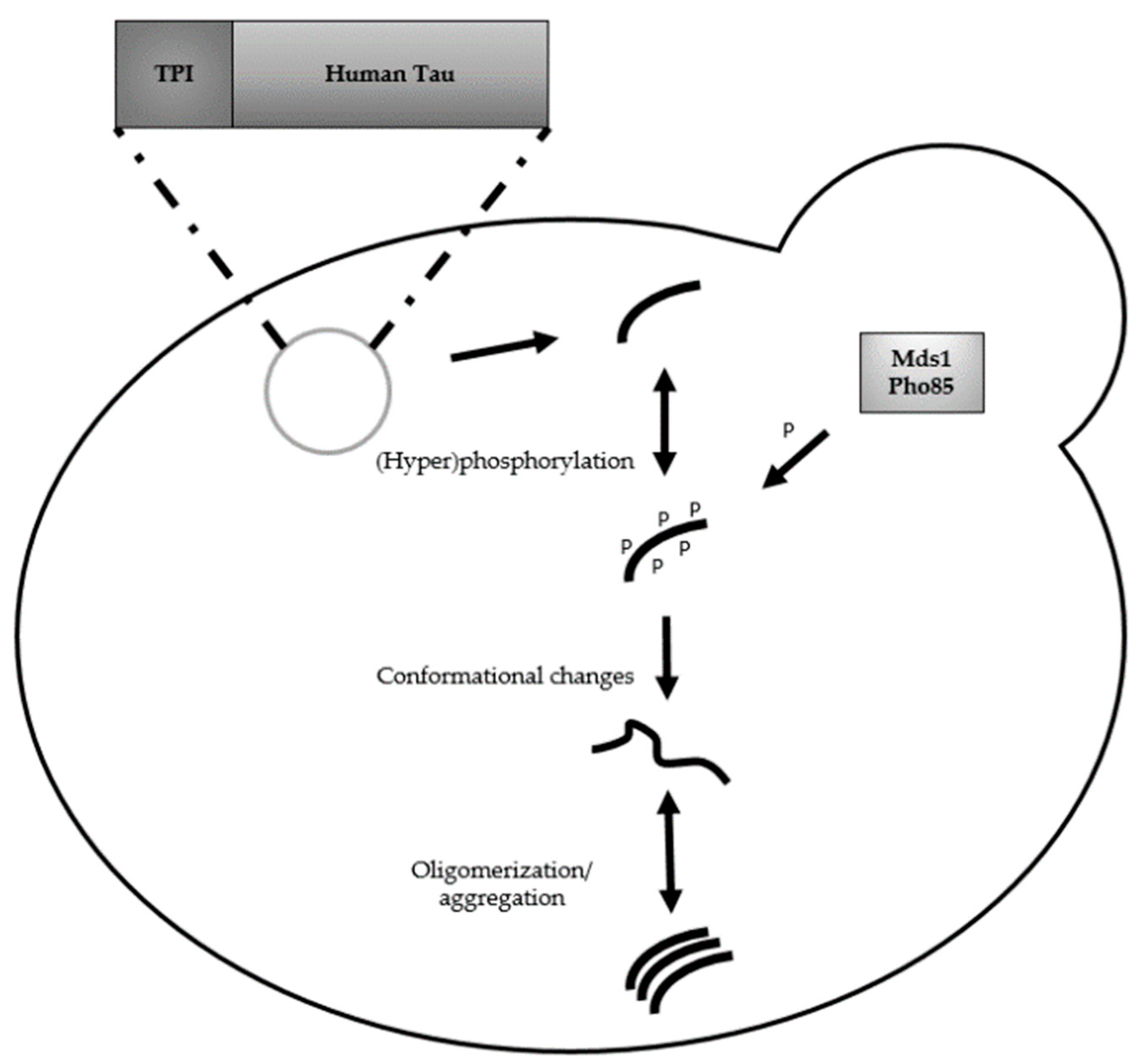
3. Humanized Yeast Models to Study Aβ Biology
3.1. Protein Aβ: Structure, Function and Aggregation
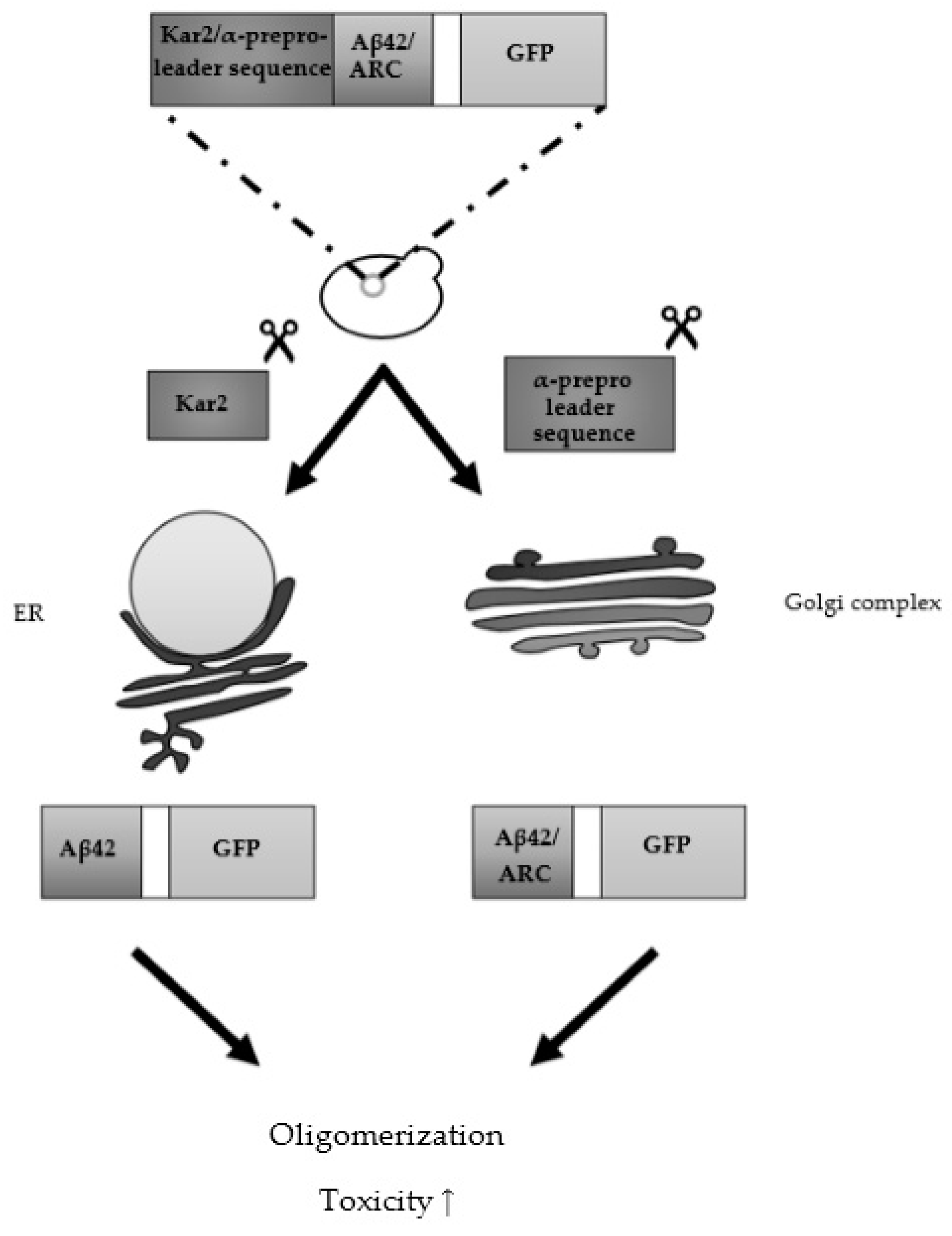
3.2. From Heterologously Expressed APP to Secretory Pathway-Targeted Aβ Peptides
4. Humanized Yeast Models to Study Frameshift Ubiquitin Mutant UBB+1 Biology
5. Studying Prion Characteristics of Aβ and Tau in Yeast
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Prince, M.; Guerchet, M.; Prina, M. The Epidemiology and Impact of Dementia: Current State and Future Trends. Available online: http://www.who.int/mental_health/neurology/en/ (accessed on 26 March 2015).
- Prince, M.; Bryce, R.; Albanese, E.; Wimo, A.; Ribeiro, W.; Ferri, C.P. The global prevalence of dementia: A systematic review and metaanalysis. Alzheimer’s Dement. 2013, 9, 63–75. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. Dementia: A problem for our age. Nature 2011, 475, S2–S4. [Google Scholar] [CrossRef] [PubMed]
- Norfray, J.F.; Provenzale, J.M. Alzheimer’s Disease: Neuropathologic Findings and Recent Advances in Imaging. Am. J. Roentgenol. 2004, 182, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Marsh, J.; Alifragis, P. Synaptic dysfunction in Alzheimer’s disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 2018, 13, 616–623. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Alzheimer’s Disease Is a Synaptic Failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Backman, L.; Jones, S.; Berger, A.-K.; Laukka, E.J.; Small, B.J. Multiple cognitive deficits during the transition to Alzheimer’s disease. J. Intern. Med. 2004, 256, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.E. Clinical trials of disease-modifying therapies for neurodegenerative diseases: The challenges and the future. Nat. Med. 2010, 16, 1223–1226. [Google Scholar] [CrossRef] [PubMed]
- Irie, K.; Murakami, K.; Masuda, Y.; Morimoto, A.; Ohigashi, H.; Ohashi, R.; Takegoshi, K.; Nagao, M.; Shimizu, T.; Shirasawa, T. Structure of β-amyloid fibrils and its relevance to their neurotoxicity: Implications for the pathogenesis of Alzheimer’s disease. J. Biosci. Bioeng. 2005, 99, 437–447. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Götz, J. Amyloid-β and tau—A toxic pas de deux in Alzheimer’s disease. Nat. Rev. Neurosci. 2011, 12, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Goffeau, A.; Barrell, B.G.; Bussey, H.; Davis, R.W.; Dujon, B.; Feldmann, H.; Galibert, F.; Hoheisel, J.D.; Jacq, C.; Johnston, M.; et al. Life with 6000 genes. Science 1996, 274, 563–567. [Google Scholar] [CrossRef]
- Botstein, D.; Chervitz, S.A.; Cherry, J.M. Yeast as a model organism. Science 1997, 277, 1259–1260. [Google Scholar] [CrossRef] [PubMed]
- Petranovic, D.; Tyo, K.; Vemuri, G.N.; Nielsen, J. Prospects of yeast systems biology for human health: Integrating lipid, protein and energy metabolism. FEMS Yeast Res. 2010, 10, 1046–1059. [Google Scholar] [CrossRef] [PubMed]
- Winderickx, J.; Delay, C.; De Vos, A.; Klinger, H.; Pellens, K.; Vanhelmont, T.; Van Leuven, F.; Zabrocki, P. Protein folding diseases and neurodegeneration: Lessons learned from yeast. Biochim. Biophys. Acta Mol. Cell Res. 2008, 1783, 1381–1395. [Google Scholar] [CrossRef] [PubMed]
- Tenreiro, S.; Munder, M.C.; Alberti, S.; Outeiro, T.F. Harnessing the power of yeast to unravel the molecular basis of neurodegeneration. J. Neurochem. 2013, 127, 438–452. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, S.; Saberidokht, B.; Subramaniam, S.; Grama, A. Scope and limitations of yeast as a model organism for studying human tissue-specific pathways. BMC Syst. Biol. 2015, 9, 96. [Google Scholar] [CrossRef] [PubMed]
- Migheli, A.; Butler, M.; Brown, K.; Shelanski, M. Light and electron microscope localization of the microtubule-associated tau protein in rat brain. J. Neurosci. 1988, 8, 1846–1851. [Google Scholar] [CrossRef] [PubMed]
- Papasozomenos, S.C.; Binder, L.I. Phosphorylation determines two distinct species of tau in the central nervous system. Cell Motil. Cytoskelet. 1987, 8, 210–226. [Google Scholar] [CrossRef] [PubMed]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsen, K.C.; Lynch, T.; Pavlou, E.; Higgins, M.; Nygaard, T.G. Localization of disinhibition-dementia-parkinsonism-amyotrophy complex to 17q21-22. Am. J. Hum. Genet. 1994, 55, 1159–1165. [Google Scholar] [PubMed]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.W.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K. Extracellular Tau and Its Potential Role in the Propagation of Tau Pathology. Front. Neurosci. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wölfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic Function of Tau Mediates Amyloid-β Toxicity in Alzheimer’s Disease Mouse Models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.A.; Cobb, M.H.; Kirschner, M.W. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [PubMed]
- Crowther, R.A. Straight and paired helical filaments in Alzheimer disease have a common structural unit. Proc. Natl. Acad. Sci. USA 1991, 88, 2288–2292. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Crowther, R.A.; Stewart, M.; Roth, M. Subunit structure of paired helical filaments in Alzheimer’s disease. J. Cell Biol. 1985, 100, 1905–1912. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, K.; Kitazawa, H.; Iguchi, Y.; Hotani, H.; Itoh, T.J. Visualization of the stop of microtubule depolymerization that occurs at the high-density region of microtubule-associated protein 2 (MAP2). J. Mol. Biol. 2001, 312, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell. Neurosci. 2014, 8. [Google Scholar] [CrossRef] [PubMed]
- Frandemiche, M.L.; De Seranno, S.; Rush, T.; Borel, E.; Elie, A.; Arnal, I.; Lante, F.; Buisson, A. Activity-Dependent Tau Protein Translocation to Excitatory Synapse Is Disrupted by Exposure to Amyloid-Beta Oligomers. J. Neurosci. 2014, 34, 6084–6097. [Google Scholar] [CrossRef] [PubMed]
- Stamer, K.; Vogel, R.; Thies, E.; Mandelkow, E.; Mandelkow, E.-M. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J. Cell Biol. 2002, 156, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L.F. Differential Regulation of Dynein and Kinesin Motor Proteins by Tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [PubMed]
- Konzack, S.; Thies, E.; Marx, A.; Mandelkow, E.-M.; Mandelkow, E. Swimming against the Tide: Mobility of the Microtubule-Associated Protein Tau in Neurons. J. Neurosci. 2007, 27, 9916–9927. [Google Scholar] [CrossRef] [PubMed]
- Vershinin, M.; Carter, B.C.; Razafsky, D.S.; King, S.J.; Gross, S.P. Multiple-motor based transport and its regulation by Tau. Proc. Natl. Acad. Sci. USA 2007, 104, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Utton, M.A.; Noble, W.J.; Hill, J.E.; Anderton, B.H.; Hanger, D.P. Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J. Cell Sci. 2005, 118, 4645–4654. [Google Scholar] [CrossRef] [PubMed]
- Kanaan, N.M.; Morfini, G.A.; LaPointe, N.E.; Pigino, G.F.; Patterson, K.R.; Song, Y.; Andreadis, A.; Fu, Y.; Brady, S.T.; Binder, L.I. Pathogenic Forms of Tau Inhibit Kinesin-Dependent Axonal Transport through a Mechanism Involving Activation of Axonal Phosphotransferases. J. Neurosci. 2011, 31, 9858–9868. [Google Scholar] [CrossRef] [PubMed]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef] [PubMed]
- Caceres, A.; Kosik, K.S. Inhibition of neurite polarity by tau antisense oligonucleotides in primary cerebellar neurons. Nature 1990, 343, 461–463. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 22–35. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Spillantini, M.G.; Potier, M.C.; Ulrich, J.; Crowther, R.A. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: Differential expression of tau protein mRNAs in human brain. EMBO J. 1989, 8, 393–399. [Google Scholar] [PubMed]
- Himmler, A.; Drechsel, D.; Kirschner, M.W.; Martin, D.W. Tau consists of a set of proteins with repeated C-terminal microtubule-binding domains and variable N-terminal domains. Mol. Cell. Biol. 1989, 9, 1381–1388. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, K.J.; Ross, J.L.; Feinstein, H.E.; Feinstein, S.C.; Israelachvili, J. Complementary dimerization of microtubule-associated tau protein: Implications for microtubule bundling and tau-mediated pathogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 7445–7450. [Google Scholar] [CrossRef] [PubMed]
- Bunker, J.M.; Wilson, L.; Jordan, M.A.; Feinstein, S.C. Modulation of microtubule dynamics by tau in living cells: Implications for development and neurodegeneration. Mol. Biol. Cell 2004, 15, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Lu, M.; Kosik, K.S. Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol. Biol. Cell 2001, 12, 171–184. [Google Scholar] [CrossRef] [PubMed]
- Derisbourg, M.; Leghay, C.; Chiappetta, G.; Fernandez-Gomez, F.-J.; Laurent, C.; Demeyer, D.; Carrier, S.; Buée-Scherrer, V.; Blum, D.; Vinh, J.; et al. Role of the Tau N-terminal region in microtubule stabilization revealed by new endogenous truncated forms. Sci. Rep. 2015, 5, 9659. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kanai, Y.; Cowan, N.J.; Hirokawa, N. Projection domains of MAP2 and tau determine spacings between microtubules in dendrites and axons. Nature 1992, 360, 674–677. [Google Scholar] [CrossRef] [PubMed]
- Sattilaro, R.F.; Dentler, W.L.; LeCluyse, E.L. Microtubule-associated proteins (MAPs) and the organization of actin filaments in vitro. J. Cell Biol. 1981, 90, 467–473. [Google Scholar] [CrossRef] [PubMed]
- Correas, I.; Padilla, R.; Avila, J. The tubulin-binding sequence of brain microtubule-associated proteins, tau and MAP-2, is also involved in actin binding. Biochem. J. 1990, 269, 61–64. [Google Scholar] [CrossRef] [PubMed]
- Rendon, A.; Jung, D.; Jancsik, V. Interaction of microtubules and microtubule-associated proteins (MAPs) with rat brain mitochondria. Biochem. J. 1990, 269, 555–556. [Google Scholar] [CrossRef] [PubMed]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, J.F.; Rivas, R.J. Actin disruption alters the localization of tau in the growth cones of cerebellar granule neurons. J. Cell Sci. 2000, 113, 2797–2809. [Google Scholar] [PubMed]
- Manczak, M.; Reddy, P.H. Abnormal interaction between the mitochondrial fission protein Drp1 and hyperphosphorylated tau in Alzheimer’s disease neurons: Implications for mitochondrial dysfunction and neuronal damage. Hum. Mol. Genet. 2012, 21, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Bhaskar, K.; Hobbs, G.A.; Yen, S.-H.; Lee, G. Tyrosine phosphorylation of tau accompanies disease progression in transgenic mouse models of tauopathy. Neuropathol. Appl. Neurobiol. 2010, 36, 462–477. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Newman, S.T.; Gard, D.L.; Band, H.; Panchamoorthy, G. Tau interacts with src-family non-receptor tyrosine kinases. J. Cell Sci. 1998, 111, 3167–3177. [Google Scholar] [PubMed]
- Lee, G.; Thangavel, R.; Sharma, V.M.; Litersky, J.M.; Bhaskar, K.; Fang, S.M.; Do, L.H.; Andreadis, A.; Van Hoesen, G.; Ksiezak-Reding, H. Phosphorylation of Tau by Fyn: Implications for Alzheimer’s Disease. J. Neurosci. 2004, 24, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Dujardin, S.; Bégard, S.; Caillierez, R.; Lachaud, C.; Delattre, L.; Carrier, S.; Loyens, A.; Galas, M.-C.; Bousset, L.; Melki, R.; et al. Ectosomes: A New Mechanism for Non-Exosomal Secretion of Tau Protein. PLoS ONE 2014, 9, e100760. [Google Scholar] [CrossRef] [PubMed]
- Calafate, S.; Buist, A.; Miskiewicz, K.; Vijayan, V.; Daneels, G.; de Strooper, B.; de Wit, J.; Verstreken, P.; Moechars, D. Synaptic Contacts Enhance Cell-to-Cell Tau Pathology Propagation. Cell Rep. 2015, 11, 1176–1183. [Google Scholar] [CrossRef] [PubMed]
- Tardivel, M.; Bégard, S.; Bousset, L.; Dujardin, S.; Coens, A.; Melki, R.; Buée, L.; Colin, M. Tunneling nanotube (TNT)-mediated neuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol. Commun. 2016, 4, 117. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E.; Mandelkow, E.-M. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 5. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.L.; Lee, V.M.Y. Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat. Med. 2014, 20, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S.C. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell 1997, 8, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Sillen, A.; Barbier, P.; Landrieu, I.; Lefebvre, S.; Wieruszeski, J.-M.; Leroy, A.; Peyrot, V.; Lippens, G. NMR Investigation of the Interaction between the Neuronal Protein Tau and the Microtubules. Biochemistry 2007. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Seereeram, A.; Noble, W. Mediators of tau phosphorylation in the pathogenesis of Alzheimer’s disease. Expert Rev. Ther. 2009, 9, 1647–1666. [Google Scholar] [CrossRef] [PubMed]
- Sergeant, N.; Bretteville, A.; Hamdane, M.; Caillet-Boudin, M.-L.; Grognet, P.; Bombois, S.; Blum, D.; Delacourte, A.; Pasquier, F.; Vanmechelen, E.; et al. Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev. Proteom. 2008, 5, 207–224. [Google Scholar] [CrossRef] [PubMed]
- Domise, M.; Didier, S.; Marinangeli, C.; Zhao, H.; Chandakkar, P.; Buée, L.; Viollet, B.; Davies, P.; Marambaud, P.; Vingtdeux, V. AMP-activated protein kinase modulates tau phosphorylation and tau pathology in vivo. Sci. Rep. 2016, 6, 26758. [Google Scholar] [CrossRef] [PubMed]
- Cavallini, A.; Brewerton, S.; Bell, A.; Sargent, S.; Glover, S.; Hardy, C.; Moore, R.; Calley, J.; Ramachandran, D.; Poidinger, M.; et al. An unbiased approach to identifying tau kinases that phosphorylate tau at sites associated with alzheimer disease. J. Biol. Chem. 2013, 288, 23331–23347. [Google Scholar] [CrossRef] [PubMed]
- Trinczek, B.; Biernat, J.; Baumann, K.; Mandelkow, E.M.; Mandelkow, E. Domains of tau protein, differential phosphorylation, and dynamic instability of microtubules. Mol. Biol. Cell 1995, 6, 1887–1902. [Google Scholar] [CrossRef] [PubMed]
- Hanger, D.P.; Anderton, B.H.; Noble, W. Tau phosphorylation: The therapeutic challenge for neurodegenerative disease. Trends Mol. Med. 2009, 15, 112–119. [Google Scholar] [CrossRef] [PubMed]
- Lu, P.-J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef] [PubMed]
- von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B.; et al. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Šimić, G.; Babić Leko, M.; Wray, S.; Harrington, C.; Delalle, I.; Jovanov-Milošević, N.; Bažadona, D.; Buée, L.; de Silva, R.; Giovanni, G.D.; et al. Tau protein hyperphosphorylation and aggregation in alzheimer’s disease and other tauopathies, and possible neuroprotective strategies. Biomolecules 2016, 6, 2–28. [Google Scholar] [CrossRef] [PubMed]
- Berger, Z.; Roder, H.; Hanna, A.; Carlson, A.; Rangachari, V.; Yue, M.; Wszolek, Z.; Ashe, K.; Knight, J.; Dickson, D.; et al. Accumulation of Pathological Tau Species and Memory Loss in a Conditional Model of Tauopathy. J. Neurosci. 2007, 27, 3650–3662. [Google Scholar] [CrossRef] [PubMed]
- Bretteville, A.; Planel, E. Tau aggregates: Toxic, inert, or protective species? J. Alzheimer’s Dis. 2008, 14, 431–436. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Sahara, N.; Saito, Y.; Murayama, M.; Yoshiike, Y.; Kim, H.; Miyasaka, T.; Murayama, S.; Ikai, A.; Takashima, A. Granular Tau Oligomers as Intermediates of Tau Filaments. Biochemistry 2007, 46, 3856–3861. [Google Scholar] [CrossRef] [PubMed]
- Lasagna-Reeves, C.A.; Castillo-Carranza, D.L.; Sengupta, U.; Sarmiento, J.; Troncoso, J.; Jackson, G.R.; Kayed, R. Identification of oligomers at early stages of tau aggregation in Alzheimer’s disease. FASEB J. 2012, 26, 1946–1959. [Google Scholar] [CrossRef] [PubMed]
- Hutton, M.; Lendon, C.L.; Rizzu, P.; Baker, M.; Froelich, S.; Houlden, H.; Pickering-Brown, S.; Chakraverty, S.; Isaacs, A.; Grover, A.; et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature 1998, 393, 702–705. [Google Scholar] [CrossRef] [PubMed]
- Poorkaj, P.; Bird, T.D.; Wijsman, E.; Nemens, E.; Garruto, R.M.; Anderson, L.; Andreadis, A.; Wiederholt, W.C.; Raskind, M.; Schellenberg, G.D. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann. Neurol. 1998, 43, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Murrell, J.R.; Goedert, M.; Farlow, M.R.; Klug, A.; Ghetti, B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc. Natl. Acad. Sci. USA 1998, 95, 7737–7741. [Google Scholar] [CrossRef] [PubMed]
- Spillantini, M.G.; Bird, T.D.; Ghetti, B. Frontotemporal dementia and Parkinsonism linked to chromosome 17: A new group of tauopathies. Brain Pathol. 1998, 8, 387–402. [Google Scholar] [CrossRef] [PubMed]
- Rademakers, R.; Cruts, M.; van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 2004, 24, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Dermaut, B.; Kumar-Singh, S.; Rademakers, R.; Theuns, J.; Cruts, M.; Van Broeckhoven, C. Tau is central in the genetic Alzheimer–frontotemporal dementia spectrum. Trends Genet. 2005, 21, 664–672. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, I.; Poorkaj, P.; Hong, M.; Nochlin, D.; Lee, V.M.; Bird, T.D.; Schellenberg, G.D. Missense and silent tau gene mutations cause frontotemporal dementia with parkinsonism-chromosome 17 type, by affecting multiple alternative RNA splicing regulatory elements. Proc. Natl. Acad. Sci. USA 1999, 96, 5598–5603. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Zheng-Fischhöfer, Q.; Ackmann, M.; Biernat, J.; von Bergen, M.; Mandelkow, E.M.; Mandelkow, E. Structure, microtubule interactions, and paired helical filament aggregation by tau mutants of frontotemporal dementias. Biochemistry 2000, 39, 11714–11721. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Zhukareva, V.; Vogelsberg-Ragaglia, V.; Wszolek, Z.; Reed, L.; Miller, B.I.; Geschwind, D.H.; Bird, T.D.; McKeel, D.; Goate, A.; et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science 1998, 282, 1914–1917. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.-Q.; Mucke, L. Reducing Endogenous Tau Ameliorates Amyloid β-Induced Deficits in an Alzheimer’s Disease Mouse Model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [PubMed]
- Vandebroek, T.; Vanhelmont, T.; Terwel, D.; Borghgraef, P.; Lemaire, K.; Snauwaert, J.; Wera, S.; Van Leuven, F.; Winderickx, J. Identification and isolation of a hyperphosphorylated, conformationally changed intermediate of human protein tau expressed in yeast. Biochemistry 2005, 44, 11466–11475. [Google Scholar] [CrossRef] [PubMed]
- Vanhelmont, T.; Vandebroek, T.; De Vos, A.; Terwel, D.; Lemaire, K.; Anandhakumar, J.; Franssens, V.; Swinnen, E.; Van Leuven, F.; Winderickx, J. Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast. FEMS Yeast Res. 2010, 10, 992–1005. [Google Scholar] [CrossRef] [PubMed]
- Vandebroek, T.; Terwel, D.; Vanhelmont, T.; Gysemans, M.; Van Haesendonck, C.; Engelborghs, Y.; Winderickx, J.; Van Leuven, F. Microtubule binding and clustering of human Tau-4R and Tau-P301L proteins isolated from yeast deficient in orthologues of glycogen synthase kinase-3beta or cdk5. J. Biol. Chem. 2006, 281, 25388–25397. [Google Scholar] [CrossRef] [PubMed]
- De Vos, A.; Anandhakumar, J.; Van den Brande, J.; Verduyckt, M.; Franssens, V.; Winderickx, J.; Swinnen, E. Yeast as a model system to study tau biology. Int. J. Alzheimers Dis. 2011, 2011, 428970. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, C.A.; Egaña, J.T.; Núñez, M.T.; Maccioni, R.B.; González-Billault, C. Oxidative stress promotes tau dephosphorylation in neuronal cells: The roles of cdk5 and PP1. Free Radic. Biol. Med. 2004, 36, 1393–1402. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.W.; Kim, S.J.; Kim, M.S. Oxidative stress with tau hyperphosphorylation in memory impaired 1,2-diacetylbenzene-treated mice. Toxicol. Lett. 2017, 279, 53–59. [Google Scholar] [CrossRef] [PubMed]
- De Vos, A.; Bynens, T.; Rosseels, J.; Coun, C.; Ring, J.; Madeo, F.; Galas, M.-C.; Winderickx, J.; Franssens, V. The peptidyl prolyl cis/trans isomerase Pin1/Ess1 inhibits phosphorylation and toxicity of tau in a yeast model for Alzheimer’s disease. AIMS Mol. Sci. 2015, 2, 144–160. [Google Scholar] [CrossRef]
- Su, B.; Wang, X.; Lee, H.; Tabaton, M.; Perry, G.; Smith, M.A.; Zhu, X. Chronic oxidative stress causes increased tau phosphorylation in M17 neuroblastoma cells. Neurosci. Lett. 2010, 468, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Rosseels, J.; Van Den Brande, J.; Violet, M.; Jacobs, D.; Grognet, P.; Lopez, J.; Huvent, I.; Caldara, M.; Swinnen, E.; Papegaey, A.; et al. Tau monoclonal antibody generation based on humanized yeast models: Impact on tau oligomerization and diagnostics. J. Biol. Chem. 2015, 290, 4059–4074. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Carranza, D.L.; Gerson, J.E.; Sengupta, U.; Guerrero-Muñoz, M.J.; Lasagna-Reeves, C.A.; Kayed, R. Specific Targeting of Tau Oligomers in Htau Mice Prevents Cognitive Impairment and Tau Toxicity Following Injection with Brain-Derived Tau Oligomeric Seeds. J. Alzheimer Dis. 2014, 40, S97–S111. [Google Scholar] [CrossRef] [PubMed]
- Patterson, K.R.; Remmers, C.; Fu, Y.; Brooker, S.; Kanaan, N.M.; Vana, L.; Ward, S.; Reyes, J.F.; Philibert, K.; Glucksman, M.J.; et al. Characterization of prefibrillar Tau oligomers in vitro and in Alzheimer disease. J. Biol. Chem. 2011, 286, 23063–23076. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ruiz, E.; Decrop, D.; Ven, K.; Tripodi, L.; Leirs, K.; Rosseels, J.; van de Wouwer, M.; Geukens, N.; De Vos, A.; Vanmechelen, E.; et al. Digital ELISA for the quantification of attomolar concentrations of Alzheimer’s disease biomarker protein Tau in biological samples. Anal. Chim. Acta 2018, 1015, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M.Y. Initiation and synergistic fibrillization of tau and alpha-synuctein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [PubMed]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Hojrup, P.; Gliemann, J.; Jakes, R. alpha-synuclein binds to Tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [PubMed]
- Duka, T.; Rusnak, M.; Drolet, R.E.; Duka, V.; Wersinger, C.; Goudreau, J.L.; Sidhu, A. Alpha-Synuclein Induces Hyperphosphorylation of Au in the Mptp Model of Parkinsonism. FASEB J. 2006, 20, 2302–2312. [Google Scholar] [CrossRef] [PubMed]
- Waxman, E.A.; Giasson, B.I. Induction of Intracellular Tau Aggregation Is Promoted by α-Synuclein Seeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci. 2011, 31, 7604–7618. [Google Scholar] [CrossRef] [PubMed]
- Frasier, M.; Walzer, M.; McCarthy, L.; Magnuson, D.; Lee, J.M.; Haas, C.; Kahle, P.; Wolozin, B. Tau phosphorylation increases in symptomatic mice overexpressing A30P α-synuclein. Exp. Neurol. 2005, 192, 274–287. [Google Scholar] [CrossRef] [PubMed]
- Zabrocki, P.; Pellens, K.; Vanhelmont, T.; Vandebroek, T.; Griffioen, G.; Wera, S.; Van Leuven, F.; Winderickx, J. Characterization of α-synuclein aggregation and synergistic toxicity with protein tau in yeast. FEBS J. 2005, 272, 1386–1400. [Google Scholar] [CrossRef] [PubMed]
- Ciaccioli, G.; Martins, A.; Rodrigues, C.; Vieira, H.; Calado, P. A powerful yeast model to investigate the synergistic interaction of α-synuclein and tau in neurodegeneration. PLoS ONE 2013, 8, e55848. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, C.S.; Wood, V.; Fantes, P.A. An Ancient Yeast for Young Geneticists: A Primer on the Schizosaccharomyces pombe Model System. Genetics 2015, 201, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, M.K.; Bi, E.; Glotzer, M. Comparative analysis of cytokinesis in budding yeast, fission yeast and animal cells. Curr. Biol. 2004, 14, R806–R818. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, T.; Pearce, A. Cell cycle molecules and mechanisms of the budding and fission yeasts. Methods Mol. Biol. 2005, 296, 3–29. [Google Scholar] [PubMed]
- Heinisch, J.J.; Brandt, R. Signaling pathways and posttranslational modifications of tau in Alzheimer’s disease: The humanization of yeast cells. Microb. Cell 2016, 3, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Rodicio, R.; Heinisch, J.J. Yeast on the milky way: Genetics, physiology and biotechnology of Kluyveromyces lactis. Yeast 2013, 30, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Barnard, E.; McFerran, N.V.; Trudgett, A.; Nelson, J.; Timson, D.J. Development and implementation of split-GFP-based bimolecular fluorescence complementation (BiFC) assays in yeast. Biochem. Soc. Trans. 2008, 36, 479–482. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Nguyen, H.B.; Pedelacq, J.D.; Koraïchi, F.; Chaudhary, A.; Ganguly, K.; Lockard, M.A.; Favre, G.; Terwilliger, T.C.; Waldo, G.S. A new protein-protein interaction sensor based on tripartite split-GFP association. Sci. Rep. 2013, 3, 02854. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Terwilliger, T.C.; Waldo, G.S. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 2005, 23, 102–107. [Google Scholar] [CrossRef] [PubMed]
- Cabantous, S.; Waldo, G.S. In vivo and in vitro protein solubility assays using split GFP. Nat. Methods 2006, 3, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Foglieni, C.; Papin, S.; Salvadè, A.; Afroz, T.; Pinton, S.; Pedrioli, G.; Ulrich, G.; Polymenidou, M.; Paganetti, P. Split GFP technologies to structurally characterize and quantify functional biomolecular interactions of FTD-related proteins. Sci. Rep. 2017, 7, 14013. [Google Scholar] [CrossRef] [PubMed]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse Formation and Function Is Modulated by the Amyloid Precursor Protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Duce, J.A.; Tsatsanis, A.; Cater, M.A.; James, S.A.; Robb, E.; Wikhe, K.; Leong, S.L.; Perez, K.; Johanssen, T.; Greenough, M.A.; et al. Iron-Export Ferroxidase Activity of β-Amyloid Precursor Protein Is Inhibited by Zinc in Alzheimer’s Disease. Cell 2010, 142, 857–867. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Xu, T.; Yan, Y.; Zhou, Y.; Jiang, Y.; Melcher, K.; Xu, H.E. Amyloid beta: Structure, biology and structure-based therapeutic development. Acta Pharmacol. Sin. 2017, 38, 1205–1235. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Koo, E.H.; Mellon, A.; Hung, A.Y.; Selkoe, D.J. Targeting of cell-surface β-amyloid precursor protein to lysosomes: Alternative processing into amyloid-bearing fragments. Nature 1992, 357, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Joshi, G.; Wang, Y. Golgi defects enhance APP amyloidogenic processing in Alzheimer’s disease. BioEssays 2015, 37, 240–247. [Google Scholar] [CrossRef] [PubMed]
- Olsson, F.; Schmidt, S.; Althoff, V.; Munter, L.M.; Jin, S.; Rosqvist, S.; Lendahl, U.; Multhaup, G.; Lundkvist, J. Characterization of Intermediate Steps in Amyloid Beta (Aβ) Production under Near-native Conditions. J. Biol. Chem. 2014, 289, 1540–1550. [Google Scholar] [CrossRef] [PubMed]
- Takami, M.; Nagashima, Y.; Sano, Y.; Ishihara, S.; Morishima-Kawashima, M.; Funamoto, S.; Ihara, Y. γ-Secretase: Successive Tripeptide and Tetrapeptide Release from the Transmembrane Domain of β-Carboxyl Terminal Fragment. J. Neurosci. 2009, 29, 13042–13052. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Inhibition and modulation of γ-secretase for Alzheimer’s disease. Neurotherapeutics 2008, 5, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Hensley, K.; Carney, J.M.; Mattson, M.P.; Aksenova, M.; Harris, M.; Wu, J.F.; Floyd, R.A.; Butterfield, D.A. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: Relevance to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 3270–3274. [Google Scholar] [CrossRef] [PubMed]
- .Benzinger, T.L.; Gregory, D.M.; Burkoth, T.S.; Miller-Auer, H.; Lynn, D.G.; Botto, R.E.; Meredith, S.C. Propagating structure of Alzheimer’s beta-amyloid(10-35) is parallel beta-sheet with residues in exact register. Proc. Natl. Acad. Sci. USA 1998, 95, 13407–13412. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Edalji, R.; Harlan, J.E.; Holzman, T.F.; Lopez, A.P.; Labkovsky, B.; Hillen, H.; Barghorn, S.; Ebert, U.; Richardson, P.L.; et al. Structural Characterization of a Soluble Amyloid β-Peptide Oligomer. Biochemistry 2009, 48, 1870–1877. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Schlossmacher, M.G.; Hung, A.Y.; Vigo-Pelfrey, C.; Mellon, A.; Ostaszewski, B.L.; Lieberburg, I.; Koo, E.H.; Schenk, D.; Teplow, D.B.; et al. Amyloid β-peptide is produced by cultured cells during normal metabolism. Nature 1992, 359, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Terry, R.D.; Masliah, E.; Salmon, D.P.; Butters, N.; DeTeresa, R.; Hill, R.; Hansen, L.A.; Katzman, R. Physical basis of cognitive alterations in alzheimer’s disease: Synapse loss is the major correlate of cognitive impairment. Ann. Neurol. 1991, 30, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Terry, R.D.; Alford, M.; DeTeresa, R.; Hansen, L.A. Cortical and subcortical patterns of synaptophysinlike immunoreactivity in Alzheimer’s disease. Am. J. Pathol. 1991, 138, 235–246. [Google Scholar] [PubMed]
- Masliah, E.; Achim, C.L.; Ge, N.; DeTeresa, R.; Terry, R.D.; Wiley, C.A. Spectrum of human immunodeficiency virus-associated neocortical damage. Ann. Neurol. 1992, 32, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Mucke, L.; Masliah, E.; Yu, G.Q.; Mallory, M.; Rockenstein, E.M.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Johnson-Wood, K.; McConlogue, L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: Synaptotoxicity without plaque formation. J. Neurosci. 2000, 20, 4050–4058. [Google Scholar] [CrossRef] [PubMed]
- Hsia, A.Y.; Masliah, E.; McConlogue, L.; Yu, G.Q.; Tatsuno, G.; Hu, K.; Kholodenko, D.; Malenka, R.C.; Nicoll, R.A.; Mucke, L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc. Natl. Acad. Sci. USA 1999, 96, 3228–3233. [Google Scholar] [CrossRef] [PubMed]
- Canevari, L.; Abramov, A.Y.; Duchen, M.R. Toxicity of amyloid beta peptide: Tales of calcium, mitochondria, and oxidative stress. Neurochem. Res. 2004, 29, 637–650. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Weggen, S.; Eriksen, J.L.; Das, P.; Sagi, S.A.; Wang, R.; Pietrzik, C.U.; Findlay, K.A.; Smith, T.E.; Murphy, M.P.; Bulter, T.; et al. A subset of NSAIDs lower amyloidogenic Aβ42 independently of cyclooxygenase activity. Nature 2001, 414, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Neniskyte, U.; Neher, J.J.; Brown, G.C. Neuronal Death Induced by Nanomolar Amyloid β Is Mediated by Primary Phagocytosis of Neurons by Microglia. J. Biol. Chem. 2011, 286, 39904–39913. [Google Scholar] [CrossRef] [PubMed]
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Watanabe, K.; Sekiguchi, M.; Hosoki, E.; Kawashima-Morishima, M.; Lee, H.-J.; Hama, E.; Sekine-Aizawa, Y.; et al. Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat. Med. 2000, 6, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Iwata, N.; Tsubuki, S.; Takaki, Y.; Shirotani, K.; Lu, B.; Gerard, N.P.; Gerard, C.; Hama, E.; Lee, H.J.; Saido, T.C. Metabolic Regulation of Brain Abeta by Neprilysin. Science 2001, 292, 1550–1552. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Clearing the brain’s amyloid cobwebs. Neuron 2001, 32, 177–180. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.-C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-Aβ antibody alters CNS and plasma Aβ clearance and decreases brain Aβ burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Yamada, S.; Holtzman, D.; Ghiso, J.; Frangione, B. Clearance of amyloid beta-peptide from brain: Transport or metabolism? Nat. Med. 2000, 6, 718. [Google Scholar] [CrossRef] [PubMed]
- Martel, C.L.; Mackic, J.B.; Matsubara, E.; Governale, S.; Miguel, C.; Miao, W.; McComb, J.G.; Frangione, B.; Ghiso, J.; Zlokovic, B. V Isoform-specific effects of apolipoproteins E2, E3, and E4 on cerebral capillary sequestration and blood-brain barrier transport of circulating Alzheimer’s amyloid beta. J. Neurochem. 1997, 69, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Ghersi-Egea, J.F.; Gorevic, P.D.; Ghiso, J.; Frangione, B.; Patlak, C.S.; Fenstermacher, J.D. Fate of cerebrospinal fluid-borne amyloid beta-peptide: Rapid clearance into blood and appreciable accumulation by cerebral arteries. J. Neurochem. 1996, 67, 880–883. [Google Scholar] [CrossRef] [PubMed]
- Maness, L.M.; Banks, W.A.; Podlisny, M.B.; Selkoe, D.J.; Kastin, A.J. Passage of human amyloid beta-protein 1–40 across the murine blood-brain barrier. Life Sci. 1994, 55, 1643–1650. [Google Scholar] [CrossRef]
- Zlokovic, B.V.; Martel, C.L.; Mackic, J.B.; Matsubara, E.; Wisniewski, T.; Mccomb, J.G.; Frangione, B.; Ghiso, J. Brain Uptake of Circulating Apolipoproteins J and E Complexed to Alzheimer′s Amyloid β. Biochem. Biophys. Res. Commun. 1994, 205, 1431–1437. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Selkoe, D.J. The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Tanzi, R.E.; Moir, R.D.; Wagner, S.L. Clearance of Alzheimer’s Aβ Peptide. Neuron 2004, 43, 605–608. [Google Scholar] [CrossRef] [PubMed]
- Wyss-Coray, T.; Loike, J.D.; Brionne, T.C.; Lu, E.; Anankov, R.; Yan, F.; Silverstein, S.C.; Husemann, J. Adult mouse astrocytes degrade amyloid-β in vitro and in situ. Nat. Med. 2003, 9, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Koistinaho, M.; Lin, S.; Wu, X.; Esterman, M.; Koger, D.; Hanson, J.; Higgs, R.; Liu, F.; Malkani, S.; Bales, K.R.; et al. Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-β peptides. Nat. Med. 2004, 10, 719–726. [Google Scholar] [CrossRef] [PubMed]
- Zlokovic, B.V.; Deane, R.; Sallstrom, J.; Chow, N.; Miano, J.M. Neurovascular pathways and Alzheimer amyloid beta-peptide. Brain Pathol. 2005, 15, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, A.M.; Hamilton, D.J.; Barnett, J.L.; Paskevich, P.A.; Nixon, R.A. Properties of the endosomal-lysosomal system in the human central nervous system: Disturbances mark most neurons in populations at risk to degenerate in Alzheimer’s disease. J. Neurosci. 1996, 16, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Komano, H.; Fuller, R.S.; Gandy, S.E.; Frail, D.E. Proteolytic processing and secretion of human beta-amyloid precursor protein in yeast. Evidence for a yeast secretase activity. J. Biol. Chem. 1994, 269, 27799–27802. [Google Scholar] [PubMed]
- Hines, V.; Zhang, W.; Ramakrishna, N.; Styles, J.; Mehta, P.; Kim, K.S.; Innis, M.; Miller, D.L. The expression and processing of human beta-amyloid peptide precursors in Saccharomyces cerevisiae: Evidence for a novel endopeptidase in the yeast secretory system. Cell. Mol. Biol. Res. 1994, 40, 273–284. [Google Scholar] [PubMed]
- Zhang, W.; Espinoza, D.; Hines, V.; Innis, M.; Mehta, P.; Miller, D.L. Characterization of β-amyloid peptide precursor processing by the yeast Yap3 and Mkc7 proteases. Biochim. Biophys. Acta Mol. Cell Res. 1997, 1359, 110–122. [Google Scholar] [CrossRef]
- Komano, H.; Seeger, M.; Gandy, S.; Wang, G.T.; Krafft, G.A.; Fuller, R.S. Involvement of cell surface glycosyl-phosphatidylinositol-linked aspartyl proteases in alpha-secretase-type cleavage and ectodomain solubilization of human Alzheimer beta-amyloid precursor protein in yeast. J. Biol. Chem. 1998, 273, 31648–31651. [Google Scholar] [CrossRef] [PubMed]
- Edbauer, D.; Winkler, E.; Regula, J.T.; Pesold, B.; Steiner, H.; Haass, C. Reconstitution of γ-secretase activity. Nat. Cell Biol. 2003, 5, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Yagishita, S.; Futai, E.; Ishiura, S. In vitro reconstitution of gamma-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 2008, 377, 141–145. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, Y.; Futai, E.; Yagishita, S.; Suo, S.; Tomita, T.; Iwatsubo, T.; Ishiura, S. Comparison of presenilin 1 and presenilin 2 γ-secretase activities using a yeast reconstitution system. J. Biol. Chem. 2011, 286, 44569–44575. [Google Scholar] [CrossRef] [PubMed]
- Yonemura, Y.; Futai, E.; Yagishita, S.; Kaether, C.; Ishiura, S. Specific combinations of presenilins and Aph1s affect the substrate specificity and activity of γ-secretase. Biochem. Biophys. Res. Commun. 2016, 478, 1751–1757. [Google Scholar] [CrossRef] [PubMed]
- Futai, E.; Yagishita, S.; Ishiura, S. Nicastrin is dispensable for gamma-secretase protease activity in the presence of specific presenilin mutations. J. Biol. Chem. 2009, 284, 13013–13022. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Kryndushkin, D.; McGlinchey, R.; Bateman, D.; Kelly, A. Prion diseases of yeast: Amyloid structure and biology. Semin. Cell Dev. Biol. 2011, 22, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Bagriantsev, S.; Liebman, S. Modulation of Abeta42 low-n oligomerization using a novel yeast reporter system. BMC Biol. 2006, 4, 32. [Google Scholar] [CrossRef] [PubMed]
- von der Haar, T.; Jossé, L.; Wright, P.; Zenthon, J.; Tuite, M.F. Development of a novel yeast cell-based system for studying the aggregation of Alzheimer’s disease-associated Abeta peptides in vivo. Neurodegener. Dis. 2007, 4, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Caine, J.; Sankovich, S.; Antony, H.; Waddington, L.; Macreadie, P.; Varghese, J.; Macreadie, I. Alzheimer’s Abeta fused to green fluorescent protein induces growth stress and a heat shock response. FEMS Yeast Res. 2007, 7, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Hamos, J.E.; Oblas, B.; Pulaski-Salo, D.; Welch, W.J.; Bole, D.G.; Drachman, D.A. Expression of heat shock proteins in Alzheimer’s disease. Neurology 1991, 41, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Macreadie, I.; Lotfi-Miri, M.; Mohotti, S.; Shapira, D.; Bennett, L.; Varghese, J. Validation of folate in a convenient yeast assay suited for identification of inhibitors of Alzheimer’s amyloid-beta aggregation. J. Alzheimer’s Dis. 2008, 15, 391–396. [Google Scholar] [CrossRef]
- Rajasekhar, K.; Suresh, S.N.; Manjithaya, R.; Govindaraju, T. Rationally Designed Peptidomimetic Modulators of Aβ Toxicity in Alzheimer’s Disease. Sci. Rep. 2015, 5, 8139. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.R.; Verdile, G.; Barr, R.K.; Gupta, V.; Steele, J.W.; Lachenmayer, M.L.; Yue, Z.; Ehrlich, M.E.; Petsko, G.; Ju, S.; et al. Latrepirdine (dimebon) enhances autophagy and reduces intracellular GFP-Aβ42 levels in yeast. J. Alzheimer’s Dis. 2012, 32, 949–967. [Google Scholar] [CrossRef] [PubMed]
- .Nixon, R.A.; Wegiel, J.; Kumar, A.; Yu, W.H.; Peterhoff, C.; Cataldo, A.; Cuervo, A.M. Extensive involvement of autophagy in Alzheimer disease: An immuno-electron microscopy study. J. Neuropathol. Exp. Neurol. 2005, 64, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Cuervo, A.M.; Kumar, A.; Peterhoff, C.M.; Schmidt, S.D.; Lee, J.-H.; Mohan, P.S.; Mercken, M.; Farmery, M.R.; Tjernberg, L.O.; et al. Macroautophagy—A novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. J. Cell Biol. 2005, 171, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-H.; Yu, W.H.; Kumar, A.; Lee, S.; Mohan, P.S.; Peterhoff, C.M.; Wolfe, D.M.; Martinez-Vicente, M.; Massey, A.C.; Sovak, G.; et al. Lysosomal Proteolysis and Autophagy Require Presenilin 1 and Are Disrupted by Alzheimer-Related PS1 Mutations. Cell 2010, 141, 1146–1158. [Google Scholar] [CrossRef] [PubMed]
- Steele, J.W.; Lachenmayer, M.L.; Ju, S.; Stock, A.; Liken, J.; Kim, S.H.; Delgado, L.M.; Alfaro, I.E.; Bernales, S.; Verdile, G.; et al. Latrepirdine improves cognition and arrests progression of neuropathology in an Alzheimer’s mouse model. Mol. Psychiatry 2013, 18, 889–897. [Google Scholar] [CrossRef] [PubMed]
- Matlack, K.E.S.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef] [PubMed]
- Tardiff, D.F.; Brown, L.E.; Yan, X.; Trilles, R.; Jui, N.T.; Barrasa, M.I.; Caldwell, K.A.; Caldwell, G.A.; Schaus, S.E.; Lindquist, S. Dihydropyrimidine-Thiones and Clioquinol Synergize To Target β-Amyloid Cellular Pathologies through a Metal-Dependent Mechanism. ACS Chem. Neurosci. 2017, 8, 2039–2055. [Google Scholar] [CrossRef] [PubMed]
- Cherny, R.A.; Atwood, C.S.; Xilinas, M.E.; Gray, D.N.; Jones, W.D.; McLean, C.A.; Barnham, K.J.; Volitakis, I.; Fraser, F.W.; Kim, Y.-S.; et al. Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits β-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice. Neuron 2001, 30, 665–676. [Google Scholar] [CrossRef]
- Adlard, P.A.; Cherny, R.A.; Finkelstein, D.I.; Gautier, E.; Robb, E.; Cortes, M.; Volitakis, I.; Liu, X.; Smith, J.P.; Perez, K.; et al. Rapid Restoration of Cognition in Alzheimer’s Transgenic Mice with 8-Hydroxy Quinoline Analogs Is Associated with Decreased Interstitial Aβ. Neuron 2008, 59, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Aβ as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- LaFerla, F.M.; Green, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007, 8, 499–509. [Google Scholar] [CrossRef] [PubMed]
- .Treusch, S.; Hamamichi, S.; Goodman, J.L.; Matlack, K.E.S.; Chung, C.Y.; Baru, V.; Shulman, J.M.; Parrado, A.; Bevis, B.J.; Valastyan, J.S.; et al. Functional links between Aβ toxicity, endocytic trafficking, and Alzheimer’s disease risk factors in yeast. Science 2011, 334, 1241–1245. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, F.; Vignaud, H.; Di Martino, J.; Salin, B.; Devin, A.; Cullin, C.; Marchal, C. A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity. Dis. Model. Mech. 2013, 6, 206–216. [Google Scholar] [CrossRef] [PubMed]
- Park, S.-K.; Ratia, K.; Ba, M.; Valencik, M.; Liebman, S.W. Inhibition of Aβ42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs. Microb. Cell 2016, 3, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Petranovic, D. Amyloid-β peptide-induced cytotoxicity and mitochondrial dysfunction in yeast. FEMS Yeast Res. 2015, 15, fov061. [Google Scholar] [CrossRef] [PubMed]
- Fukui, H.; Moraes, C.T. The mitochondrial impairment, oxidative stress and neurodegeneration connection: Reality or just an attractive hypothesis? Trends Neurosci. 2008, 31, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Bisschops, M.M.M.; Agarwal, N.R.; Ji, B.; Shanmugavel, K.P.; Petranovic, D. Interplay of Energetics and ER Stress Exacerbates Alzheimer’s Amyloid-β (Aβ) Toxicity in Yeast. Front. Mol. Neurosci. 2017, 10, 232. [Google Scholar] [CrossRef] [PubMed]
- Stahl, A.; Moberg, P.; Ytterberg, J.; Panfilov, O.; Brockenhuus Von Lowenhielm, H.; Nilsson, F.; Glaser, E. Isolation and identification of a novel mitochondrial metalloprotease (PreP) that degrades targeting presequences in plants. J. Biol. Chem. 2002, 277, 41931–41939. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, N.; Berglund, A.-K.; Engmann, T.; Spånning, E.; Vögtle, F.-N.; Pavlov, P.; Meisinger, C.; Langer, T.; Glaser, E. Targeting Capacity and Conservation of PreP Homologues Localization in Mitochondria of Different Species. J. Mol. Biol. 2011, 410, 400–410. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, N.; Guo, L.; Yan, S.; Du, H.; Pinho, C.M.; Chen, J.X.; Glaser, E.; Yan, S.S. Decreased proteolytic activity of the mitochondrial amyloid-β degrading enzyme, PreP peptidasome, in Alzheimer’s disease brain mitochondria. J. Alzheimer’s Dis. 2011, 27, 75–87. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, P.F.; Glaser, E. Processing peptidases in mitochondria and chloroplasts. Biochim. Biophys. Acta-Mol. Cell Res. 2013, 1833, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Mossmann, D.; Vögtle, F.-N.; Taskin, A.A.; Teixeira, P.F.; Ring, J.; Burkhart, J.M.; Burger, N.; Pinho, C.M.; Tadic, J.; Loreth, D.; et al. Amyloid-β Peptide Induces Mitochondrial Dysfunction by Inhibition of Preprotein Maturation. Cell Metab. 2014, 20, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Brunetti, D.; Torsvik, J.; Dallabona, C.; Teixeira, P.; Sztromwasser, P.; Fernandez-Vizarra, E.; Cerutti, R.; Reyes, A.; Preziuso, C.; D’Amati, G.; et al. Defective PITRM1 mitochondrial peptidase is associated with A amyloidotic neurodegeneration. EMBO Mol. Med. 2016, 8, 176–190. [Google Scholar] [CrossRef] [PubMed]
- Cenini, G.; Rub, C.; Bruderek, M.; Voos, W. Amyloid β-peptides interfere with mitochondrial preprotein import competence by a coaggregation process. Mol. Biol. Cell 2016, 27, 3257–3272. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Waddington, L.; Varghese, J.; Macreadie, I.G. A new method to measure cellular toxicity of non-fibrillar and fibrillar Alzheimer’s Abeta using yeast. J. Alzheimer’s Dis. 2008, 13, 147–150. [Google Scholar] [CrossRef]
- Dubey, A.K.; Bharadwaj, P.R.; Varghese, J.N.; Macreadie, I.G. Alzheimer’s Amyloid-β Rescues Yeast from Hydroxide Toxicity. J. Alzheimer’s Dis. 2009, 18, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Liou, Y.-C.; Sun, A.; Ryo, A.; Zhou, X. Z.; Yu, Z.-X.; Huang, H.-K.; Uchida, T.; Bronson, R.; Bing, G.; Li, X.; et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature 2003, 424, 556–561. [Google Scholar] [CrossRef] [PubMed]
- Kondo, A.; Albayram, O.; Zhou, X.Z.; Lu, K.P. Pin1 Knockout Mice: A Model for the Study of Tau Pathology in Alzheimer’s Disease. Methods Mol Biol. 2017, 1523, 415–425. [Google Scholar] [PubMed]
- van Leeuwen, F.W.; de Kleijn, D.P.; van den Hurk, H.H.; Neubauer, A.; Sonnemans, M.A.; Sluijs, J.A.; Köycü, S.; Ramdjielal, R.D.; Salehi, A.; Martens, G.J.; et al. Frameshift mutants of beta amyloid precursor protein and ubiquitin-B in Alzheimer’s and Down patients. Science 1998, 279, 242–247. [Google Scholar] [CrossRef] [PubMed]
- Morishima-Kawashima, M.; Hasegawa, M.; Takio, K.; Suzuki, M.; Titani, K.; Ihara, Y. Ubiquitin is conjugated with amino-terminally processed tau in paired helical filaments. Neuron 1993, 10, 1151–1160. [Google Scholar] [CrossRef]
- Hilt, W.; Wolf, D.H. Proteasomes: Destruction as a programme. Trends Biochem. Sci. 1996, 21, 96–102. [Google Scholar] [CrossRef]
- van Tijn, P.; de Vrij, F.M.S.; Schuurman, K.G.; Dantuma, N.P.; Fischer, D.F.; van Leeuwen, F.W.; Hol, E.M. Dose-dependent inhibition of proteasome activity by a mutant ubiquitin associated with neurodegenerative disease. J. Cell Sci. 2007, 120, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Lindsten, K.; de Vrij, F.M.S.; Verhoef, L.G.G.C.; Fischer, D.F.; van Leeuwen, F.W.; Hol, E.M.; Masucci, M.G.; Dantuma, N.P. Mutant ubiquitin found in neurodegenerative disorders is a ubiquitin fusion degradation substrate that blocks proteasomal degradation. J. Cell Biol. 2002, 157, 417–427. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.J.; Sommer, C.; Leibiger, C.; Gentier, R.J.G.; Dumit, V.I.; Paduch, K.; Eisenberg, T.; Habernig, L.; Trausinger, G.; Magnes, C.; et al. Accumulation of Basic Amino Acids at Mitochondria Dictates the Cytotoxicity of Aberrant Ubiquitin. Cell Rep. 2015, 10, 1557–1571. [Google Scholar] [CrossRef] [PubMed]
- Tank, E.M.H.; True, H.L. Disease-Associated Mutant Ubiquitin Causes Proteasomal Impairment and Enhances the Toxicity of Protein Aggregates. PLoS Genet. 2009, 5, e1000382. [Google Scholar] [CrossRef] [PubMed]
- Krutauz, D.; Reis, N.; Nakasone, M.A.; Siman, P.; Zhang, D.; Kirkpatrick, D.S.; Gygi, S.P.; Brik, A.; Fushman, D.; Glickman, M.H. Extended ubiquitin species are protein-based DUB inhibitors. Nat. Chem. Biol. 2014, 10, 664–670. [Google Scholar] [CrossRef] [PubMed]
- De Vrij, F.M.S.; Sluijs, J.A.; Gregori, L.; Fischer, D.F.; Hermens, W.T.J.M.C.; Goldgaber, D.; Verhaagen, J.; Van Leeuwen, F.W.; Hol, E.M. Mutant ubiquitin expressed in Alzheimer’s disease causes neuronal death. FASEB J. 2001, 15, 2680–2688. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Sun, X.; Hou, F.-S.; Oh, H.-W.; Hilgenberg, L.G.W.; Hol, E.M.; van Leeuwen, F.W.; Smith, M.A.; O’Dowd, D.K.; Schreiber, S.S. Mutant ubiquitin found in Alzheimer’s disease causes neuritic beading of mitochondria in association with neuronal degeneration. Cell Death Differ. 2007, 14, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Braun, R.J. Ubiquitin-dependent proteolysis in yeast cells expressing neurotoxic proteins. Front. Mol. Neurosci. 2015, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb. Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. Creutzfeldt-Jakob disease and scrapie prions. Alzheimer Dis. Assoc. Disord. 1989, 3, 52–78. [Google Scholar] [CrossRef] [PubMed]
- Safar, J.G. Molecular pathogenesis of sporadic prion diseases in man. Prion 2012, 6, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Shewmaker, F.P.; Bateman, D.A.; Edskes, H.K.; Gorkovskiy, A.; Dayani, Y.; Bezsonov, E.E. Yeast Prions: Structure, Biology, and Prion-Handling Systems. Microbiol. Mol. Biol. Rev. 2015, 79, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Edskes, H.K.; Son, M.; Bezsonov, E.E.; DeWilde, M.; Ducatez, M. Yeast Prions Compared to Functional Prions and Amyloids. J. Mol. Biol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Riek, R.; Saupe, S.J. The HET-S/s Prion Motif in the Control of Programmed Cell Death. Cold Spring Harb. Perspect. Biol. 2016, 8, a023515. [Google Scholar] [CrossRef] [PubMed]
- Coustou, V.; Deleu, C.; Saupe, S.; Begueret, J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc. Natl. Acad. Sci. USA 1997, 94, 9773–9778. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Bezsonov, E.E.; Son, M.; Ducatez, M.; DeWilde, M.; Edskes, H.K. Anti-Prion Systems in Yeast and Inositol Polyphosphates. Biochemistry 2018, 57, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-X.; Greene, L.E.; Masison, D.C.; Eisenberg, E. Curing of yeast [PSI+] prion by guanidine inactivation of Hsp104 does not require cell division. Proc. Natl. Acad. Sci. USA 2005, 102, 12789–12794. [Google Scholar] [CrossRef] [PubMed]
- Wegrzyn, R.D.; Bapat, K.; Newnam, G.P.; Zink, A.D.; Chernoff, Y.O. Mechanism of prion loss after Hsp104 inactivation in yeast. Mol. Cell. Biol. 2001, 21, 4656–4669. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, H.; Edskes, H.K.; Wickner, R.B. [URE3] prion propagation in Saccharomyces cerevisiae: Requirement for chaperone Hsp104 and curing by overexpressed chaperone Ydj1p. Mol. Cell. Biol. 2000, 20, 8916–8922. [Google Scholar] [CrossRef] [PubMed]
- Kryndushkin, D.S.; Shewmaker, F.; Wickner, R.B. Curing of the [URE3] prion by Btn2p, a Batten disease-related protein. EMBO J. 2008, 27, 2725–2735. [Google Scholar] [CrossRef] [PubMed]
- Wickner, R.B.; Bezsonov, E.; Bateman, D.A. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc. Natl. Acad. Sci. USA 2014, 111, E2711–E2720. [Google Scholar] [CrossRef] [PubMed]
- O’Driscoll, J.; Clare, D.; Saibil, H. Prion aggregate structure in yeast cells is determined by the Hsp104-Hsp110 disaggregase machinery. J. Cell Biol. 2015, 211, 145–158. [Google Scholar] [CrossRef] [PubMed]
- Romanova, N.V.; Chernoff, Y.O. Hsp104 and prion propagation. Protein Pept. Lett. 2009, 16, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, A.; Soto, C. Prion-Like Protein Aggregates and Type 2 Diabetes. Cold Spring Harb. Perspect. Med. 2017, 7, a024315. [Google Scholar] [CrossRef] [PubMed]
- Moore, R.C.; Xiang, F.; Monaghan, J.; Han, D.; Zhang, Z.; Edström, L.; Anvret, M.; Prusiner, S.B. Huntington Disease Phenocopy Is a Familial Prion Disease. Am. J. Hum. Genet. 2001, 69, 1385–1388. [Google Scholar] [CrossRef] [PubMed]
- Abbott, A. The red-hot debate about transmissible Alzheimer’s. Nature 2016, 531, 294–297. [Google Scholar] [CrossRef] [PubMed]
- Olsson, T.T.; Klementieva, O.; Gouras, G.K. Prion-like seeding and nucleation of intracellular amyloid-β. Neurobiol. Dis. 2018, 113, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.-X.; Qiang, W.; Yau, W.-M.; Schwieters, C.D.; Meredith, S.C.; Tycko, R. Molecular Structure of β-Amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [Google Scholar] [CrossRef] [PubMed]
- Coalier, K.A.; Paranjape, G.S.; Karki, S.; Nichols, M.R. Stability of early-stage amyloid-β(1–42) aggregation species. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.B.; Diamond, M.I. Prion-like properties of Tau protein: The importance of extracellular Tau as a therapeutic target. J. Biol. Chem. 2014, 289, 19855–19861. [Google Scholar] [CrossRef] [PubMed]
- .Kaufman, S.K.; Sanders, D.W.; Thomas, T.L.; Ruchinskas, A.J.; Vaquer-Alicea, J.; Sharma, A.M.; Miller, T.M.; Diamond, M.I. Tau Prion Strains Dictate Patterns of Cell Pathology, Progression Rate, and Regional Vulnerability In Vivo. Neuron 2016, 92, 796–812. [Google Scholar] [CrossRef] [PubMed]
- Fruhmann, G.; Seynnaeve, D.; Zheng, J.; Ven, K.; Molenberghs, S.; Wilms, T.; Liu, B.; Winderickx, J.; Franssens, V. Yeast buddies helping to unravel the complexity of neurodegenerative disorders. Mech. Ageing Dev. 2017, 161, 288–305. [Google Scholar] [CrossRef] [PubMed]
- Brachmann, A.; Toombs, J.A.; Ross, E.D. Reporter assay systems for [URE3] detection and analysis. Methods 2006, 39, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Schlumpberger, M.; Prusiner, S.B.; Herskowitz, I. Induction of distinct [URE3] yeast prion strains. Mol. Cell. Biol. 2001, 21, 7035–7046. [Google Scholar] [CrossRef] [PubMed]
- Newby, G.A.; Kiriakov, S.; Hallacli, E.; Kayatekin, C.; Tsvetkov, P.; Mancuso, C.P.; Bonner, J.M.; Hesse, W.R.; Chakrabortee, S.; Manogaran, A.L.; et al. A Genetic Tool to Track Protein Aggregates and Control Prion Inheritance. Cell 2017, 171, 966–979.e18. [Google Scholar] [CrossRef] [PubMed]
- Chun, W.; Waldo, G.S.; Johnson, G.V.W. Split GFP complementation assay: a novel approach to quantitatively measure aggregation of tau in situ: Effects of GSK3β activation and caspase 3 cleavage. J. Neurochem. 2007, 103, 2529–2539. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Yang, Q.; Zheng, J.; Zhu, X.; Hao, X.; Song, J.; Lebacq, T.; Franssens, V.; Winderickx, J.; Nystrom, T.; et al. A genome-wide imaging-based screening to identify genes involved in synphilin-1 inclusion formation in Saccharomyces cerevisiae. Sci. Rep. 2016, 6, 30134. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Li, Y.; Zhang, Y. Screening of APP interaction proteins by DUALmembrane yeast two-hybrid system. Int. J. Clin. Exp. Pathol. 2015, 8, 2802–2808. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Toxicity Modifiers | Description | Other Models | References |
|---|---|---|---|---|
| Tau | Pin1 (yeast homologue Ess1) | Depletion of Pin1 isomerase activity results in reduced growth of Tau expressing yeast cells. | mouse model | [93,196,197] |
| Aβ | peptidomimetic inhibitors | Inhibition of Aβ42 aggregation by peptidomimetics. | - | [169] |
| Aβ | latrepirdine (Dimebon™) | Latrepirdine induces autophagy and decreases the intracellular GFP-Aβ42 levels in yeast. | Hela cells, mouse model | [170,174] |
| Aβ | clioquinol | Small molecule screen identified several 8-hydroxyquinolines, including clioquinol, that ameliorate Aβ toxicity. | mouse model, nematode model | [175,176,177,178,179] |
| Aβ | dihydropyrimidine-thiones | Phenotypic small molecule yeast screen identified dihydropyrimidine-thiones that rescue Aβ-induced toxicity in a metal dependent manner. | nematode model | [176] |
| Aβ | PICALM (yeast homologues Yap1801, Yap1802) | Screening of overexpression library yielded suppressors and enhancers of Aβ42 toxicity, including the PICALM suppressor. | rat cortical neurons | [181,182,183] |
| Technique | Used for | Description |
|---|---|---|
| Split-GFP system [111,112,113,114,115,236] | Protein–protein interaction | GFP fluorescence is reconstituted when its two subunits are in close proximity. |
| Synthetic genetic array [237] | Synthetic lethality | Approach for the systematic construction of double mutants for large-scale mapping of synthetic genetic interactions. |
| Yeast two-hybrid [238] | Protein–protein interaction | Protein interaction leads to reporter gene expression. |
| Prion-forming assay [233] | Prion forming | The prion domain of the yeast Ure2 prion is replaced by a potential prion domain of any protein. Reporter gene expression is induced if this domain can complement for the Ure2 prion domain. |
| Yeast transcriptional reporting aggregating proteins (yTRAP) [235] | Prion forming | High-throughput quantitative prion forming assay. Uses fluorescence as quantifiable reporter. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Seynnaeve, D.; Vecchio, M.D.; Fruhmann, G.; Verelst, J.; Cools, M.; Beckers, J.; Mulvihill, D.P.; Winderickx, J.; Franssens, V. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. Int. J. Mol. Sci. 2018, 19, 1947. https://doi.org/10.3390/ijms19071947
Seynnaeve D, Vecchio MD, Fruhmann G, Verelst J, Cools M, Beckers J, Mulvihill DP, Winderickx J, Franssens V. Recent Insights on Alzheimer’s Disease Originating from Yeast Models. International Journal of Molecular Sciences. 2018; 19(7):1947. https://doi.org/10.3390/ijms19071947
Chicago/Turabian StyleSeynnaeve, David, Mara Del Vecchio, Gernot Fruhmann, Joke Verelst, Melody Cools, Jimmy Beckers, Daniel P. Mulvihill, Joris Winderickx, and Vanessa Franssens. 2018. "Recent Insights on Alzheimer’s Disease Originating from Yeast Models" International Journal of Molecular Sciences 19, no. 7: 1947. https://doi.org/10.3390/ijms19071947
APA StyleSeynnaeve, D., Vecchio, M. D., Fruhmann, G., Verelst, J., Cools, M., Beckers, J., Mulvihill, D. P., Winderickx, J., & Franssens, V. (2018). Recent Insights on Alzheimer’s Disease Originating from Yeast Models. International Journal of Molecular Sciences, 19(7), 1947. https://doi.org/10.3390/ijms19071947