Functional Regulation of PPARs through Post-Translational Modifications


Abstract
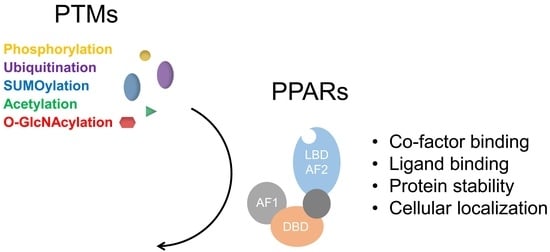
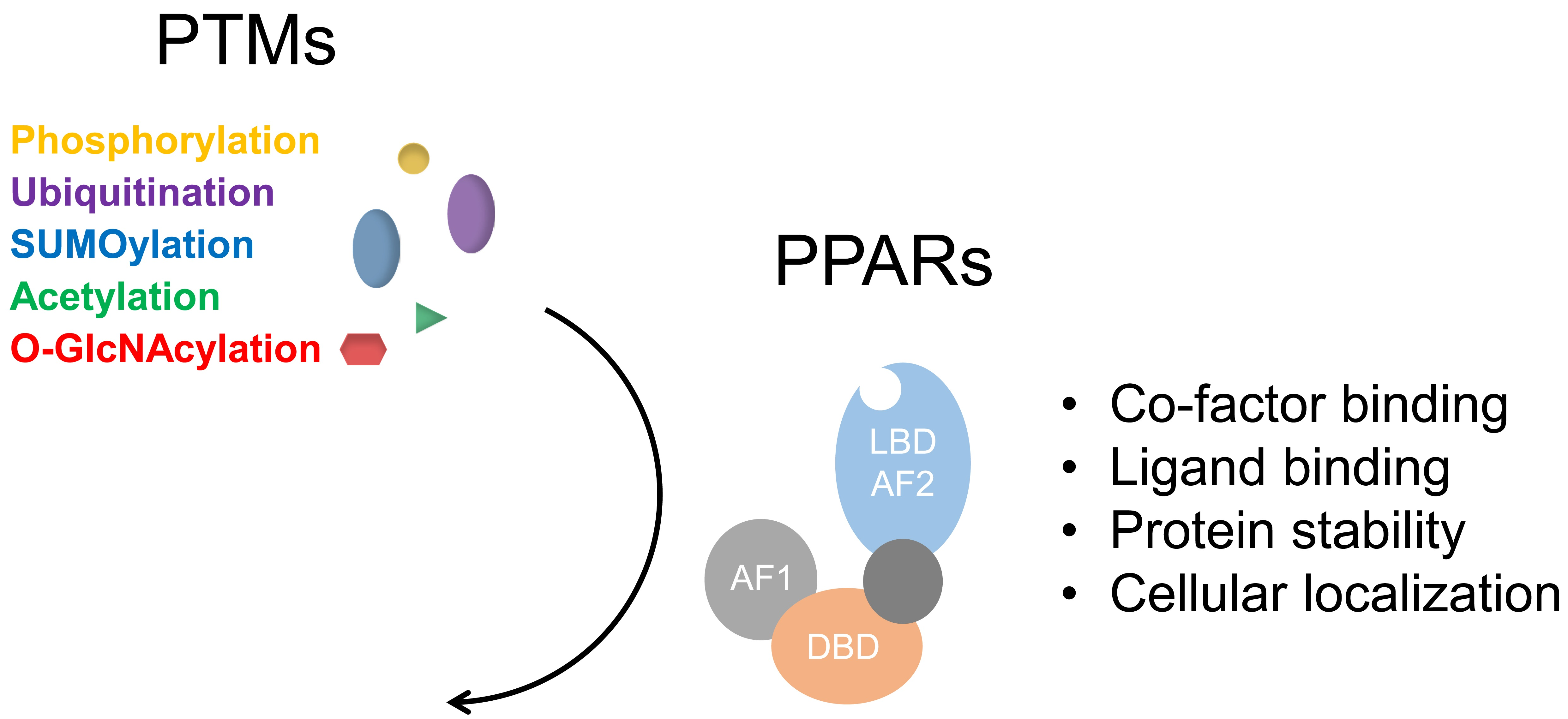
1. Introduction
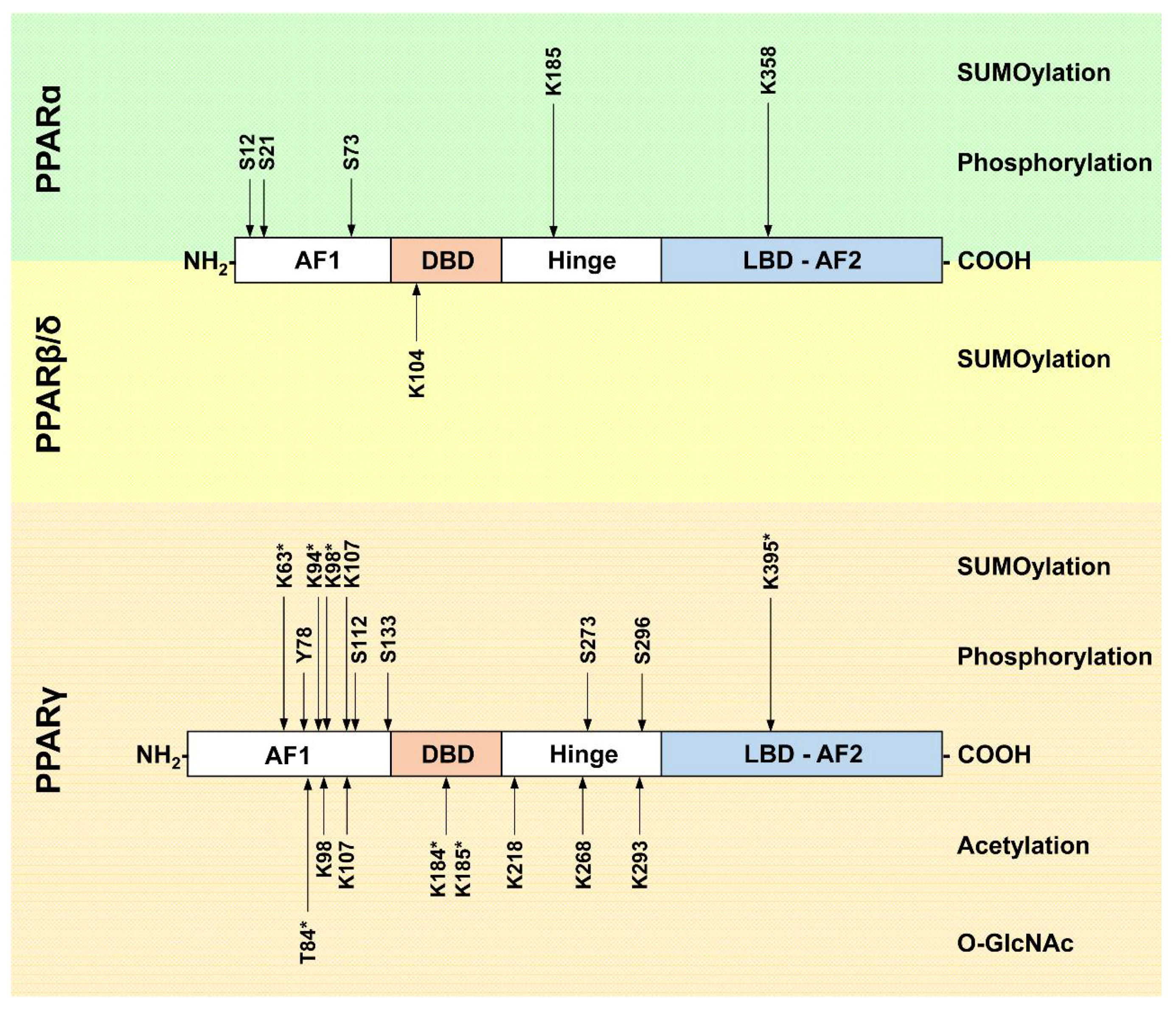
2. Post-Translational Modifications of PPARα
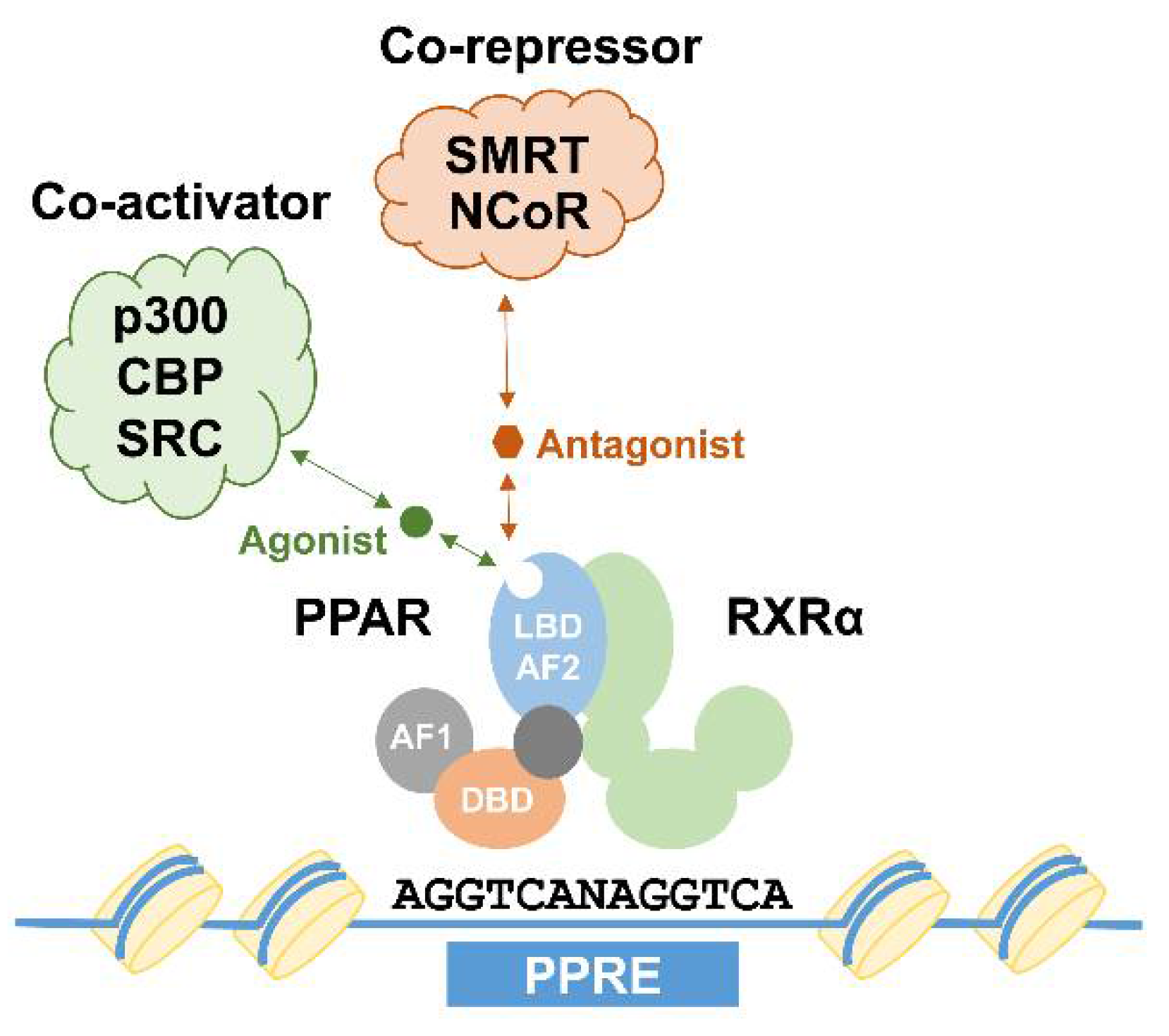
2.1. Phosphorylation
2.2. SUMOylation
2.3. Ubiquitination
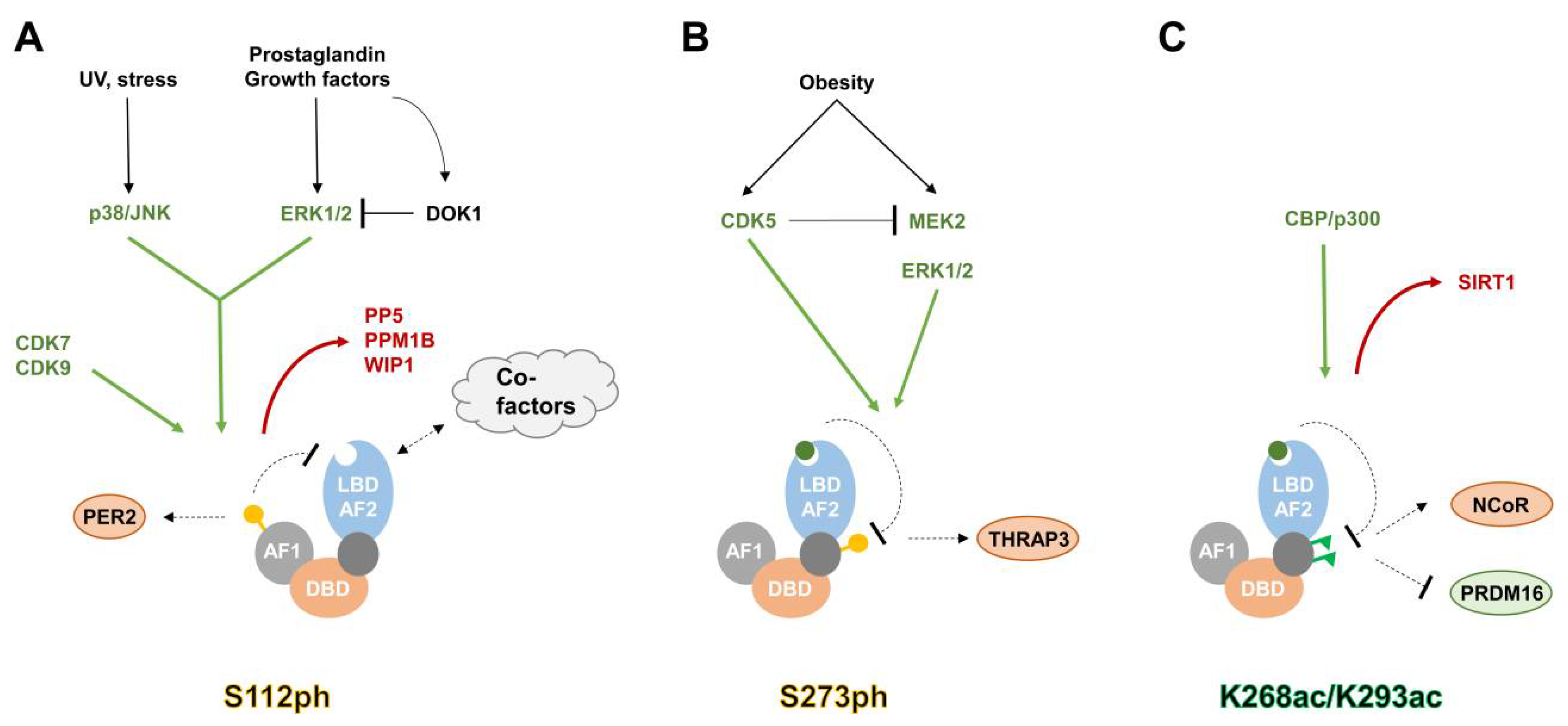
3. Post-Translational Modifications of PPARγ
3.1. Phosphorylation
3.2. SUMOylation
3.3. Acetylation
3.4. Ubiquitination
3.5. O-GlcNAcylation
4. Post-Translational Modifications of PPARβ/δ
SUMOylation
5. Outlook/Perspective
Acknowledgments
Conflicts of Interest
Abbreviations
| PPAR | Peroxisome Proliferator Activated Receptor |
| NR | Nuclear receptor |
| TF | Transcription factor |
| AF-1 | Activation-function 1 |
| DBD | DNA-binding domain |
| LBD | Ligand-binding domain |
| RXRα | Retinoic acid receptor α |
| PPRE | PPAR response element |
| NCoR | Nuclear receptor corepressor |
| SMART | Silencing mediator of retinoid and thyroid hormone receptor |
| CBP | CREB-binding protein |
| SRC1 | Steroid receptor coactivator 1 |
| T2D | Type 2 diabetes |
| NAFLD | Nonalcoholic fatty liver disease |
| NASH | Nonalcoholic steatosis |
| TZD | Thiazolidinedione |
| BAT | Brown adipose tissue |
| FAO | Fatty acid oxidation |
| WAT | White adipose tissue |
| PTM | Post-translational modification |
| MAPK | Mitogen-activated protein kinase |
| CDK | Cyclin-dependent kinase |
| PGC1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| XPD | Xeroderma pigmentosum group D |
| TFIIH | Transcription factor II H |
| GSKβ | Glycogen synthase kinase β |
| SUMO | Small ubiquitin-like modifier |
| GABP | GA-binding protein |
| MuRF1 | Muscle-specific RING finger protein 1 |
| EGF | Epidermal growth factor |
| PDGF | Platelet-derived growth factor |
| TGFβ | Transforming growth factor β |
| TPA | 12-O-tetradecanoyl-13-phorbol acetate |
| PGF2α | Prostaglandin F2α |
| ERK | Extracellular signal–regulated kinase |
| MEK2 | Dual specificity mitogen-activated protein kinase kinase 2 |
| JNK | c-Jun N-terminal kinase |
| DOK1 | Docking protein 1 |
| HFD | High fat diet |
| PP5 | Protein phosphatase 5 |
| PPM1B | Protein phosphatase Mg2+/Mn2+ dependent 1B |
| WIP1 | Wild-type p53-induced phosphatase 1 |
| PER2 | Period circadian regulator 2 |
| GWAS | Genome-wide association study |
| THRAP3 | Thyroid hormone receptor associated protein 3 |
| LC-MS/MS | Liquid chromatography-tandem mass spectrometry |
| c-SRC | SRC proto-oncogene, non-receptor tyrosine kinase |
| PTP-1B | Protein-tyrosine phosphatase 1B |
| UBC9 | Ubiquitin conjugating enzyme 9 |
| PIAS | Protein inhibitor of activated STAT |
| SENP2 | SUMO-specific protease 2 |
| FGF21 | Fibroblast growth factor 21 |
| GDF11 | Growth differentiation factor 11 |
| SIRT1 | NAD-dependent deacetylase sirtuin-1 |
| PRDM16 | PR domain containing 16 |
| SIAH2 | Seven in absentia homolog 2 |
| MKRN1 | Makorin RING finger protein 1 |
| TRIM23 | Tripartite motif containing 23 |
| NEDD4 | Neural precursor cell expressed, developmentally down-regulated 4 |
| O-GlcNAc | β-O-linked N-acetylglucosamine |
| ChIP-seq | Chromatin immunoprecipitation-sequencing |
| SNP | Single-nucleotide polymorphism |
References
- Monsalve, F.A.; Pyarasani, R.D.; Delgado-Lopez, F.; Moore-Carrasco, R. Peroxisome proliferator-activated receptor targets for the treatment of metabolic diseases. Mediat. Inflamm. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Maccallini, C.; Mollica, A.; Amoroso, R. The positive regulation of enos signaling by PPAR agonists in cardiovascular diseases. Am. J. Cardiovasc. Drugs 2017, 17, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Tenenbaum, A.; Fisman, E.Z. Fibrates are an essential part of modern anti-dyslipidemic arsenal: Spotlight on atherogenic dyslipidemia and residual risk reduction. Cardiovasc. Diabetol. 2012, 11. [Google Scholar] [CrossRef] [PubMed]
- Davidson, M.A.; Mattison, D.R.; Azoulay, L.; Krewski, D. Thiazolidinedione drugs in the treatment of type 2 diabetes mellitus: Past, present and future. Crit. Rev. Toxicol. 2018, 48, 52–108. [Google Scholar] [CrossRef] [PubMed]
- Bortolini, M.; Wright, M.B.; Bopst, M.; Balas, B. Examining the safety of PPAR agonists—Current trends and future prospects. Expert Opin. Drug Saf. 2013, 12, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Dubois, V.; Eeckhoute, J.; Lefebvre, P.; Staels, B. Distinct but complementary contributions of PPAR isotypes to energy homeostasis. J. Clin. Investig. 2017, 127, 1202–1214. [Google Scholar] [CrossRef] [PubMed]
- Tontonoz, P.; Hu, E.; Spiegelman, B.M. Stimulation of adipogenesis in fibroblasts by PPARγ 2, a lipid-activated transcription factor. Cell 1994, 79, 1147–1156. [Google Scholar] [CrossRef]
- Gross, B.; Pawlak, M.; Lefebvre, P.; Staels, B. PPARs in obesity-induced T2dm, dyslipidaemia and NAFLD. Nat. Rev. Endocrinol. 2017, 13, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.K.; Zhuang, Y.; Wahli, W. Synthetic and natural peroxisome proliferator-activated receptor (PPAR) agonists as candidates for the therapy of the metabolic syndrome. Expert Opin. Ther. Targets 2017, 21, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Shalev, A.; Siegrist-Kaiser, C.A.; Yen, P.M.; Wahli, W.; Burger, A.G.; Chin, W.W.; Meier, C.A. The peroxisome proliferator-activated receptor α is a phosphoprotein: Regulation by insulin. Endocrinology 1996, 137, 4499–4502. [Google Scholar] [CrossRef] [PubMed]
- Passilly, P.; Schohn, H.; Jannin, B.; Cherkaoui Malki, M.; Boscoboinik, D.; Dauca, M.; Latruffe, N. Phosphorylation of peroxisome proliferator-activated receptor α in rat FAO cells and stimulation by ciprofibrate. Biochem. Pharmacol. 1999, 58, 1001–1008. [Google Scholar] [CrossRef]
- Barger, P.M.; Browning, A.C.; Garner, A.N.; Kelly, D.P. P38 mitogen-activated protein kinase activates peroxisome proliferator-activated receptor α: A potential role in the cardiac metabolic stress response. J. Biol. Chem. 2001, 276, 44495–44501. [Google Scholar] [CrossRef] [PubMed]
- Juge-Aubry, C.E.; Hammar, E.; Siegrist-Kaiser, C.; Pernin, A.; Takeshita, A.; Chin, W.W.; Burger, A.G.; Meier, C.A. Regulation of the transcriptional activity of the peroxisome proliferator-activated receptor α by phosphorylation of a ligand-independent trans-activating domain. J. Biol. Chem. 1999, 274, 10505–10510. [Google Scholar] [CrossRef] [PubMed]
- Compe, E.; Drane, P.; Laurent, C.; Diderich, K.; Braun, C.; Hoeijmakers, J.H.; Egly, J.M. Dysregulation of the peroxisome proliferator-activated receptor target genes by xpd mutations. Mol. Cell Biol. 2005, 25, 6065–6076. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Burns, K.A.; Hosick, P.A.; McBeth, L.; Nestor-Kalinoski, A.; Drummond, H.A.; AlAmodi, A.A.; Hankins, M.W.; Vanden Heuvel, J.P.; Stec, D.E. Biliverdin reductase a attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3β phosphorylation of serine 73 of peroxisome proliferator-activated receptor (PPAR) α. J. Biol. Chem. 2016, 291, 25179–25191. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Hosick, P.A.; Chen, S.; Tukey, R.H.; Hankins, M.W.; Nestor-Kalinoski, A.; Stec, D.E. Mice with hyperbilirubinemia due to gilbert’s syndrome polymorphism are resistant to hepatic steatosis by decreased serine 73 phosphorylation of PPARα. Am. J. Physiol. Endocrinol. Metab. 2017, 312, E244–E252. [Google Scholar] [CrossRef] [PubMed]
- Molzer, C.; Wallner, M.; Kern, C.; Tosevska, A.; Schwarz, U.; Zadnikar, R.; Doberer, D.; Marculescu, R.; Wagner, K.H. Features of an altered AMPK metabolic pathway in gilbert’s syndrome, and its role in metabolic health. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Flotho, A.; Melchior, F. SUMOylation: A regulatory protein modification in health and disease. Annu. Rev. Biochem. 2013, 82, 357–385. [Google Scholar] [CrossRef] [PubMed]
- Pourcet, B.; Pineda-Torra, I.; Derudas, B.; Staels, B.; Glineur, C. SUMOylation of human peroxisome proliferator-activated receptor α inhibits its trans-activity through the recruitment of the nuclear corepressor ncor. J. Biol. Chem. 2010, 285, 5983–5992. [Google Scholar] [CrossRef] [PubMed]
- Leuenberger, N.; Pradervand, S.; Wahli, W. Sumoylated PPARα mediates sex-specific gene repression and protects the liver from estrogen-induced toxicity in mice. J. Clin. Investig. 2009, 119, 3138–3148. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Willis, M.S. The story so far: Post-translational regulation of peroxisome proliferator-activated receptors by ubiquitination and SUMOylation. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H515–H526. [Google Scholar] [CrossRef] [PubMed]
- Gopinathan, L.; Hannon, D.B.; Peters, J.M.; Vanden Heuvel, J.P. Regulation of peroxisome proliferator-activated receptor-α by MDM2. Toxicol. Sci. 2009, 108, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.E.; Liao, J.Y.; He, J.; Schisler, J.C.; Newgard, C.B.; Drujan, D.; Glass, D.J.; Frederick, C.B.; Yoder, B.C.; Lalush, D.S.; et al. The ubiquitin ligase MURF1 regulates PPARα activity in the heart by enhancing nuclear export via monoubiquitination. Mol. Cell Endocrinol. 2015, 413, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Hu, E.; Kim, J.B.; Sarraf, P.; Spiegelman, B.M. Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 1996, 274, 2100–2103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Berger, J.; Zhou, G.; Elbrecht, A.; Biswas, S.; White-Carrington, S.; Szalkowski, D.; Moller, D.E. Insulin- and mitogen-activated protein kinase-mediated phosphorylation and activation of peroxisome proliferator-activated receptor γ. J. Biol. Chem. 1996, 271, 31771–31774. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Reginato, M.J.; Shao, D.; Lazar, M.A.; Chatterjee, V.K. Transcriptional activation by peroxisome proliferator-activated receptor γ is inhibited by phosphorylation at a consensus mitogen-activated protein kinase site. J. Biol. Chem. 1997, 272, 5128–5132. [Google Scholar] [CrossRef] [PubMed]
- Reginato, M.J.; Krakow, S.L.; Bailey, S.T.; Lazar, M.A. Prostaglandins promote and block adipogenesis through opposing effects on peroxisome proliferator-activated receptor γ. J. Biol. Chem. 1998, 273, 1855–1858. [Google Scholar] [CrossRef] [PubMed]
- Camp, H.S.; Tafuri, S.R. Regulation of peroxisome proliferator-activated receptor γ activity by mitogen-activated protein kinase. J. Biol. Chem. 1997, 272, 10811–10816. [Google Scholar] [CrossRef] [PubMed]
- Camp, H.S.; Tafuri, S.R.; Leff, T. c-Jun N-terminal kinase phosphorylates peroxisome proliferator-activated receptor-γ1 and negatively regulates its transcriptional activity. Endocrinology 1999, 140, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Shao, D.; Rangwala, S.M.; Bailey, S.T.; Krakow, S.L.; Reginato, M.J.; Lazar, M.A. Interdomain communication regulating ligand binding by PPAR-γ. Nature 1998, 396, 377–380. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Muller-Wieland, D.; Pfeiffer, A.; Krone, W.; Kahn, C.R. Obesity associated with a mutation in a genetic regulator of adipocyte differentiation. N. Engl. J. Med. 1998, 339, 953–959. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Haruta, T.; Usui, I.; Takata, Y.; Takano, A.; Uno, T.; Kawahara, J.; Ueno, E.; Sasaoka, T.; Ishibashi, O.; et al. Pioglitazone ameliorates tumor necrosis factor-α-induced insulin resistance by a mechanism independent of adipogenic activity of peroxisome proliferator—Activated receptor-γ. Diabetes 2001, 50, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Werman, A.; Hollenberg, A.; Solanes, G.; Bjorbaek, C.; Vidal-Puig, A.J.; Flier, J.S. Ligand-independent activation domain in the n terminus of peroxisome proliferator-activated receptor γ (PPARγ). Differential activity of PPARγ1 and -2 isoforms and influence of insulin. J. Biol. Chem. 1997, 272, 20230–20235. [Google Scholar] [CrossRef] [PubMed]
- Stechschulte, L.A.; Czernik, P.J.; Rotter, Z.C.; Tausif, F.N.; Corzo, C.A.; Marciano, D.P.; Asteian, A.; Zheng, J.; Bruning, J.B.; Kamenecka, T.M.; et al. PPARg post-translational modifications regulate bone formation and bone resorption. EBioMedicine 2016, 10, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Ge, C.; Cawthorn, W.P.; Li, Y.; Zhao, G.; Macdougald, O.A.; Franceschi, R.T. Reciprocal control of osteogenic and adipogenic differentiation by ERK/MAP kinase phosphorylation of RUNX2 and PPARγ transcription factors. J. Cell Physiol. 2016, 231, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Hosooka, T.; Noguchi, T.; Kotani, K.; Nakamura, T.; Sakaue, H.; Inoue, H.; Ogawa, W.; Tobimatsu, K.; Takazawa, K.; Sakai, M.; et al. DOK1 mediates high-fat diet-induced adipocyte hypertrophy and obesity through modulation of PPAR-γ phosphorylation. Nat. Med. 2008, 14, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Hinds, T.D., Jr.; Stechschulte, L.A.; Cash, H.A.; Whisler, D.; Banerjee, A.; Yong, W.; Khuder, S.S.; Kaw, M.K.; Shou, W.; Najjar, S.M.; et al. Protein phosphatase 5 mediates lipid metabolism through reciprocal control of glucocorticoid receptor and peroxisome proliferator-activated receptor-γ (PPARγ). J. Biol. Chem. 2011, 286, 42911–42922. [Google Scholar] [CrossRef] [PubMed]
- Tasdelen, I.; van Beekum, O.; Gorbenko, O.; Fleskens, V.; van den Broek, N.J.; Koppen, A.; Hamers, N.; Berger, R.; Coffer, P.J.; Brenkman, A.B.; et al. The serine/threonine phosphatase PPM1B (PP2Cβ) selectively modulates PPARγ activity. Biochem. J. 2013, 451, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, L.; Xu, L.; Liu, L.; He, Y.; Zhang, Y.; Huang, X.; Zhao, T.; Wu, L.; Zhao, Y.; et al. WIP1 phosphatase is a critical regulator of adipogenesis through dephosphorylating PPARγ serine 112. Cell Mol. Life Sci. 2017, 74, 2067–2079. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, B.; Bellet, M.M.; Katada, S.; Astarita, G.; Hirayama, J.; Amin, R.H.; Granneman, J.G.; Piomelli, D.; Leff, T.; Sassone-Corsi, P. PER2 controls lipid metabolism by direct regulation of PPARγ. Cell Metab. 2010, 12, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Helenius, K.; Yang, Y.; Alasaari, J.; Makela, T.P. MAT1 inhibits peroxisome proliferator-activated receptor γ-mediated adipocyte differentiation. Mol. Cell Biol. 2009, 29, 315–323. [Google Scholar] [CrossRef] [PubMed]
- Iankova, I.; Petersen, R.K.; Annicotte, J.S.; Chavey, C.; Hansen, J.B.; Kratchmarova, I.; Sarruf, D.; Benkirane, M.; Kristiansen, K.; Fajas, L. Peroxisome proliferator-activated receptor γ recruits the positive transcription elongation factor b complex to activate transcription and promote adipogenesis. Mol. Endocrinol. 2006, 20, 1494–1505. [Google Scholar] [CrossRef] [PubMed]
- Rangwala, S.M.; Rhoades, B.; Shapiro, J.S.; Rich, A.S.; Kim, J.K.; Shulman, G.I.; Kaestner, K.H.; Lazar, M.A. Genetic modulation of PPARγ phosphorylation regulates insulin sensitivity. Dev. Cell 2003, 5, 657–663. [Google Scholar] [CrossRef]
- Gouda, H.N.; Sagoo, G.S.; Harding, A.H.; Yates, J.; Sandhu, M.S.; Higgins, J.P. The association between the peroxisome proliferator-activated receptor-γ2 (PPARγ2) Pro12Ala gene variant and type 2 diabetes mellitus: A huge review and meta-analysis. Am. J. Epidemiol. 2010, 171, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Bluher, M.; Paschke, R. Analysis of the relationship between PPAR-γ 2 gene variants and severe insulin resistance in obese patients with impaired glucose tolerance. Exp. Clin. Endocrinol. Diabetes 2003, 111, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Estall, J.L.; Kajimura, S.; Bostrom, P.; Laznik, D.; Ruas, J.L.; Chalmers, M.J.; Kamenecka, T.M.; Bluher, M.; et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARγ by CDK5. Nature 2010, 466, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Choi, S.S.; Kim, E.S.; Jedrychowski, M.P.; Yang, Y.R.; Jang, H.J.; Suh, P.G.; Banks, A.S.; Gygi, S.P.; Spiegelman, B.M. THRAP3 docks on phosphoserine 273 of PPARγ and controls diabetic gene programming. Genes Dev. 2014, 28, 2361–2369. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.H.; Banks, A.S.; Kamenecka, T.M.; Busby, S.A.; Chalmers, M.J.; Kumar, N.; Kuruvilla, D.S.; Shin, Y.; He, Y.; Bruning, J.B.; et al. Antidiabetic actions of a non-agonist PPARγ ligand blocking CDK5-mediated phosphorylation. Nature 2011, 477, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Amato, A.A.; Rajagopalan, S.; Lin, J.Z.; Carvalho, B.M.; Figueira, A.C.; Lu, J.; Ayers, S.D.; Mottin, M.; Silveira, R.L.; Souza, P.C.; et al. Gq-16, a novel peroxisome proliferator-activated receptor γ (PPARγ) ligand, promotes insulin sensitization without weight gain. J. Biol. Chem. 2012, 287, 28169–28179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Qiu, L.; Wang, R.; Feng, X.; Han, Y.; Zhu, Y.; Chen, D.; Liu, Y.; Jin, L.; Li, Y. Selective targeting of PPARγ by the natural product chelerythrine with a unique binding mode and improved antidiabetic potency. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Fan, W.; Xu, J.; Lu, M.; Yamamoto, H.; Auwerx, J.; Sears, D.D.; Talukdar, S.; Oh, D.; Chen, A.; et al. Adipocyte NCOR knockout decreases PPARγ phosphorylation and enhances PPARγ activity and insulin sensitivity. Cell 2011, 147, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391. [Google Scholar] [CrossRef] [PubMed]
- Banks, A.S.; McAllister, F.E.; Camporez, J.P.; Zushin, P.J.; Jurczak, M.J.; Laznik-Bogoslavski, D.; Shulman, G.I.; Gygi, S.P.; Spiegelman, B.M. An ERK/CDK5 axis controls the diabetogenic actions of PPARγ. Nature 2015, 517, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Liu, L.; Lin, J.Z.; Aprahamian, T.R.; Farmer, S.R. Browning of white adipose tissue with roscovitine induces a distinct population of UCP1+ adipocytes. Cell Metab. 2016, 24, 835–847. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.; Jung, J.E.; Yang, Y.R.; Kim, E.S.; Jang, H.J.; Kim, E.K.; Kim, I.S.; Lee, J.Y.; Kim, J.K.; Seo, J.K.; et al. Novel phosphorylation of PPARγ ameliorates obesity-induced adipose tissue inflammation and improves insulin sensitivity. Cell Signal 2015, 27, 2488–2495. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, D.; Yamaguchi, T.; Shimizu, M.; Nakata, N.; Hirose, F.; Osumi, T. The transactivating function of peroxisome proliferator-activated receptor γ is negatively regulated by SUMO conjugation in the amino-terminal domain. Genes Cells 2004, 9, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Ohshima, T.; Koga, H.; Shimotohno, K. Transcriptional activity of peroxisome proliferator-activated receptor γ is modulated by SUMO-1 modification. J. Biol. Chem. 2004, 279, 29551–29557. [Google Scholar] [CrossRef] [PubMed]
- Floyd, Z.E.; Stephens, J.M. Control of peroxisome proliferator-activated receptor γ2 stability and activity by SUMOylation. Obes. Res. 2004, 12, 921–928. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M.; Yamashita, D.; Yamaguchi, T.; Hirose, F.; Osumi, T. Aspects of the regulatory mechanisms of PPAR functions: Analysis of a bidirectional response element and regulation by SUMOylation. Mol. Cell Biochem. 2006, 286, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Jennewein, C.; Kuhn, A.M.; Schmidt, M.V.; Meilladec-Jullig, V.; von Knethen, A.; Gonzalez, F.J.; Brune, B. SUMOylation of peroxisome proliferator-activated receptor γ by apoptotic cells prevents lipopolysaccharide-induced ncor removal from kappab binding sites mediating transrepression of proinflammatory cytokines. J. Immunol. 2008, 181, 5646–5652. [Google Scholar] [CrossRef] [PubMed]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Diezko, R.; Suske, G. Ligand binding reduces SUMOylation of the peroxisome proliferator-activated receptor γ (PPARγ) activation function 1 (AF1) domain. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Ghisletti, S.; Huang, W.; Ogawa, S.; Pascual, G.; Lin, M.E.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. Parallel SUMOylation-dependent pathways mediate gene- and signal-specific transrepression by lxrs and PPARγ. Mol. Cell 2007, 25, 57–70. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Zhou, Q.; Shi, Y.; Liu, J.; Zhong, F.; Hao, X.; Li, C.; Chen, N.; Wang, W. SUMOylation of PPARγ by rosiglitazone prevents lps-induced ncor degradation mediating down regulation of chemokines expression in renal proximal tubular cells. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Chung, S.S.; Ahn, B.Y.; Kim, M.; Kho, J.H.; Jung, H.S.; Park, K.S. Sumo modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor γ in c2c12 myotubes. Biochem. J. 2011, 433, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Gregoire, S. A recurrent phospho-Sumoyl switch in transcriptional repression and beyond. Mol. Cell 2006, 23, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Dutchak, P.A.; Katafuchi, T.; Bookout, A.L.; Choi, J.H.; Yu, R.T.; Mangelsdorf, D.J.; Kliewer, S.A. Fibroblast growth factor-21 regulates PPARγ activity and the antidibetic actions of thiazolidinediones. Cell 2012, 148, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Shao, J.; Wang, Z.; Yang, T.; Liu, S.; Liu, Y.; Fan, X.; Ye, W. Growth differentiation factor 11 is a protective factor for osteoblastogenesis by targeting PPARγ. Gene 2015, 557, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Mikkonen, L.; Hirvonen, J.; Janne, O.A. Sumo-1 regulates body weight and adipogenesis via PPARγ in male and female mice. Endocrinology 2013, 154, 698–708. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhou, R.; Niu, J.; McNutt, M.A.; Wang, P.; Tong, T. Sirt1 is regulated by a PPARγ-SIRT1 negative feedback loop associated with senescence. Nucleic Acids Res. 2010, 38, 7458–7471. [Google Scholar] [CrossRef] [PubMed]
- Qiang, L.; Wang, L.; Kon, N.; Zhao, W.; Lee, S.; Zhang, Y.; Rosenbaum, M.; Zhao, Y.; Gu, W.; Farmer, S.R.; et al. Brown remodeling of white adipose tissue by SIRT1-dependent deacetylation of PPARγ. Cell 2012, 150, 620–632. [Google Scholar] [CrossRef] [PubMed]
- Tian, L.; Wang, C.; Hagen, F.K.; Gormley, M.; Addya, S.; Soccio, R.; Casimiro, M.C.; Zhou, J.; Powell, M.J.; Xu, P.; et al. Acetylation-defective mutant of PPARγ is associated with decreased lipid synthesis in breast cancer cells. Oncotarget 2014, 5, 7303–7315. [Google Scholar] [CrossRef] [PubMed]
- Kilroy, G.; Kirk-Ballard, H.; Carter, L.E.; Floyd, Z.E. The ubiquitin ligase SIAH2 regulates PPARγ activity in adipocytes. Endocrinology 2012, 153, 1206–1218. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Park, K.W.; Lee, E.W.; Jang, W.S.; Seo, J.; Shin, S.; Hwang, K.A.; Song, J. Suppression of PPARγ through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ. 2014, 21, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Takahashi, H.; Saeki, Y.; Ozaki, T.; Itoh, S.; Suzuki, M.; Mizushima, W.; Tanaka, K.; Hatakeyama, S. The E3 ubiquitin ligase TRIM23 regulates adipocyte differentiation via stabilization of the adipogenic activator PPARγ. ELife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Wang, R.; Lama, R.; Wang, X.; Floyd, Z.E.; Park, E.A.; Liao, F.F. Ubiquitin ligase NEDD4 regulates PPARγ stability and adipocyte differentiation in 3t3-l1 cells. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.; Park, S.Y.; Roth, J.; Kim, H.S.; Cho, J.W. O-Glcnac modification of PPARγ reduces its transcriptional activity. Biochem. Biophys. Res. Commun. 2012, 417, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Koo, Y.D.; Choi, J.W.; Kim, M.; Chae, S.; Ahn, B.Y.; Oh, B.C.; Hwang, D.; Seol, J.H.; Kim, Y.B.; Park, Y.J.; et al. Sumo-specific protease 2 (SENP2) is an important regulator of fatty acid metabolism in skeletal muscle. Diabetes 2015, 64, 2420–2431. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Modification | Enzyme | Target-Site | References |
|---|---|---|---|
| Phosphorylation | ERK1/2 | PPARα S12, S21 PPARγ S112, S273, S133 | [13,24,26,29,53,68] |
| p38-α | PPARα S12, S21 | [12] | |
| CDK7 | PPARα S12, S21 PPARγ S112 | [14,41] | |
| GSKβ | PPARα S73 | [15] | |
| JNK | PPARγ S112 | [26] | |
| CDK9 | PPARγ S112 | [42] | |
| CDK5 | PPARγ S112, S273, S296 | [46,53] | |
| MEK2 | PPARγ S133 | [53] | |
| c-SRC | PPARγ Y78 | [55] | |
| PP5 | PPARγ S112 | [37] | |
| PPM1B | PPARγ S112 | [38] | |
| WIP1 | PPARγ S112 | [39] | |
| PTB-1B | PPARγ Y78 | [55] | |
| Acetylation | CBP | PPARγ K98, K107, K218, K268, K293 | [71] |
| p300 | PPARγ K? | [70] | |
| SIRT1 | PPARγ K184/185 *, K268, K293 | [70,71,72] | |
| SUMOylation | PIAS1/PIASxβ | PPARα K358 PPARγ K107, K395 * | [20,57,61,63,64] |
| PIASy | PPARα K185 | [19] | |
| UBC9 | PPARα K185 PPARγ K107, K395 * | [19,56,59,61] | |
| SENP2 | PPARγ K107 PPARβ/δ K104 | [65,78] | |
| Ubiquitination | MKRN1 | PPARγ K184/185 | [74] |
| SIAH2 | PPARγ K? | [73] | |
| NEDD4 | PPARγ K? | [76] | |
| TRIM23 | PPARγ K? | [75] | |
| MDM2 | PPARα K? | [22] | |
| MuRF | PPARα K? | [23] | |
| O-GlcNAcylation | OGT | PPARγ T84 * | [77] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brunmeir, R.; Xu, F. Functional Regulation of PPARs through Post-Translational Modifications. Int. J. Mol. Sci. 2018, 19, 1738. https://doi.org/10.3390/ijms19061738
Brunmeir R, Xu F. Functional Regulation of PPARs through Post-Translational Modifications. International Journal of Molecular Sciences. 2018; 19(6):1738. https://doi.org/10.3390/ijms19061738
Chicago/Turabian StyleBrunmeir, Reinhard, and Feng Xu. 2018. "Functional Regulation of PPARs through Post-Translational Modifications" International Journal of Molecular Sciences 19, no. 6: 1738. https://doi.org/10.3390/ijms19061738
APA StyleBrunmeir, R., & Xu, F. (2018). Functional Regulation of PPARs through Post-Translational Modifications. International Journal of Molecular Sciences, 19(6), 1738. https://doi.org/10.3390/ijms19061738