Bimodal Function of Anti-TNF Treatment: Shall We Be Concerned about Anti-TNF Treatment in Patients with Rheumatoid Arthritis and Heart Failure?


Abstract
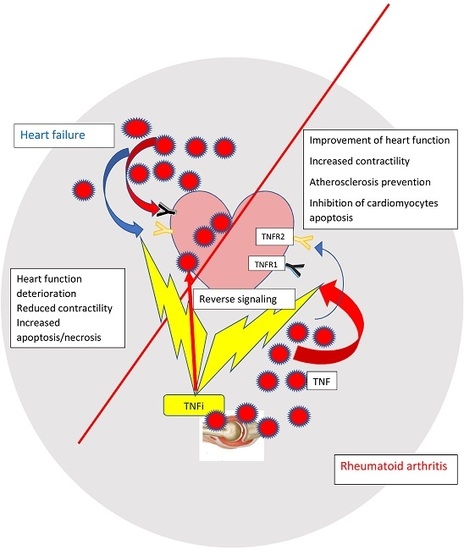
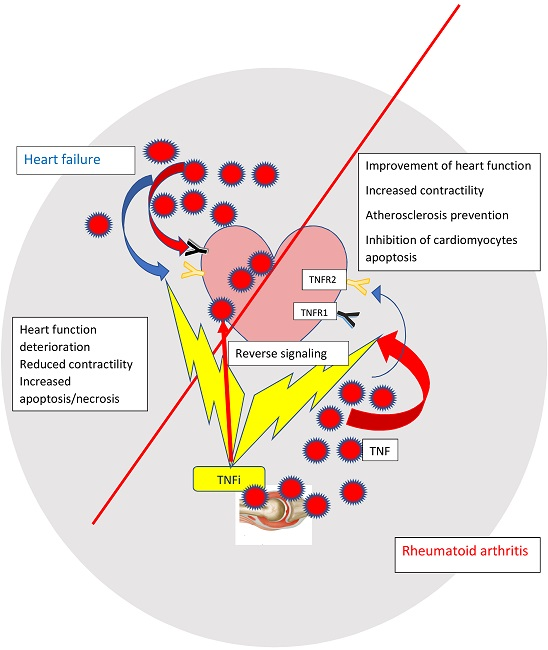
:
{kind=link}
1. Introduction
2. The Role of TNF in Physiological and Pathological Conditions
3. The Role of TNF in Heart Failure
4. Types of Heart Failure
5. The Role of TNF in Rheumatoid Arthritis and Heart Failure
6. Anti-TNF Treatment in a Real World Clinic
7. Conclusions
- Heart failure in the general population and in inflammatory conditions vary with regard to the potential mechanisms and the source of inflammatory cytokines.
- Contrary to heart failure in the general population where TNF is produced by the failing heart and in some extent may play a cardioprotective role, in RA patients it is synthesized by synoviocytes and immunocompetent cells and may exert a detrimental effect on heart function.
- TNF blockade in RA patients suppresses the inflammatory response and directly translates to the improvement of heart function.
- Direct translation of negative data in HF patients treated with anti-TNF and should be done with caution as it is not possible to compare two different diseases and the doses of anti-TNF agents in HF studies were 2–3 times higher than those used for the treatment of RA
- It is plausible that TNF inhibition brings different pathophysiological consequences in HF and RA patients as the result of crosstalk between TNF inflammatory cytokines, adipokines and apoptotic, and necroptotic signaling molecules.
Acknowledgments
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet 2016, 388, 2023–2038. [Google Scholar] [CrossRef] [Green Version]
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 american college of rheumatology criteria: A systematic review. Semin. Arthritis Rheum. 2006, 36, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Firestein, G.S.; McInnes, I.B. Immunopathogenesis of rheumatoid arthritis. Immunity 2017, 46, 183–196. [Google Scholar] [CrossRef] [PubMed]
- Taverner, D.; Vallve, J.C.; Ferre, R.; Paredes, S.; Masana, L.; Castro, A. Variables associated with subclinical atherosclerosis in a cohort of rheumatoid arthritis patients: Sex-specific associations and differential effects of disease activity and age. PLoS ONE 2018, 13, e0193690. [Google Scholar] [CrossRef] [PubMed]
- Kasselman, L.J.; Vernice, N.A.; DeLeon, J.; Reiss, A.B. The gut microbiome and elevated cardiovascular risk in obesity and autoimmunity. Atherosclerosis 2018, 271, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Jantan, I.; Bukhari, S.N.A. Rheumatoid arthritis: Recent advances on its etiology, role of cytokines and pharmacotherapy. Biomed. Pharmacother. 2017, 92, 615–633. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. The pathogenesis of rheumatoid arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, M.; Maini, S.R. Role of cytokines in rheumatoid arthritis: An education in pathophysiology and therapeutics. Immunol. Rev. 2008, 223, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Ryu, H.; Lim, H.; Choi, G.; Park, Y.J.; Cho, M.; Na, H.; Ahn, C.W.; Kim, Y.C.; Kim, W.U.; Lee, S.H.; et al. Atherogenic dyslipidemia promotes autoimmune follicular helper T cell responses via IL-27. Nat. Immunol. 2018, 19, 583–593. [Google Scholar] [CrossRef] [PubMed]
- Kremers, H.M.; Crowson, C.S.; Therneau, T.M.; Roger, V.L.; Gabriel, S.E. High ten-year risk of cardiovascular disease in newly diagnosed rheumatoid arthritis patients: A population-based cohort study. Arthritis Rheum. 2008, 58, 2268–2274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naranjo, A.; Sokka, T.; Descalzo, M.A.; Calvo-Alén, J.; Hørslev-Petersen, K.; Luukkainen, R.K.; Combe, B.; Burmester, G.R.; Devlin, J.; Ferraccioli, G.; et al. Cardiovascular disease in patients with rheumatoid arthritis: Results from the QUEST-RA study. Arthritis Res. Ther. 2008, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolfe, F.; Michaud, K. The risk of myocardial infarction and pharmacologic and nonpharmacologic myocardial infarction predictors in rheumatoid arthritis: A cohort and nested case-control analysis. Arthritis Rheum. 2008, 58, 2612–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bengtsson, K.; Jacobsson, L.T.; Rydberg, B.; Kvist, G.; Torstenson, T.; Dehlin, M.; Hilme, E.; Lindhe, A.; Wallerstedt, S.M.; Forsblad-d'Elia, H. Comparisons between comorbid conditions and health care consumption in rheumatoid arthritis patients with or without biological disease-modifying anti-rheumatic drugs: A register-based study. BMC Musculoskelet. Disord. 2016, 17, 499. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Ganeshalingam, K.; Semb, A.G.; Szekanecz, Z.; Nurmohamed, M. Cardiovascular risk in rheumatoid arthritis: Recent advances in the understanding of the pivotal role of inflammation, risk predictors and the impact of treatment. Rheumatology 2014, 53, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Meune, C.; Touze, E.; Trinquart, L.; Allanore, Y. Trends in cardiovascular mortality in patients with rheumatoid arthritis over 50 years: A systematic review and meta-analysis of cohort studies. Rheumatology 2009, 48, 1309–1313. [Google Scholar] [CrossRef] [PubMed]
- Meune, C.; Touze, E.; Trinquart, L.; Allanore, Y. High risk of clinical cardiovascular events in rheumatoid arthritis: Levels of associations of myocardial infarction and stroke through a systematic review and meta-analysis. Arch. Cardiovasc. Dis. 2010, 103, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Avina-Zubieta, J.A.; Choi, H.K.; Sadatsafavi, M.; Etminan, M.; Esdaile, J.M.; Lacaille, D. Risk of cardiovascular mortality in patients with rheumatoid arthritis: A meta-analysis of observational studies. Arthritis Rheum. 2008, 59, 1690–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Widdifield, J.; Paterson, J.M.; Huang, A.; Bernatsky, S. Causes of death in rheumatoid arthritis: How do they compare to the general population? Arthritis Care Res. 2018. [Google Scholar] [CrossRef] [PubMed]
- Logstrup, B.B.; Ellingsen, T.; Pedersen, A.B.; Kjaersgaard, A.; Botker, H.E.; Maeng, M. Development of heart failure in patients with rheumatoid arthritis: A danish population-based study. Eur. J. Clin. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Libby, P. Role of inflammation in atherosclerosis associated with rheumatoid arthritis. Am. J. Med. 2008, 121, S21–31. [Google Scholar] [CrossRef] [PubMed]
- Dessein, P.H.; Joffe, B.I.; Veller, M.G.; Stevens, B.A.; Tobias, M.; Reddi, K.; Stanwix, A.E. Traditional and nontraditional cardiovascular risk factors are associated with atherosclerosis in rheumatoid arthritis. J. Rheumatol. 2005, 32, 435–442. [Google Scholar] [PubMed]
- Ku, I.A.; Imboden, J.B.; Hsue, P.Y.; Ganz, P. Rheumatoid arthritis: Model of systemic inflammation driving atherosclerosis. Circ. J. 2009, 73, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Innala, L.; Moller, B.; Ljung, L.; Magnusson, S.; Smedby, T.; Sodergren, A.; Ohman, M.L.; Rantapaa-Dahlqvist, S.; Wallberg-Jonsson, S. Cardiovascular events in early RA are a result of inflammatory burden and traditional risk factors: A five year prospective study. Arthritis Res. Ther. 2011, 13, R131. [Google Scholar] [CrossRef] [PubMed]
- Maradit-Kremers, H.; Crowson, C.S.; Nicola, P.J.; Ballman, K.V.; Roger, V.L.; Jacobsen, S.J.; Gabriel, S.E. Increased unrecognized coronary heart disease and sudden deaths in rheumatoid arthritis: A population-based cohort study. Arthritis Rheum. 2005, 52, 402–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicola, P.J.; Maradit-Kremers, H.; Roger, V.L.; Jacobsen, S.J.; Crowson, C.S.; Ballman, K.V.; Gabriel, S.E. The risk of congestive heart failure in rheumatoid arthritis: A population-based study over 46 years. Arthritis Rheum. 2005, 52, 412–420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, J.M., 3rd; Roger, V.L.; Crowson, C.S.; Kremers, H.M.; Therneau, T.M.; Gabriel, S.E. The presentation and outcome of heart failure in patients with rheumatoid arthritis differs from that in the general population. Arthritis Rheum. 2008, 58, 2603–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crowson, C.S.; Liao, K.P.; Davis, J.M., 3rd; Solomon, D.H.; Matteson, E.L.; Knutson, K.L.; Hlatky, M.A.; Gabriel, S.E. Rheumatoid arthritis and cardiovascular disease. Am. Heart J. 2013, 166, 622–628. [Google Scholar] [CrossRef] [PubMed]
- Han, C.; Robinson, D.W., Jr.; Hackett, M.V.; Paramore, L.C.; Fraeman, K.H.; Bala, M.V. Cardiovascular disease and risk factors in patients with rheumatoid arthritis, psoriatic arthritis, and ankylosing spondylitis. J. Rheumatol. 2006, 33, 2167–2172. [Google Scholar] [PubMed]
- Gerli, R.; Sherer, Y.; Bocci, E.B.; Vaudo, G.; Moscatelli, S.; Shoenfeld, Y. Precocious atherosclerosis in rheumatoid arthritis: Role of traditional and disease-related cardiovascular risk factors. Ann. N. Y. Acad. Sci. 2007, 1108, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, M.M.; Benfaremo, D.; Gabrielli, A. Biologics in inflammatory and immunomediated arthritis. Curr. Pharm. Biotechnol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.H.; Bernasconi, C.; Aassi, M.; Molina, J.F.; Epis, O.M. Treatment of rheumatoid arthritis with anti-tumor necrosis factor or tocilizumab therapy as first biologic agent in a global comparative observational study. Arthritis Care Res. 2017, 69, 1484–1494. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulos, C.E.; Orfanos, P.; Manoussakis, M.N.; Tzioufas, A.G.; Moutsopoulos, H.M.; Vlachoyiannopoulos, P.G. Treat-to-target biologic therapy in patients with rheumatoid arthritis is more efficacious and safe compared to delayed initiation of biologics: A real-world study. Clin. Exp. Rheumatol. 2017, 35, 192–200. [Google Scholar] [PubMed]
- Narazaki, M.; Tanaka, T.; Kishimoto, T. The role and therapeutic targeting of IL-6 in rheumatoid arthritis. Expert. Rev. Clin. Immunol. 2017, 13, 535–551. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Landewe, R.; Bijlsma, J.; Burmester, G.; Chatzidionysiou, K.; Dougados, M.; Nam, J.; Ramiro, S.; Voshaar, M.; van Vollenhoven, R.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2016 Update. Ann. Rheum. Dis. 2017, 76, 960–977. [Google Scholar] [CrossRef] [PubMed]
- Kotyla, P.J.; Kucharz, E.J. Who might be predisposed to the development of serious side effects when treated with TNF-α antagonist? Clin. Exp. Rheumatol. 2006, 24, 211, author reply 211. [Google Scholar] [PubMed]
- Kotyla, P.J.; Sliwinska-Kotyla, B.; Kucharz, E.J. Treatment with infliximab may contribute to the development of peripheral neuropathy among the patients with rheumatoid arthritis. Clin. Rheumatol. 2007, 26, 1595–1596. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.; Lago, P.; Faria, R.; Torres, T. Safety of anti-TNF therapies in immune-mediated inflammatory diseases: Focus on infections and malignancy. Drug Dev. Res. 2015, 76, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Canete, J.D.; Hernandez, M.V.; Sanmarti, R. Safety profile of biological therapies for treating rheumatoid arthritis. Expert. Opin. Biol. Ther. 2017, 17, 1089–1103. [Google Scholar] [CrossRef] [PubMed]
- Harrold, L.R.; Litman, H.J.; Saunders, K.C.; Dandreo, K.J.; Gershenson, B.; Greenberg, J.D.; Low, R.; Stark, J.; Suruki, R.; Jaganathan, S.; et al. One-year risk of serious infection in patients treated with certolizumab pegol as compared with other TNF inhibitors in a real-world setting: Data from a national U.S. Rheumatoid arthritis registry. Arthritis Res. Ther. 2018, 20, 2. [Google Scholar] [CrossRef] [PubMed]
- Rutherford, A.I.; Patarata, E.; Subesinghe, S.; Hyrich, K.L.; Galloway, J.B. Opportunistic infections in rheumatoid arthritis patients exposed to biologic therapy: Results from the British society for rheumatology biologics register for rheumatoid arthritis. Rheumatology 2018. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Shang, Q.; Tam, L.S. Targeting inflammation in the prevention of cardiovascular disease in patients with inflammatory arthritis. Transl. Res. 2016, 167, 138–151. [Google Scholar] [CrossRef] [PubMed]
- McInnes, I.B.; Schett, G. Pathogenetic insights from the treatment of rheumatoid arthritis. Lancet 2017, 389, 2328–2337. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, M.J.; Tardif, J.C. Inflammation and beyond: New directions and emerging drugs for treating atherosclerosis. Expert. Opin. Emerg. Drugs 2017, 22, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Tousoulis, D.; Economou, E.K.; Oikonomou, E.; Papageorgiou, N.; Siasos, G.; Latsios, G.; Kokkou, E.; Mourouzis, K.; Papaioannou, S.; Deftereos, S.; et al. The role and predictive value of cytokines in atherosclerosis and coronary artery disease. Curr. Med. Chem. 2015, 22, 2636–2650. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F. Tumour necrosis factor and cancer. Nat. Rev. Cancer 2009, 9, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.M. Antitumor necrosis factor-α therapy and potential cancer inhibition. Eur. J. Cancer Prev. 2008, 17, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Ham, B.; Fernandez, M.C.; D'Costa, Z.; Brodt, P. The diverse roles of the TNF axis in cancer progression and metastasis. Trends Cancer Res. 2016, 11, 1–27. [Google Scholar] [PubMed]
- Varfolomeev, E.; Vucic, D. Intracellular regulation of TNF activity in health and disease. Cytokine 2018, 101, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.K.; Gracias, D.T.; Croft, M. TNF activity and T cells. Cytokine 2018, 101, 14–18. [Google Scholar] [CrossRef] [PubMed]
- Falvo, J.V.; Tsytsykova, A.V.; Goldfeld, A.E. Transcriptional control of the TNF gene. Curr. Dir. Autoimmun. 2010, 11, 27–60. [Google Scholar] [PubMed]
- Zhang, H.; Park, Y.; Wu, J.; Chen, X.; Lee, S.; Yang, J.; Dellsperger, K.C.; Zhang, C. Role of TNF-α in vascular dysfunction. Clin. Sci. 2009, 116, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Black, R.A.; Rauch, C.T.; Kozlosky, C.J.; Peschon, J.J.; Slack, J.L.; Wolfson, M.F.; Castner, B.J.; Stocking, K.L.; Reddy, P.; Srinivasan, S.; et al. A metalloproteinase disintegrin that releases tumour-necrosis factor-α from cells. Nature 1997, 385, 729–733. [Google Scholar] [CrossRef] [PubMed]
- Tartaglia, L.A.; Weber, R.F.; Figari, I.S.; Reynolds, C.; Palladino, M.A., Jr.; Goeddel, D.V. The two different receptors for tumor necrosis factor mediate distinct cellular responses. Proc. Natl. Acad. Sci. USA 1991, 88, 9292–9296. [Google Scholar] [CrossRef] [PubMed]
- Sedger, L.M.; McDermott, M.F. TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants—past, present and future. Cytokine Growth Factor Rev. 2014, 25, 453–472. [Google Scholar] [CrossRef] [PubMed]
- Grell, M.; Douni, E.; Wajant, H.; Löhden, M.; Clauss, M.; Maxeiner, B.; Georgopoulos, S.; Lesslauer, W.; Kollias, G.; Pfizenmaier, K.; et al. The transmembrane form of tumor necrosis factor is the prime activating ligand of the 80 kDa tumor necrosis factor receptor. Cell 1995, 83, 793–802. [Google Scholar] [CrossRef]
- Doss, G.P.; Agoramoorthy, G.; Chakraborty, C. TNF/TNFr: Drug target for autoimmune diseases and immune-mediated inflammatory diseases. Front. Biosci. 2014, 19, 1028–1040. [Google Scholar] [CrossRef]
- Faustman, D.L.; Davis, M. TNF receptor 2 and disease: Autoimmunity and regenerative medicine. Front. Immunol. 2013, 4, 478. [Google Scholar] [CrossRef] [PubMed]
- Nikoletopoulou, V.; Markaki, M.; Palikaras, K.; Tavernarakis, N. Crosstalk between apoptosis, necrosis and autophagy. Biochim. Biophys. Acta 2013, 1833, 3448–3459. [Google Scholar] [CrossRef] [PubMed]
- Puimege, L.; Libert, C.; Van Hauwermeiren, F. Regulation and dysregulation of tumor necrosis factor receptor-1. Cytokine Growth Factor Rev. 2014, 25, 285–300. [Google Scholar] [CrossRef] [PubMed]
- Bradley, J.R.; Thiru, S.; Pober, J.S. Disparate localization of 55-kd and 75-kd tumor necrosis factor receptors in human endothelial cells. Am. J. Pathol. 1995, 146, 27–32. [Google Scholar] [PubMed]
- Tartaglia, L.A.; Goeddel, D.V.; Reynolds, C.; Figari, I.S.; Weber, R.F.; Fendly, B.M.; Palladino, M.A., Jr. Stimulation of human T-cell proliferation by specific activation of the 75-kda tumor necrosis factor receptor. J. Immunol. 1993, 151, 4637–4641. [Google Scholar] [PubMed]
- Carpentier, I.; Coornaert, B.; Beyaert, R. Function and regulation of tumor necrosis factor receptor type 2. Curr. Med. Chem. 2004, 11, 2205–2212. [Google Scholar] [CrossRef] [PubMed]
- Hamid, T.; Xu, Y.; Ismahil, M.A.; Li, Q.; Jones, S.P.; Bhatnagar, A.; Bolli, R.; Prabhu, S.D. TNF receptor signaling inhibits cardiomyogenic differentiation of cardiac stem cells and promotes a neuroadrenergic-like fate. Am. J. Physiol. Heart Circ. Physiol. 2016, 311, H1189–H1201. [Google Scholar] [CrossRef] [PubMed]
- Borghi, A.; Verstrepen, L.; Beyaert, R. TRAF2 multitasking in TNF receptor-induced signaling to Nf-κB, map kinases and cell death. Biochem. Pharmacol. 2016, 116, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Oppenheim, J.J. The phenotypic and functional consequences of tumour necrosis factor receptor type 2 expression on CD4+ FoxP3+ regulatory T cells. Immunology 2011, 133, 426–433. [Google Scholar] [CrossRef] [PubMed]
- Gullestad, L.; Ueland, T.; Vinge, L.E.; Finsen, A.; Yndestad, A.; Aukrust, P. Inflammatory cytokines in heart failure: Mediators and markers. Cardiology 2012, 122, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ueland, T.; Gullestad, L.; Nymo, S.H.; Yndestad, A.; Aukrust, P.; Askevold, E.T. Inflammatory cytokines as biomarkers in heart failure. Clin. Chim. Acta 2015, 443, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Kalman, J.; Mayer, L.; Fillit, H.M.; Packer, M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N. Engl. J. Med. 1990, 323, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Meldrum, D.R. Tumor necrosis factor in the heart. Am. J. Physiol. 1998, 274, R577–595. [Google Scholar] [CrossRef] [PubMed]
- Berges, A.; Van Nassauw, L.; Bosmans, J.; Timmermans, J.P.; Vrints, C. Role of nitric oxide and oxidative stress in ischaemic myocardial injury and preconditioning. Acta Cardiol. 2003, 58, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Thielmann, M.; Dorge, H.; Martin, C.; Belosjorow, S.; Schwanke, U.; van De Sand, A.; Konietzka, I.; Buchert, A.; Kruger, A.; Schulz, R.; et al. Myocardial dysfunction with coronary microembolization: Signal transduction through a sequence of nitric oxide, tumor necrosis factor-α, and sphingosine. Circ. Res. 2002, 90, 807–813. [Google Scholar] [CrossRef] [PubMed]
- Prabhu, S.D. Cytokine-induced modulation of cardiac function. Circ. Res. 2004, 95, 1140–1153. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L. Inflammatory mediators and the failing heart: Past, present, and the foreseeable future. Circ. Res. 2002, 91, 988–998. [Google Scholar] [CrossRef] [PubMed]
- Guggilam, A.; Cardinale, J.P.; Mariappan, N.; Sriramula, S.; Haque, M.; Francis, J. Central TNF inhibition results in attenuated neurohumoral excitation in heart failure: A role for superoxide and nitric oxide. Basic Res. Cardiol. 2011, 106, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Shimano, M.; Ouchi, N.; Walsh, K. Cardiokines: Recent progress in elucidating the cardiac secretome. Circulation 2012, 126, e327–332. [Google Scholar] [CrossRef] [PubMed]
- Kleinbongard, P.; Schulz, R.; Heusch, G. TNFα in myocardial ischemia/reperfusion, remodeling and heart failure. Heart Fail. Rev. 2011, 16, 49–69. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.S.; Nackley, A.G.; Satterfield, K.; Maixner, W.; Diatchenko, L.; Flood, P.M. β2 Adrenergic receptor activation stimulates pro-inflammatory cytokine production in macrophages via PKA- and NF-κB-independent mechanisms. Cell Signal. 2007, 19, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.X.; Kimura, S.; Nishiyama, A.; Shokoji, T.; Rahman, M.; Yao, L.; Nagai, Y.; Fujisawa, Y.; Miyatake, A.; Abe, Y. Cardiac oxidative stress in acute and chronic isoproterenol-infused rats. Cardiovasc. Res. 2005, 65, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandrasekar, B.; Marelli-Berg, F.M.; Tone, M.; Bysani, S.; Prabhu, S.D.; Murray, D.R. β-Adrenergic stimulation induces interleukin-18 expression via β2-AR, PI3K, Akt, IKK, and NF-κB. Biochem. Biophys. Res. Commun. 2004, 319, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Murray, D.R.; Prabhu, S.D.; Chandrasekar, B. Chronic β-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation 2000, 101, 2338–2341. [Google Scholar] [CrossRef] [PubMed]
- Garlie, J.B.; Hamid, T.; Gu, Y.; Ismahil, M.A.; Chandrasekar, B.; Prabhu, S.D. Tumor necrosis factor receptor 2 signaling limits β-adrenergic receptor-mediated cardiac hypertrophy in vivo. Basic Res. Cardiol. 2011, 106, 1193–1205. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Gorog, D.A.; Bellahcene, M.; Cao, X.; Quinlan, R.A.; Marber, M.S. Tumor necrosis factor-induced protection of the murine heart is independent of p38-MAPK activation. J. Mol. Cell. Cardiol. 2003, 35, 1523–1527. [Google Scholar] [CrossRef] [PubMed]
- Asgeri, M.; Pourafkari, L.; Kundra, A.; Javadzadegan, H.; Negargar, S.; Nader, N.D. Dual effects of tumor necrosis factor α on myocardial injury following prolonged hypoperfusion of the heart. Immunol. Investig. 2015, 44, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; McMurray, J.J.; Packer, M.; Swedberg, K.; Borer, J.S.; Colucci, W.S.; Djian, J.; Drexler, H.; Feldman, A.; Kober, L.; et al. Targeted anticytokine therapy in patients with chronic heart failure: Results of the randomized etanercept worldwide evaluation (renewal). Circulation 2004, 109, 1594–1602. [Google Scholar] [CrossRef] [PubMed]
- Chung, E.S.; Packer, M.; Lo, K.H.; Fasanmade, A.A.; Willerson, J.T.; Anti, T.N.F.T.A.C.H.F.I. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-α, in patients with moderate-to-severe heart failure: Results of the anti-TNF therapy against congestive heart failure (attach) trial. Circulation 2003, 107, 3133–3140. [Google Scholar] [PubMed]
- Paulus, W.J.; Tschope, C.; Sanderson, J.E.; Rusconi, C.; Flachskampf, F.A.; Rademakers, F.E.; Marino, P.; Smiseth, O.A.; De Keulenaer, G.; Leite-Moreira, A.F.; et al. How to diagnose diastolic heart failure: A consensus statement on the diagnosis of heart failure with normal left ventricular ejection fraction by the heart failure and echocardiography associations of the european society of cardiology. Eur. Heart J. 2007, 28, 2539–2550. [Google Scholar] [CrossRef] [PubMed]
- Paulus, W.J.; Tschope, C. A novel paradigm for heart failure with preserved ejection fraction: Comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J. Am. Coll. Cardiol. 2013, 62, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Heinzel, F.R.; Hohendanner, F.; Jin, G.; Sedej, S.; Edelmann, F. Myocardial hypertrophy and its role in heart failure with preserved ejection fraction. J. Appl. Physiol. 2015, 119, 1233–1242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zile, M.R.; Gottdiener, J.S.; Hetzel, S.J.; McMurray, J.J.; Komajda, M.; McKelvie, R.; Baicu, C.F.; Massie, B.M.; Carson, P.E. Prevalence and Significance of Alterations in Cardiac Structure and Function in Patients With Heart Failure and a Preserved Ejection FractionClinical Perspective. Circulation 2011, 124, 2491–2501. [Google Scholar] [CrossRef] [PubMed]
- Mantel, A.; Holmqvist, M.; Andersson, D.C.; Lund, L.H.; Askling, J. Association between rheumatoid arthritis and risk of ischemic and nonischemic heart failure. J. Am. Coll. Cardiol. 2017, 69, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Torre-Amione, G.; Kapadia, S.; Benedict, C.; Oral, H.; Young, J.B.; Mann, D.L. Proinflammatory cytokine levels in patients with depressed left ventricular ejection fraction: A report from the studies of left ventricular dysfunction (SOLVD). J. Am. Coll. Cardiol. 1996, 27, 1201–1206. [Google Scholar] [CrossRef]
- Wislowska, M.; Jaszczyk, B.; Kochmanski, M.; Sypula, S.; Sztechman, M. Diastolic heart function in RA patients. Rheumatol. Int. 2008, 28, 513–519. [Google Scholar] [CrossRef] [PubMed]
- Birdane, A.; Korkmaz, C.; Ata, N.; Cavusoglu, Y.; Kasifoglu, T.; Dogan, S.M.; Gorenek, B.; Goktekin, O.; Unalir, A.; Timuralp, B. Tissue Doppler imaging in the evaluation of the left and right ventricular diastolic functions in rheumatoid arthritis. Echocardiography 2007, 24, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Franssen, C.; Chen, S.; Unger, A.; Korkmaz, H.I.; De Keulenaer, G.W.; Tschope, C.; Leite-Moreira, A.F.; Musters, R.; Niessen, H.W.; Linke, W.A.; et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. 2016, 4, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-T.; Chen, L.-W.; Chien, M.-Y. Effects of exercise training on anabolic and catabolic markers in patients with chronic heart failure: A systematic review. Heart Fail. Rev. 2017, 22, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Miettinen, K.H.; Lassus, J.; Harjola, V.P.; Siirila-Waris, K.; Melin, J.; Punnonen, K.R.; Nieminen, M.S.; Laakso, M.; Peuhkurinen, K.J. Prognostic role of pro- and anti-inflammatory cytokines and their polymorphisms in acute decompensated heart failure. Eur. J. Heart Fail. 2008, 10, 396–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeuchi, T.; Miyasaka, N.; Inui, T.; Yano, T.; Yoshinari, T.; Abe, T.; Koike, T. High titers of both rheumatoid factor and anti-CCP antibodies at baseline in patients with rheumatoid arthritis are associated with increased circulating baseline TNF level, low drug levels, and reduced clinical responses: A post hoc analysis of the RISING study. Arthritis Res. Ther. 2017, 19, 194. [Google Scholar] [PubMed]
- Kotyla, P.; Jankiewicz-Ziobro, K.; Owczarek, A.; Kucharz, E.J. Etanercept increases tumor necrosis factor-α level but not sFas level in patients with rheumatoid arthritis. Isr. Med. Assoc. J. 2015, 17, 14–18. [Google Scholar] [PubMed]
- Kubota, T.; Miyagishima, M.; Alvarez, R.J., Jr.; Kormos, R.; Rosenblum, W.D.; Demetris, A.J.; Semigran, M.J.; Dec, G.W.; Holubkov, R.; McTiernan, C.F.; et al. Expression of proinflammatory cytokines in the failing human heart: Comparison of recent-onset and end-stage congestive heart failure. J. Heart Lung Transplant. 2000, 19, 819–824. [Google Scholar] [CrossRef]
- Peluso, I.; Palmery, M. The relationship between body weight and inflammation: Lesson from anti-TNF-α antibody therapy. Hum. Immunol. 2016, 77, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, A.; Aoki, Y.; Sonobe, M.; Watanabe, F.; Takahashi, H.; Saito, M.; Nakagawa, K. Relative expression and correlation of tumor necrosis factor-α, interferon-γ, and interleukin-17 in the rheumatoid synovium. Clin. Rheumatol. 2016, 35, 1691–1697. [Google Scholar] [CrossRef] [PubMed]
- Al-Lamki, R.S.; Brookes, A.P.; Wang, J.; Reid, M.J.; Parameshwar, J.; Goddard, M.J.; Tellides, G.; Wan, T.; Min, W.; Pober, J.S.; et al. TNF receptors differentially signal and are differentially expressed and regulated in the human heart. Am. J. Transplant. 2009, 9, 2679–2696. [Google Scholar] [CrossRef] [PubMed]
- Hamid, T.; Gu, Y.; Ortines, R.V.; Bhattacharya, C.; Wang, G.; Xuan, Y.T.; Prabhu, S.D. Divergent tumor necrosis factor receptor-related remodeling responses in heart failure: Role of nuclear factor-κB and inflammatory activation. Circulation 2009, 119, 1386–1397. [Google Scholar] [CrossRef] [PubMed]
- Higuchi, Y.; McTiernan, C.F.; Frye, C.B.; McGowan, B.S.; Chan, T.O.; Feldman, A.M. Tumor necrosis factor receptors 1 and 2 differentially regulate survival, cardiac dysfunction, and remodeling in transgenic mice with tumor necrosis factor-α-induced cardiomyopathy. Circulation 2004, 109, 1892–1897. [Google Scholar] [CrossRef] [PubMed]
- Chytilova, A.; Borchert, G.H.; Mandikova-Alanova, P.; Hlavackova, M.; Kopkan, L.; Khan, M.A.; Imig, J.D.; Kolar, F.; Neckar, J. Tumour necrosis factor-α contributes to improved cardiac ischaemic tolerance in rats adapted to chronic continuous hypoxia. Acta Physiol. 2015, 214, 97–108. [Google Scholar] [CrossRef] [PubMed]
- Al-Lamki, R.S.; Lu, W.; Wang, J.; Yang, J.; Sargeant, T.J.; Wells, R.; Suo, C.; Wright, P.; Goddard, M.; Huang, Q.; et al. TNF, acting through inducibly expressed TNFR2, drives activation and cell cycle entry of c-kit+ cardiac stem cells in ischemic heart disease. Stem Cells 2013, 31, 1881–1892. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yu, Q.; Na, R.; Liu, B. Etanercept protects rat cardiomyocytes against hypertrophy by regulating inflammatory cytokines secretion and cell apoptosis. Braz. J. Med. Biol. Res. 2017, 50, e5868. [Google Scholar] [CrossRef] [PubMed]
- Stamm, C.; Friehs, I.; Cowan, D.B.; Moran, A.M.; Cao-Danh, H.; Duebener, L.F.; del Nido, P.J.; McGowan, F.X., Jr. Inhibition of tumor necrosis factor-α improves postischemic recovery of hypertrophied hearts. Circulation 2001, 104, I350–355. [Google Scholar] [CrossRef] [PubMed]
- Putko, B.N.; Wang, Z.; Lo, J.; Anderson, T.; Becher, H.; Dyck, J.R.; Kassiri, Z.; Oudit, G.Y. Circulating levels of tumor necrosis factor-α receptor 2 are increased in heart failure with preserved ejection fraction relative to heart failure with reduced ejection fraction: Evidence for a divergence in pathophysiology. PLoS ONE 2014, 9, e99495. [Google Scholar] [CrossRef] [PubMed]
- Suffredini, A.F.; Reda, D.; Banks, S.M.; Tropea, M.; Agosti, J.M.; Miller, R. Effects of recombinant dimeric TNF receptor on human inflammatory responses following intravenous endotoxin administration. J. Immunol. 1995, 155, 5038–5045. [Google Scholar] [PubMed]
- Utz, J.P.; Limper, A.H.; Kalra, S.; Specks, U.; Scott, J.P.; Vuk-Pavlovic, Z.; Schroeder, D.R. Etanercept for the treatment of stage II and III progressive pulmonary sarcoidosis. Chest 2003, 124, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Mann, D.L.; Bozkurt, B.; Torre-Amione, G.; Soran, O.Z.; Sivasubramanian, N. Effect of the soluble TNF-antagonist etanercept on tumor necrosis factor bioactivity and stability. Clin. Transl. Sci. 2008, 1, 142–145. [Google Scholar] [CrossRef] [PubMed]
- Edrees, A.F.; Misra, S.N.; Abdou, N.I. Anti-tumor necrosis factor (TNF) therapy in rheumatoid arthritis: Correlation of TNF-α serum level with clinical response and benefit from changing dose or frequency of infliximab infusions. Clin. Exp. Rheumatol. 2005, 23, 469–474. [Google Scholar] [PubMed]
- Eissner, G.; Kirchner, S.; Lindner, H.; Kolch, W.; Janosch, P.; Grell, M.; Scheurich, P.; Andreesen, R.; Holler, E. Reverse signaling through transmembrane TNF confers resistance to lipopolysaccharide in human monocytes and macrophages. J. Immunol. 2000, 164, 6193–6198. [Google Scholar] [CrossRef] [PubMed]
- Juhasz, K.; Buzas, K.; Duda, E. Importance of reverse signaling of the TNF superfamily in immune regulation. Expert. Rev. Clin. Immunol. 2013, 9, 335–348. [Google Scholar] [CrossRef] [PubMed]
- Al-Aly, Z.; Pan, H.; Zeringue, A.; Xian, H.; McDonald, J.R.; El-Achkar, T.M.; Eisen, S. Tumor necrosis factor-α blockade, cardiovascular outcomes, and survival in rheumatoid arthritis. Transl. Res. 2011, 157, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Michaud, K. Heart failure in rheumatoid arthritis: Rates, predictors, and the effect of anti-tumor necrosis factor therapy. Am. J. Med. 2004, 116, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Listing, J.; Strangfeld, A.; Kekow, J.; Schneider, M.; Kapelle, A.; Wassenberg, S.; Zink, A. Does tumor necrosis factor α inhibition promote or prevent heart failure in patients with rheumatoid arthritis? Arthritis Rheum. 2008, 58, 667–677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arts, E.E.; Fransen, J.; Den Broeder, A.A.; van Riel, P.; Popa, C.D. Low disease activity (DAS28≤3.2) reduces the risk of first cardiovascular event in rheumatoid arthritis: A time-dependent Cox regression analysis in a large cohort study. Ann. Rheum. Dis. 2017, 76, 1693–1699. [Google Scholar] [CrossRef] [PubMed]
- Gabriel, S.E. Tumor necrosis factor inhibition: A part of the solution or a part of the problem of heart failure in rheumatoid arthritis? Arthritis Rheum. 2008, 58, 637–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotyla, P.J.; Owczarek, A.; Rakoczy, J.; Lewicki, M.; Kucharz, E.J.; Emery, P. Infliximab treatment increases left ventricular ejection fraction in patients with rheumatoid arthritis: Assessment of heart function by echocardiography, endothelin 1, interleukin 6, and NT-pro brain natriuretic peptide. J. Rheumatol. 2012, 39, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Vizzardi, E.; Cavazzana, I.; Franceschini, F.; Bonadei, I.; Sciatti, E.; Lombardi, C.M.; Tincani, A.; Metra, M. Left ventricular function in rheumatoid arthritis during anti-TNF-α treatment: A speckle tracking prospective echocardiographic study. Monaldi Arch. Chest Dis. 2016, 84, 716. [Google Scholar] [CrossRef] [PubMed]
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kotyla, P.J. Bimodal Function of Anti-TNF Treatment: Shall We Be Concerned about Anti-TNF Treatment in Patients with Rheumatoid Arthritis and Heart Failure? Int. J. Mol. Sci. 2018, 19, 1739. https://doi.org/10.3390/ijms19061739
Kotyla PJ. Bimodal Function of Anti-TNF Treatment: Shall We Be Concerned about Anti-TNF Treatment in Patients with Rheumatoid Arthritis and Heart Failure? International Journal of Molecular Sciences. 2018; 19(6):1739. https://doi.org/10.3390/ijms19061739
Chicago/Turabian StyleKotyla, Przemyslaw J. 2018. "Bimodal Function of Anti-TNF Treatment: Shall We Be Concerned about Anti-TNF Treatment in Patients with Rheumatoid Arthritis and Heart Failure?" International Journal of Molecular Sciences 19, no. 6: 1739. https://doi.org/10.3390/ijms19061739
APA StyleKotyla, P. J. (2018). Bimodal Function of Anti-TNF Treatment: Shall We Be Concerned about Anti-TNF Treatment in Patients with Rheumatoid Arthritis and Heart Failure? International Journal of Molecular Sciences, 19(6), 1739. https://doi.org/10.3390/ijms19061739