The Contribution of EDF1 to PPARγ Transcriptional Activation in VEGF-Treated Human Endothelial Cells


Abstract
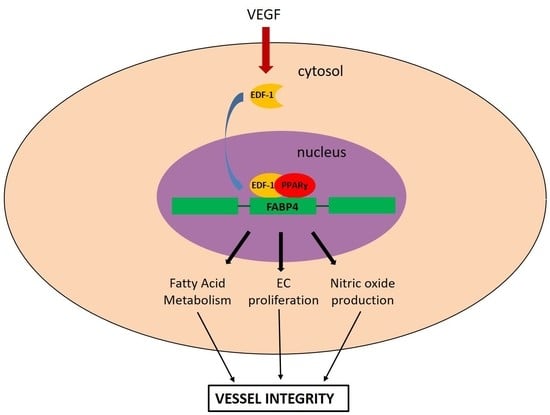
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
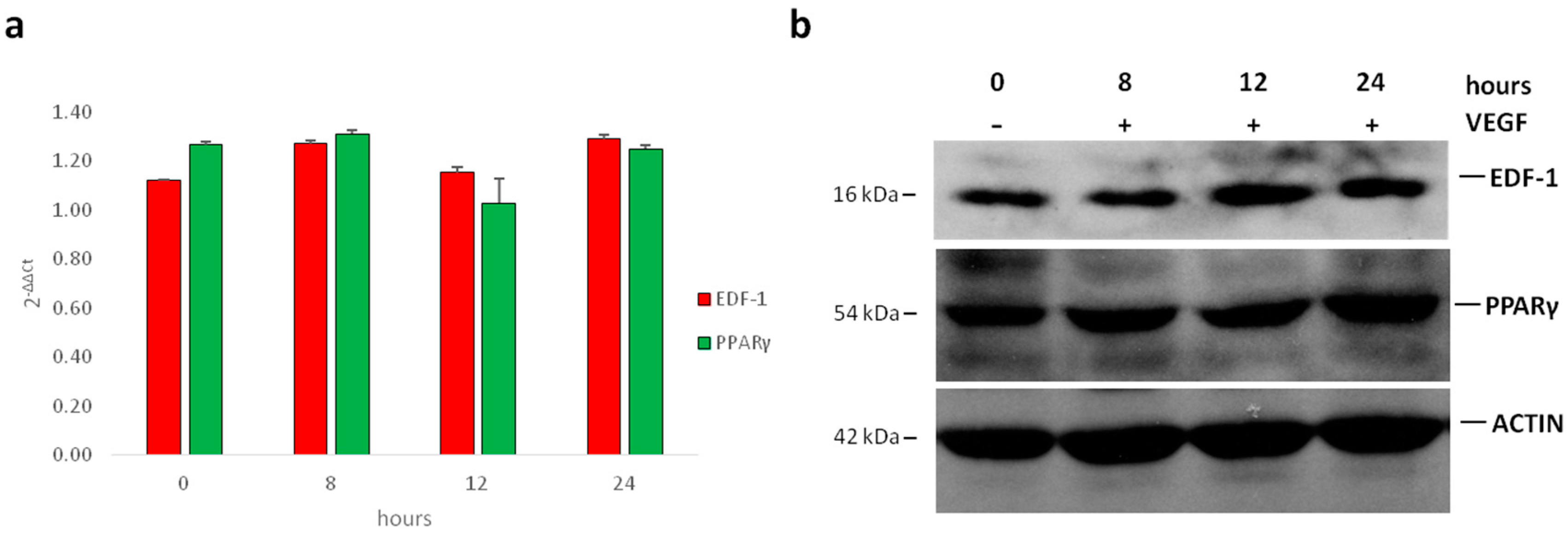
2.1. Translocation of EDF1 to the Nucleus in Response to VEGF
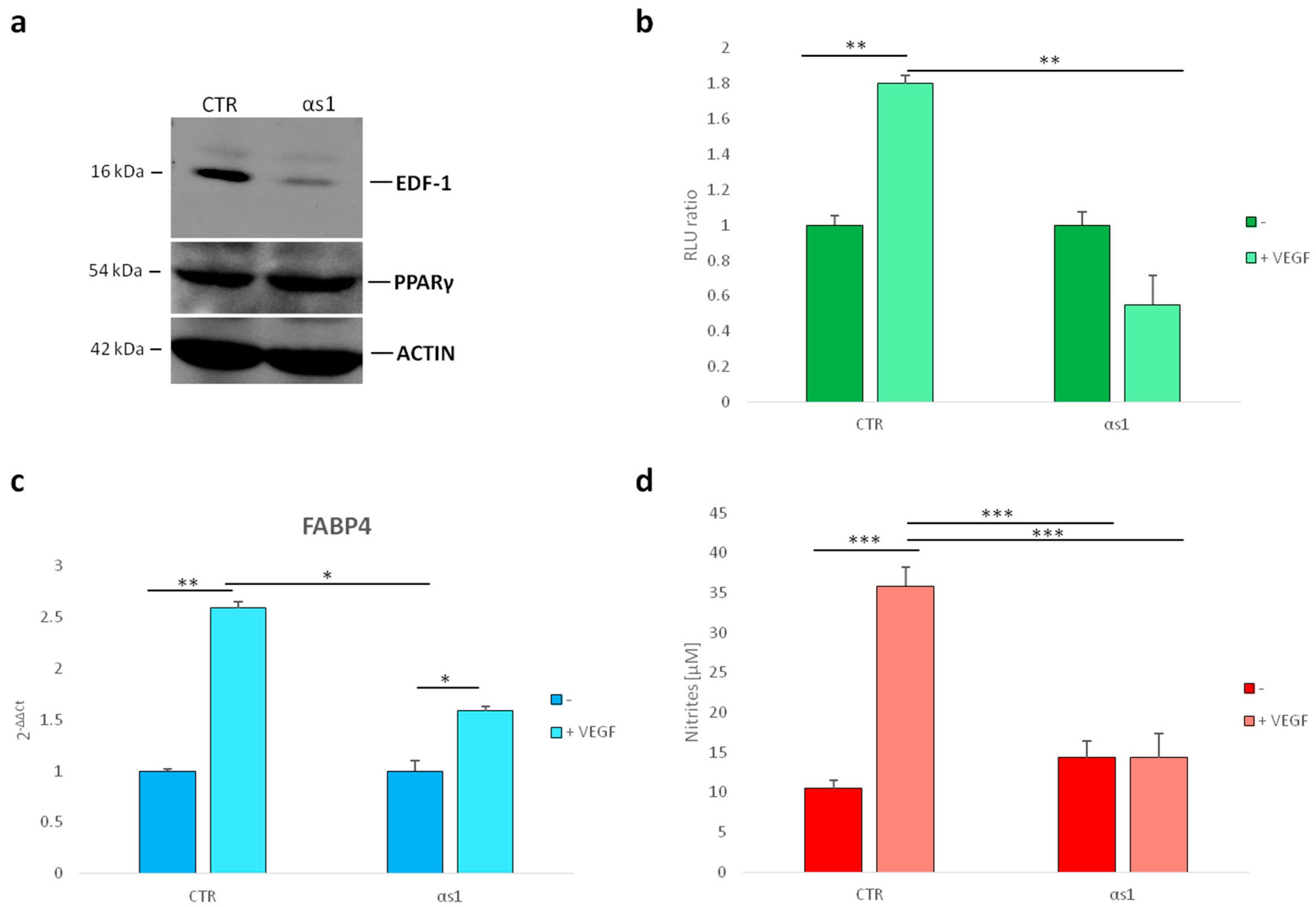
2.2. Interaction between EDF1 and PPARγ in HUVEC
2.3. Effect of Silencing EDF1 in VEGF-Induced PPARγ Activity
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Western Blot and Immunoprecipitation
4.3. Immunofluorescence Staining
4.4. Reporter Gene Assay
4.5. Real-Time-PCR
4.6. NO Release
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| VEGF | Vascular Endothelial Growth Factor |
| EDF | Endothelial Differentiation-related factor |
| PPARγ | Peroxisome proliferator-activated receptor γ |
| EC | Endothelial cells |
| NO | Nitric oxide |
References
- Duan, S.Z.; Usher, M.G.; Mortensen, R.M. Peroxisome Proliferator-Activated Receptor—Mediated Effects in the Vasculature. Circ. Res. 2008, 102, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Chawla, A.; Schwarz, E.J.; Dimaculangan, D.D.; Lazar, M.A. Peroxisome proliferator-activated receptor (PPAR) γ: Adipose predominant expression and induction early in adipocyte differentiation. Endocrinology 1994, 135, 798–800. [Google Scholar] [CrossRef] [PubMed]
- Ahmadian, M.; Suh, J.M.; Hah, N.; Liddle, C.; Atkins, A.R.; Downes, M.; Evans, R.M. PPARgamma signaling and metabolism: The good, the bad and the future. Nat. Med. 2013, 19, 557–566. [Google Scholar] [CrossRef] [PubMed]
- Imai, T.; Takakuwa, R.; Marchand, S.; Dentz, E.; Bornert, J.M.; Messaddeq, N.; Wendling, O.; Mark, M.; Desvergne, B.; Wahli, W.; et al. Peroxisome proliferator-activated receptor γ is required in mature white and brown adipocytes for their survival in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 4543–4547. [Google Scholar] [CrossRef] [PubMed]
- Tomaru, T.; Steger, D.J.; Lefterova, M.I.; Schupp, M.; Lazar, M.A. Adipocyte specific expression of murine resistin is mediated by synergism between peroxisome proliferator-activated receptor γ and CCAAT/enhancer-binding proteins. J. Biol. Chem. 2009, 284, 6116–6125. [Google Scholar] [CrossRef] [PubMed]
- Iwaki, M.; Matsuda, M.; Maeda, N.; Funahashi, T.; Matsuzawa, Y.; Makishima, M.; Shimomura, I. Induction of adiponectin, a fat-derived antidiabetic and antiatherogenic factor, by nuclear receptors. Diabetes 2003, 52, 1655–1663. [Google Scholar] [CrossRef] [PubMed]
- Halabi, C.M.; Beyer, A.M.; de Lange, W.J.; Keen, H.L.; Baumbach, G.L.; Faraci, F.M.; Sigmund, C.D. Interference with PPARγ Function in Smooth Muscle Causes Vascular Dysfunction and Hypertension. Cell Metab. 2008, 7, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Kotlinowski, J.; Jozkowicz, A. PPAR Gamma and Angiogenesis: Endothelial Cells Perspective. J. Diabetes Res. 2016, 2016, 8492353. [Google Scholar] [CrossRef] [PubMed]
- Kleinhenz, J.M.; Kleinhenz, D.J.; You, S.; Ritzenthaler, J.D.; Hansen, J.M.; Archer, D.R.; Sutliff, R.L.; Hart, C.M. Disruption of endothelial peroxisome proliferator-activated receptor-γ reduces vascular nitric oxide production. Am. J. Physiol. Heart Circ. Physiol. 2009, 297, H1647–H1654. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Ishibashi, S.; Perrey, S.; Osuga, J.; Gotoda, T.; Kitamine, T.; Tamura, Y.; Okazaki, H.; Yahagi, N.; Iizuka, Y.; et al. Troglitazone inhibits atherosclerosis in apolipoprotein E-knockout mice: Pleiotropic effects on CD36 expression and HDL. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Charbonnel, B.; Dormandy, J.; Erdmann, E.; Massi-Benedetti, M.; Skene, A.; PROactive Study Group. The prospective pioglitazone clinical trial in macrovascular events (PROactive): Can pioglitazone reduce cardiovascular events in diabetes? Study design and baseline characteristics of 5238 patients. Diabetes Care 2004, 27, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Dougherty, E.J.; Danner, R.L. PPARγ signaling and emerging opportunities for improved therapeutics. Pharmacol. Res. 2016, 111, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, M.G.; Lunyak, V.V.; Glass, C.K. Sensors and signals: A coactivator/corepressor/epigenetic code for integrating signal-dependent programs of transcriptional response. Genes Dev. 2005, 20, 1405–1428. [Google Scholar] [CrossRef] [PubMed]
- Brendel, C.; Gelman, L.; Auwerx, J. Multiprotein bridging factor-1 (MBF-1) is a cofactor for nuclear receptors that regulate lipid metabolism. Mol. Endocrinol. 2002, 16, 1367–1377. [Google Scholar] [CrossRef] [PubMed]
- Leidi, M.; Mariotti, M.; Maier, J.A. Transcriptional coactivator EDF-1 is required for PPARgamma-stimulated adipogenesis. Cell. Mol. Life Sci. 2009, 66, 2733–2742. [Google Scholar] [CrossRef] [PubMed]
- Takemaru, K.; Li, F.Q.; Ueda, H.; Hirose, S. Multiprotein bridging factor 1 (MBF1) is an evolutionarily conserved transcriptional coactivator that connects a regulatory factor and TATA element-binding protein. Proc. Natl. Acad. Sci. USA 1997, 94, 7251–7256. [Google Scholar] [CrossRef] [PubMed]
- Busk, P.K.; Wulf-Andersen, L.; Strøm, C.C.; Enevoldsen, M.; Thirstrup, K.; Haunsø, S.; Sheikh, S.P. Multiprotein bridging factor 1 cooperates with c-Jun and is necessary for cardiac hypertrophy in vitro. Exp. Cell Res. 2003, 286, 102–114. [Google Scholar] [CrossRef]
- Liu, Q.X.; Jindra, M.; Ueda, H.; Hiromi, Y.; Hirose, S. Drosophila MBF1 is a co-activator for Tracheae Defective and contributes to the formation of tracheal and nervous systems. Development 2003, 130, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, I.; Mariotti, M.; Consalez, G.G.; Soria, M.R.; Maier, J.A. EDF-1, a novel gene product down-regulated in human endothelial cell differentiation. J. Biol. Chem. 1998, 273, 31119–31124. [Google Scholar] [CrossRef] [PubMed]
- Ballabio, E.; Mariotti, M.; De Benedictis, L.; Maier, J.A. The dual role of endothelial differentiation-related factor-1 in the cytosol and nucleus: Modulation by protein kinase A. Cell. Mol. Life Sci. 2004, 61, 1069–1074. [Google Scholar] [CrossRef] [PubMed]
- Bautch, V.L. VEGF-Directed Blood Vessel Patterning: From Cells to Organism. Cold Spring Harb. Perspect. Med. 2012, 2, a006452. [Google Scholar] [CrossRef] [PubMed]
- Mariotti, M.; De Benedictis, L.; Avon, E.; Maier, J.A.M. Interaction between endothelial differentiation-related factor-1 and calmodulin in vitro and in vivo. J. Biol. Chem. 2000, 275, 24047–24051. [Google Scholar] [CrossRef] [PubMed]
- Leidi, M.; Mariotti, M.; Maier, J.A. EDF-1 contributes to the regulation of nitric oxide release in VEGF-treated human endothelial cells. Eur. J. Cell Biol. 2010, 89, 654–660. [Google Scholar] [CrossRef] [PubMed]
- Castiglioni, S.; Cazzaniga, A.; Maier, J.A. Potential interplay between NFκB and PPARγ in human dermal microvascular endothelial cells cultured in low magnesium. Magnes. Res. 2014, 27, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Furuhashi, M.; Saitoh, S.; Shimamoto, K.; Miura, T. Fatty Acid-Binding Protein 4 (FABP4): Pathophysiological Insights and Potent Clinical Biomarker of Metabolic and Cardiovascular Diseases. Clin. Med. Insights Cardiol. 2015, 8, 23–33. [Google Scholar] [CrossRef] [PubMed]
- Elmasri, H.; Ghelfi, E.; Yu, C.W.; Traphagen, S.; Cernadas, M.; Cao, H.; Shi, G.P.; Plutzky, J.; Sahin, M.; Hotamisligil, G.; et al. Endothelial cell-fatty acid binding protein 4 promotes angiogenesis: Role of stem cell factor/c-kit pathway. Angiogenesis 2012, 15, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Magri, C.J.; Gatt, N.; Xuereb, R.G.; Fava, S. Peroxisome proliferator-activated receptor-γ and the endothelium: Implications in cardiovascular disease. Expert Rev. Cardiovasc. Ther. 2011, 9, 1279–1294. [Google Scholar] [CrossRef] [PubMed]
- Aragonès, G.; Saavedra, P.; Heras, M.; Cabré, A.; Girona, J.; Masana, L. Fatty acid-binding protein 4 impairs the insulin-dependent nitric oxide pathway in vascular endothelial cells. Cardiovasc. Diabetol. 2012, 11, 72. [Google Scholar] [CrossRef] [PubMed]
- Gheibi, S.; Jeddi, S.; Kashfi, K.; Ghasemi, A. Regulation of vascular tone homeostasis by NO and H2S: Implications in hypertension. Biochem. Pharmacol. 2018, 149, 42–59. [Google Scholar] [CrossRef] [PubMed]
- Cho, D.H.; Choi, Y.J.; Jo, S.A.; Jo, I. Nitric oxide production and regulation of endothelial nitric-oxide synthase phosphorylation by prolonged treatment with troglitazone: Evidence for involvement of peroxisome proliferator-activated receptor (PPAR) gamma-dependent and PPARgamma-independent signaling pathways. J. Biol. Chem. 2004, 279, 2499–2506. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cazzaniga, A.; Locatelli, L.; Castiglioni, S.; Maier, J. The Contribution of EDF1 to PPARγ Transcriptional Activation in VEGF-Treated Human Endothelial Cells. Int. J. Mol. Sci. 2018, 19, 1830. https://doi.org/10.3390/ijms19071830
Cazzaniga A, Locatelli L, Castiglioni S, Maier J. The Contribution of EDF1 to PPARγ Transcriptional Activation in VEGF-Treated Human Endothelial Cells. International Journal of Molecular Sciences. 2018; 19(7):1830. https://doi.org/10.3390/ijms19071830
Chicago/Turabian StyleCazzaniga, Alessandra, Laura Locatelli, Sara Castiglioni, and Jeanette Maier. 2018. "The Contribution of EDF1 to PPARγ Transcriptional Activation in VEGF-Treated Human Endothelial Cells" International Journal of Molecular Sciences 19, no. 7: 1830. https://doi.org/10.3390/ijms19071830
APA StyleCazzaniga, A., Locatelli, L., Castiglioni, S., & Maier, J. (2018). The Contribution of EDF1 to PPARγ Transcriptional Activation in VEGF-Treated Human Endothelial Cells. International Journal of Molecular Sciences, 19(7), 1830. https://doi.org/10.3390/ijms19071830