Identification of Differentially Expressed Genes Induced by Aberrant Methylation in Oral Squamous Cell Carcinomas Using Integrated Bioinformatic Analysis




Abstract
:1. Introduction
2. Results
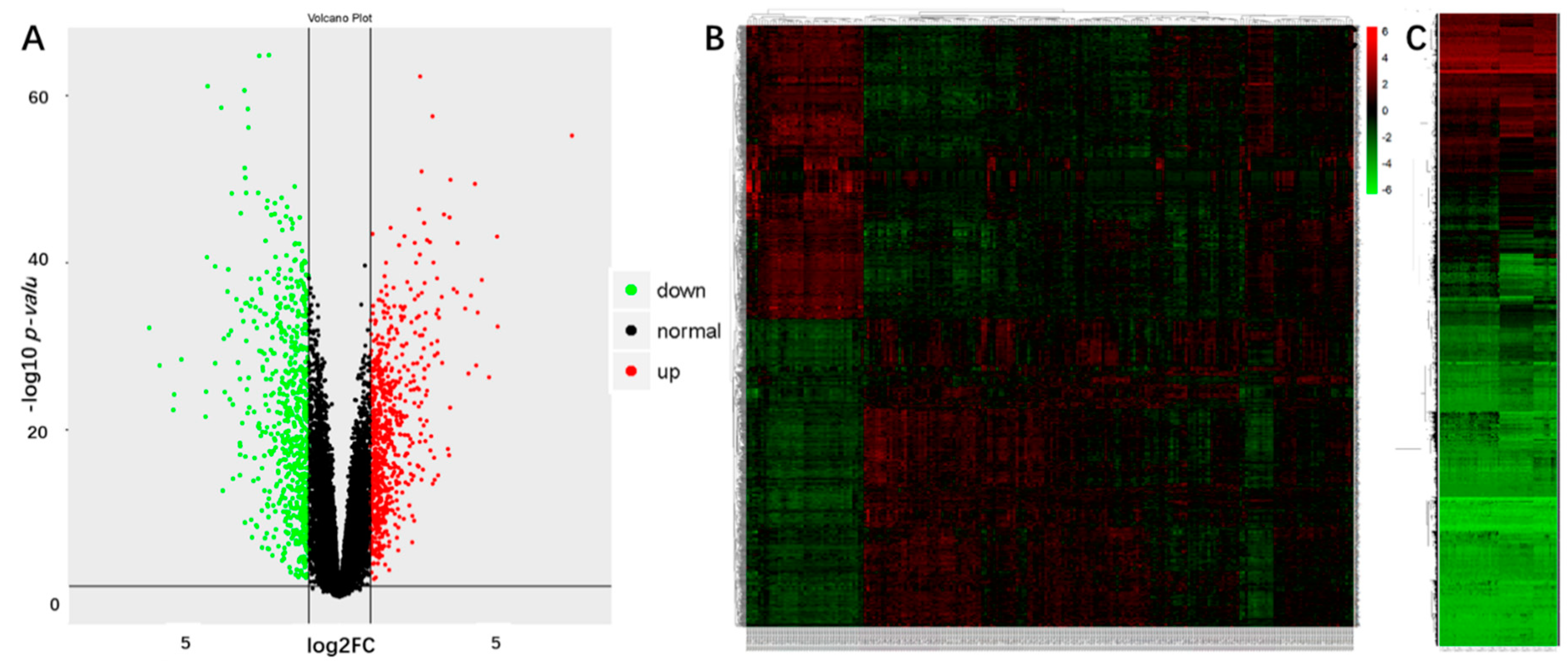
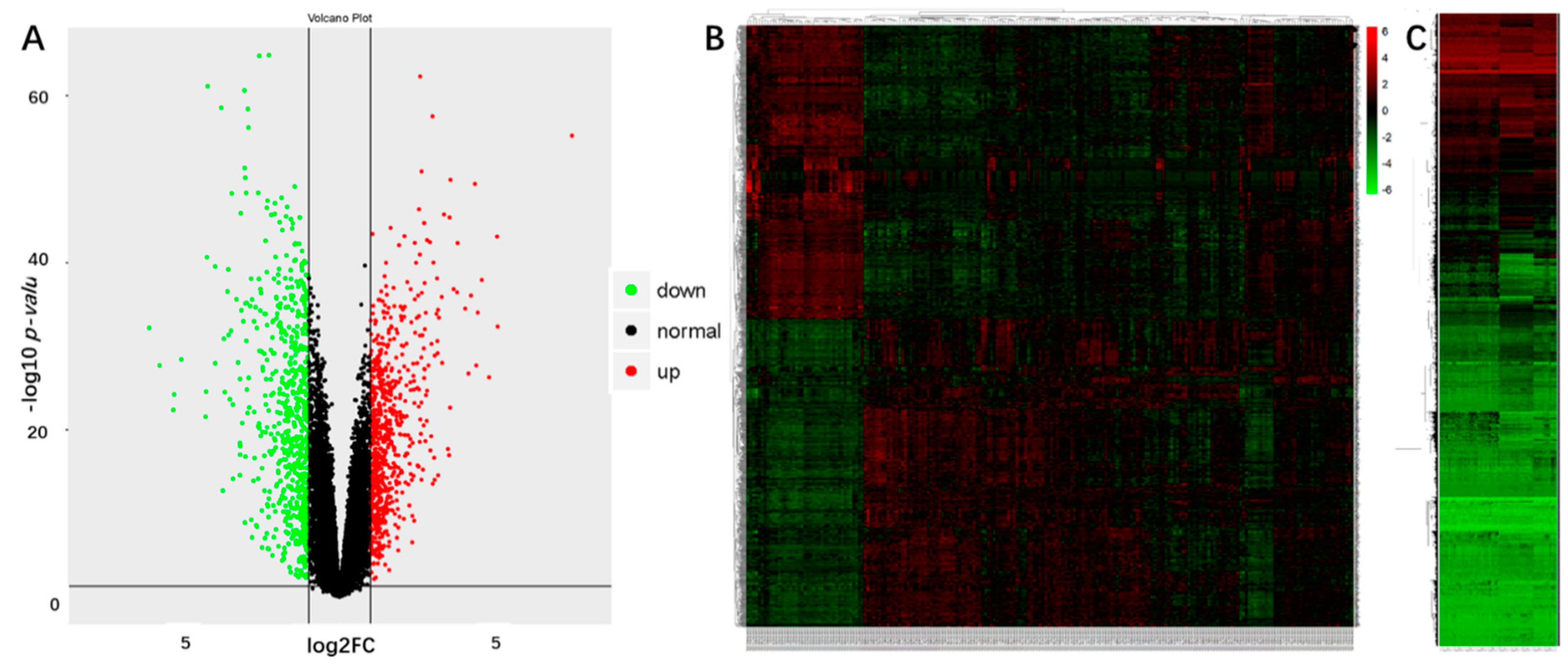
2.1. Identification of the DEGs and DMGs in OSCCs
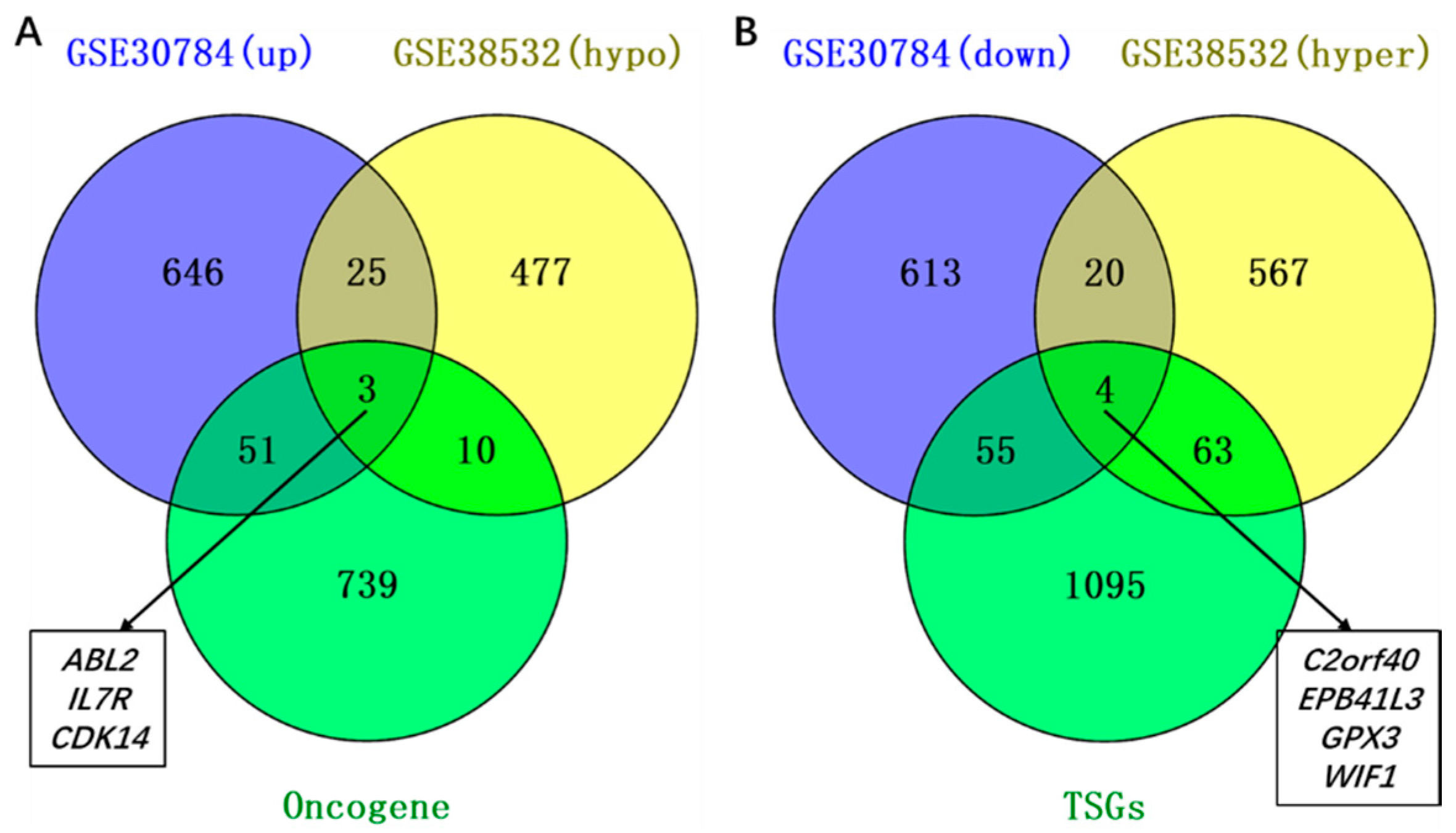
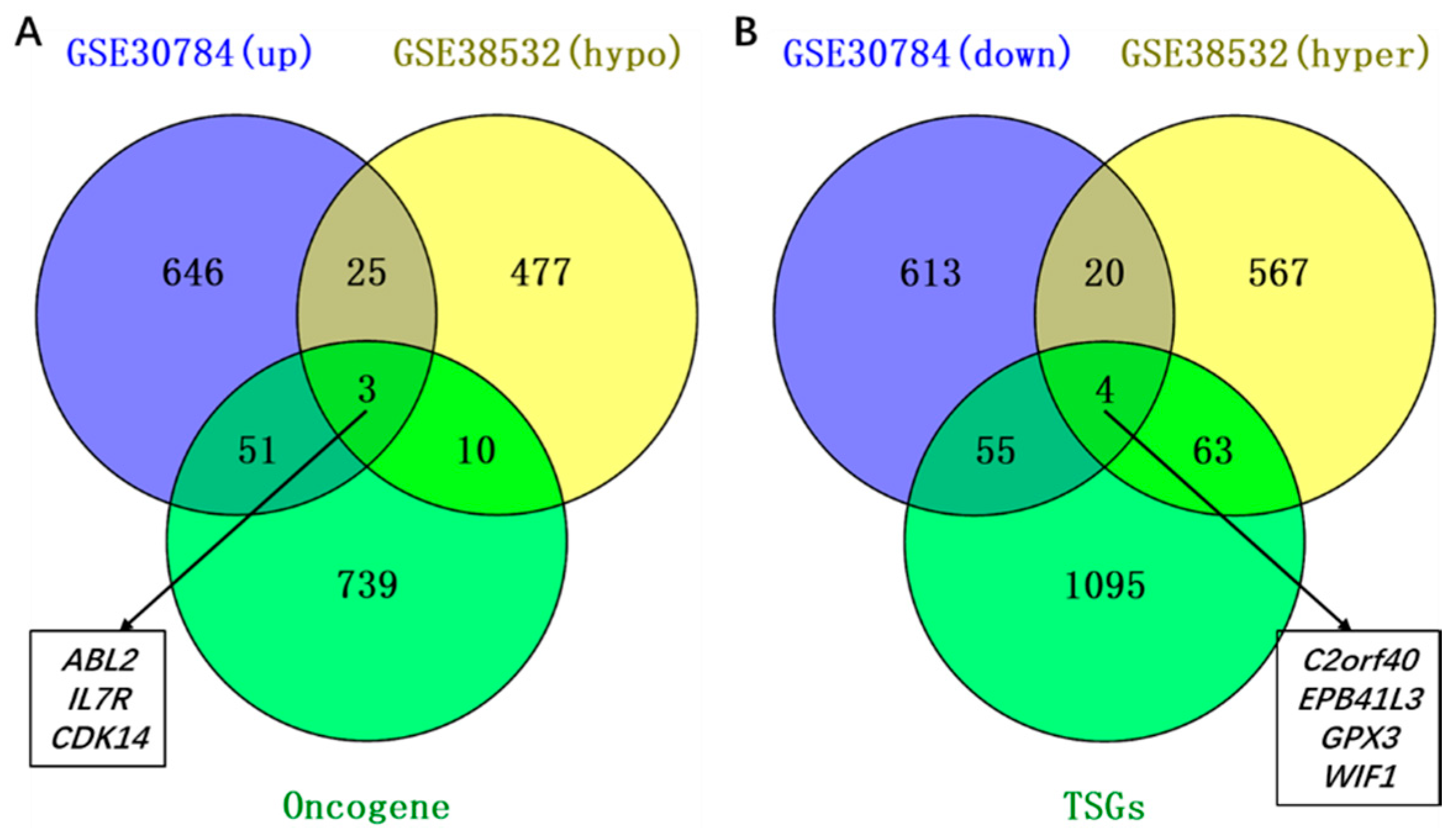
2.2. Identification of Aberrantly Methylated Differentially Expressed Genes
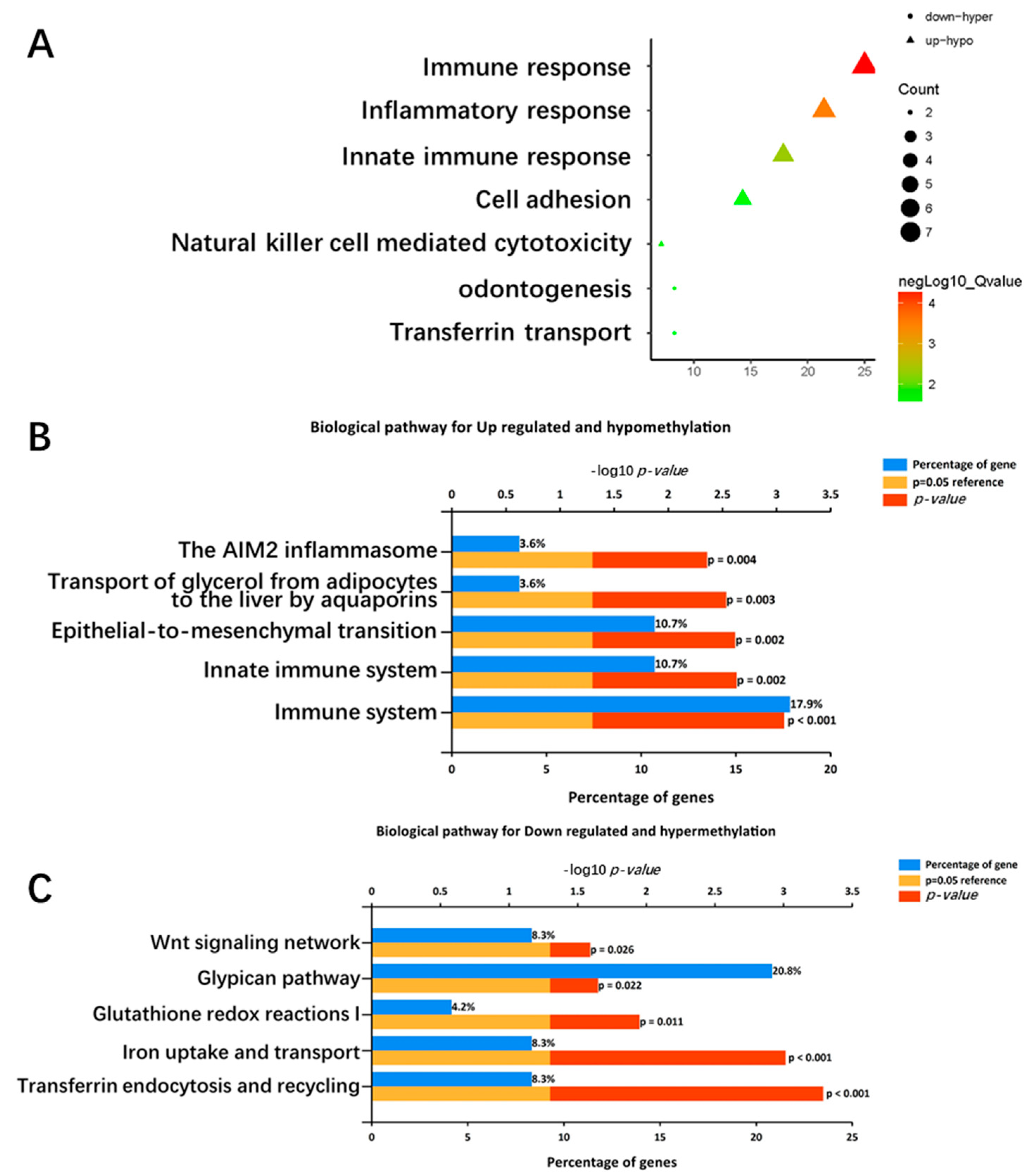
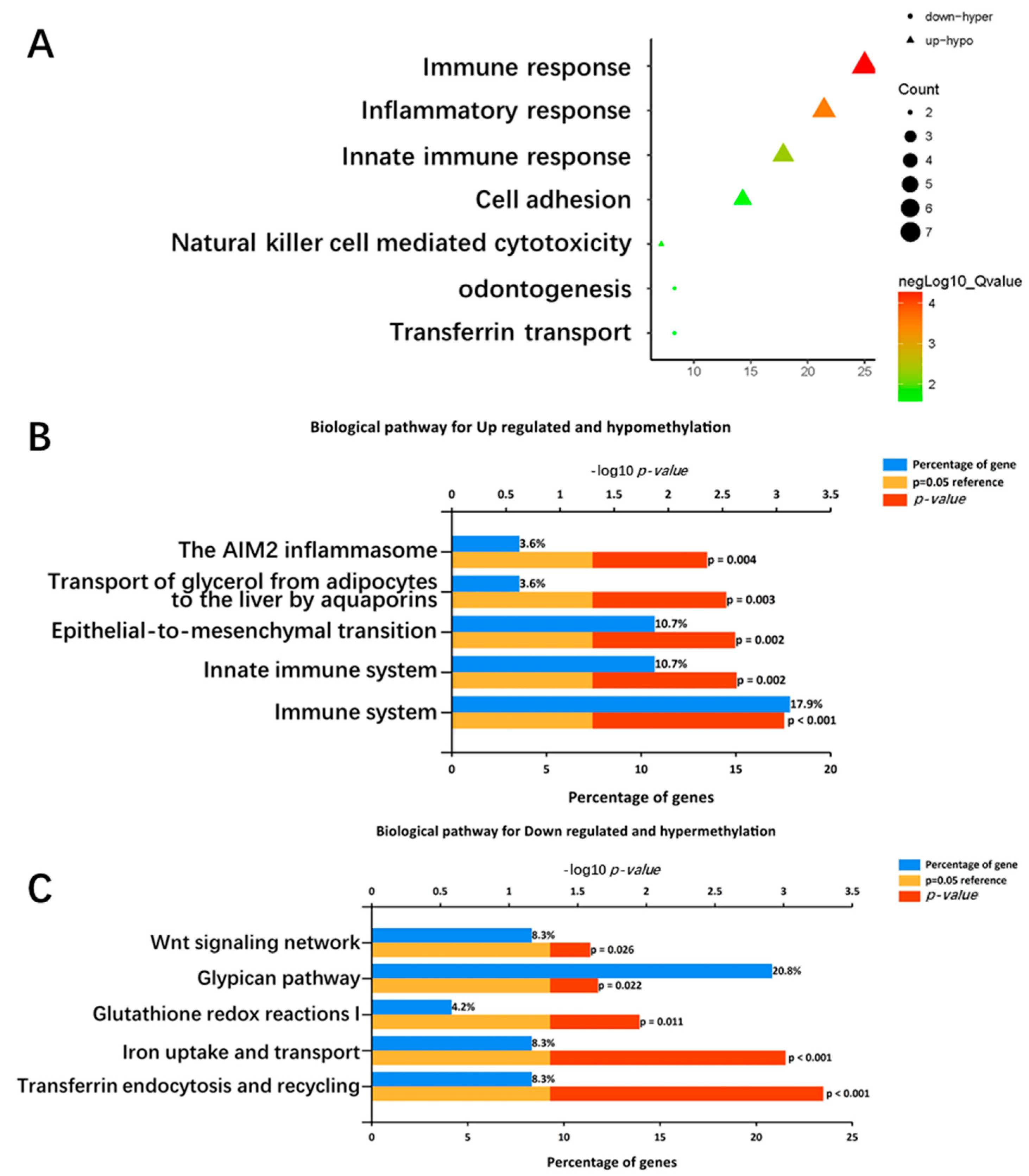
2.3. Functional Enrichment Analysis
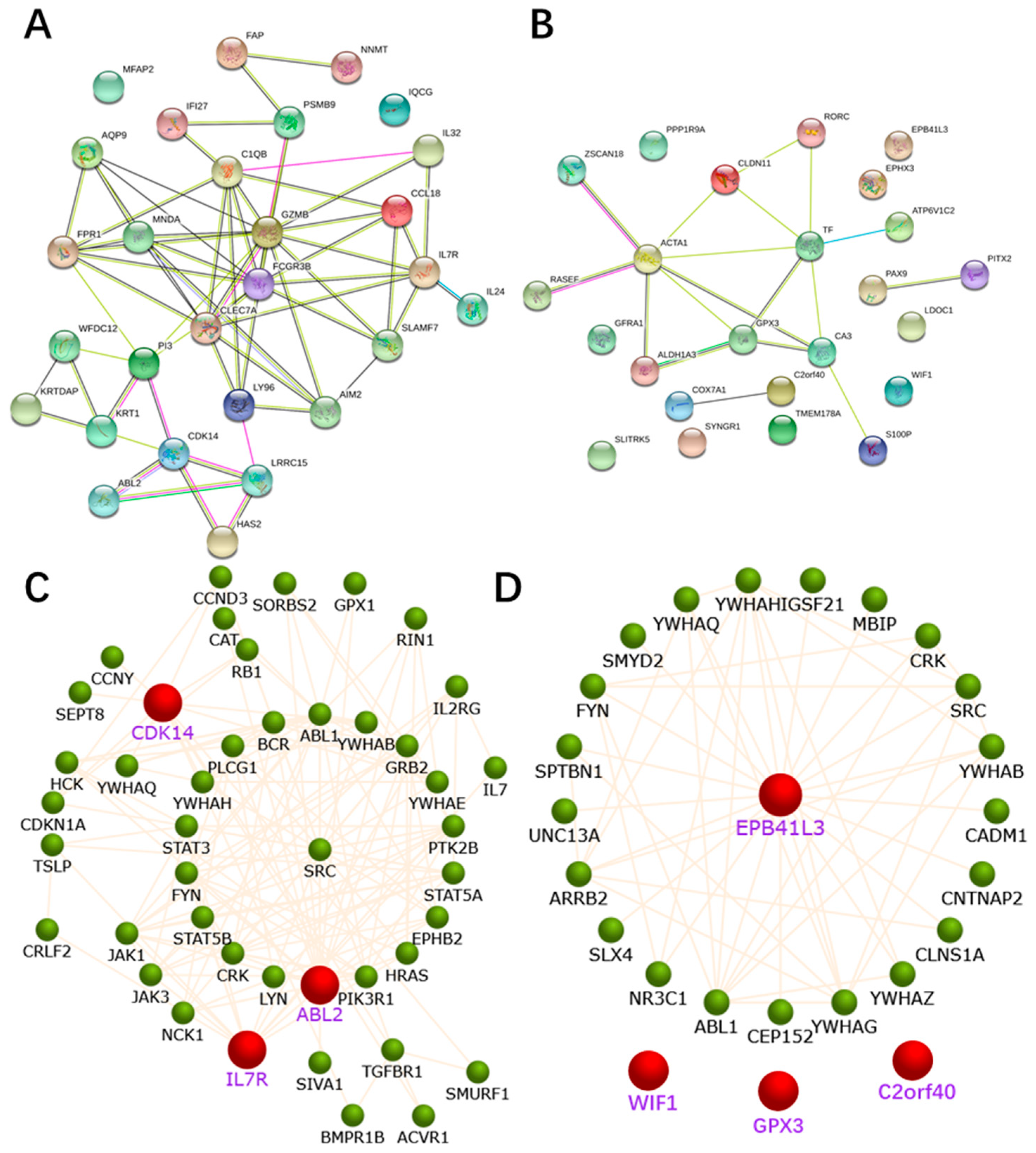
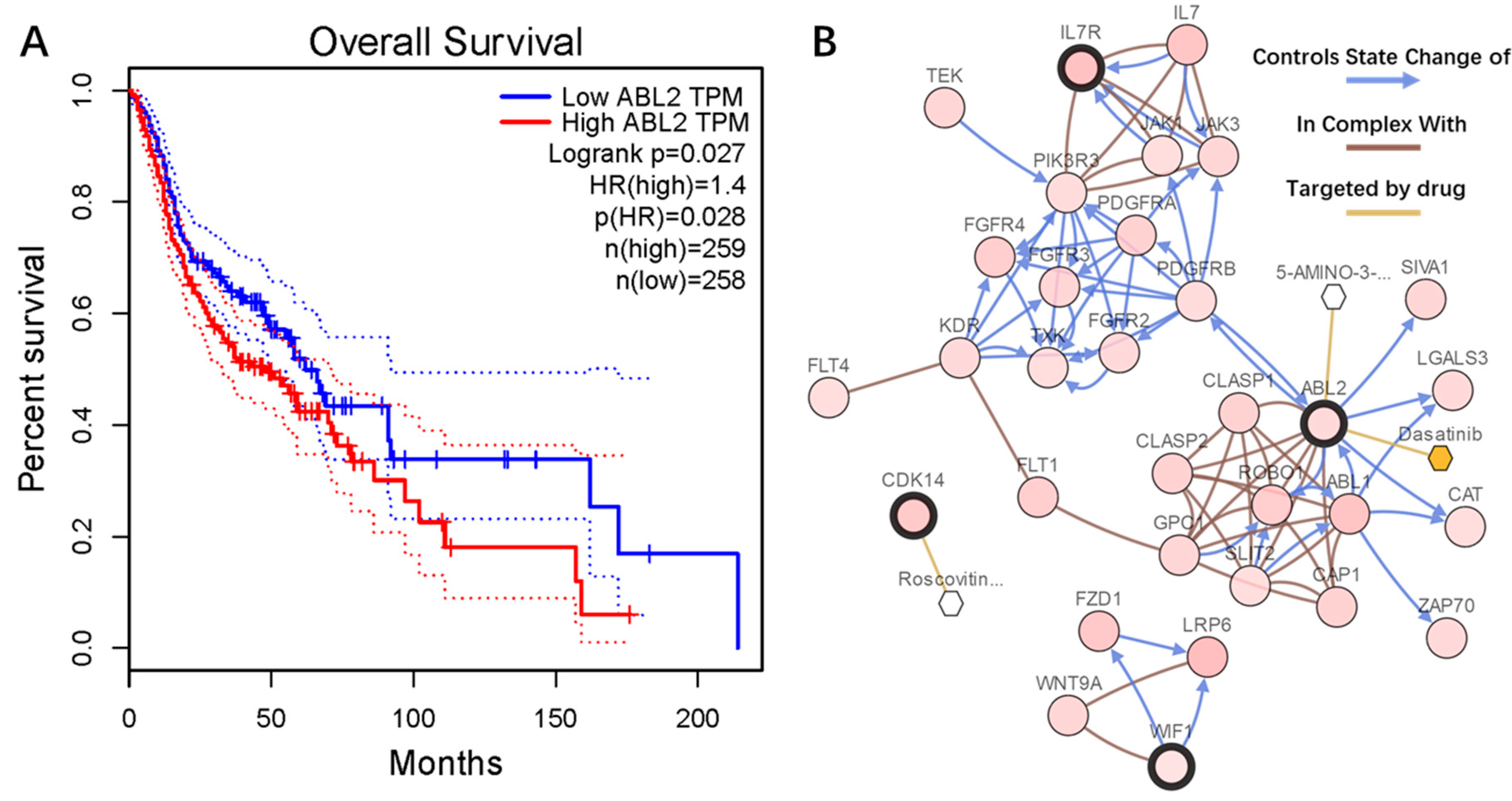
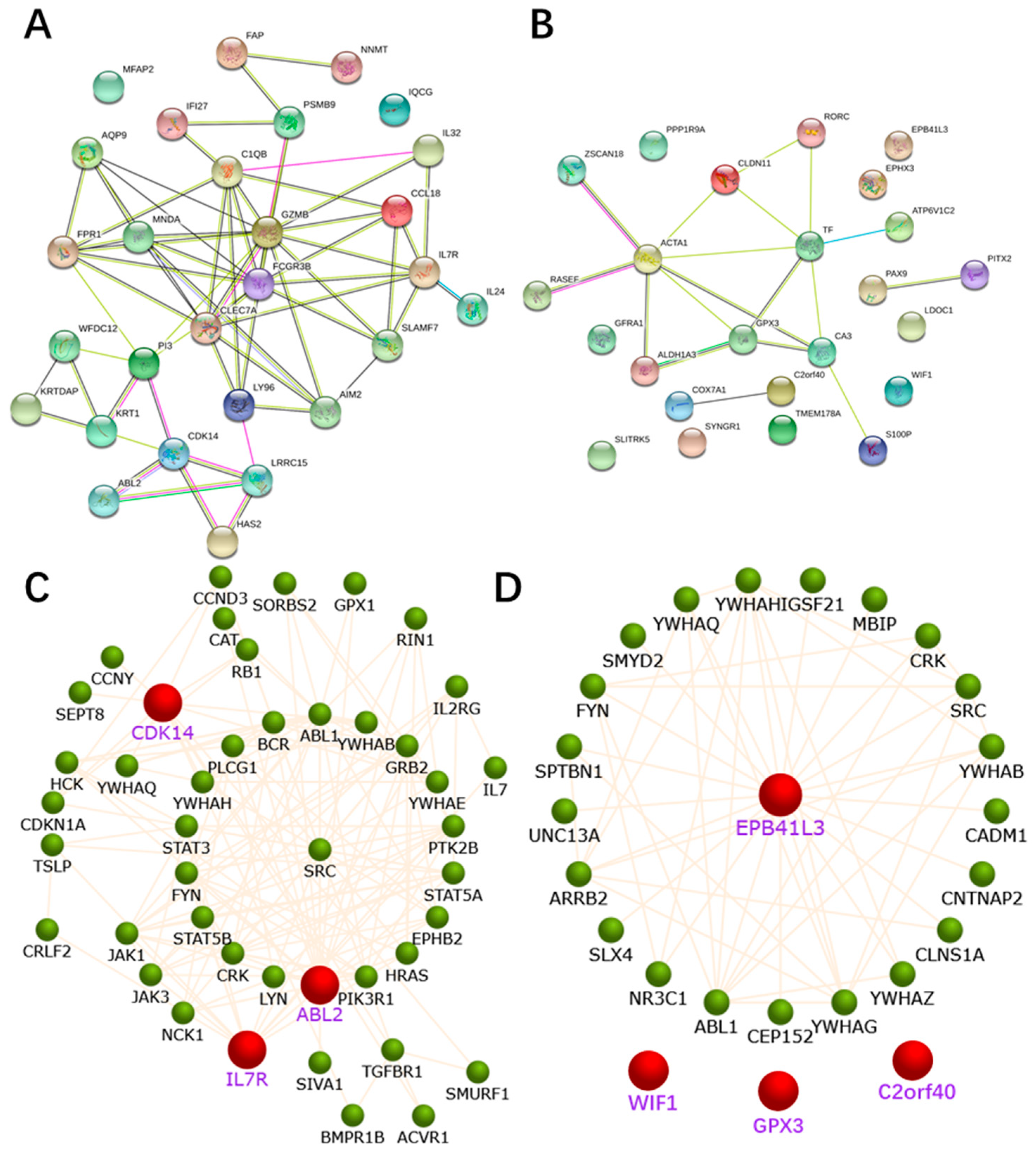
2.4. PPI Network Construction
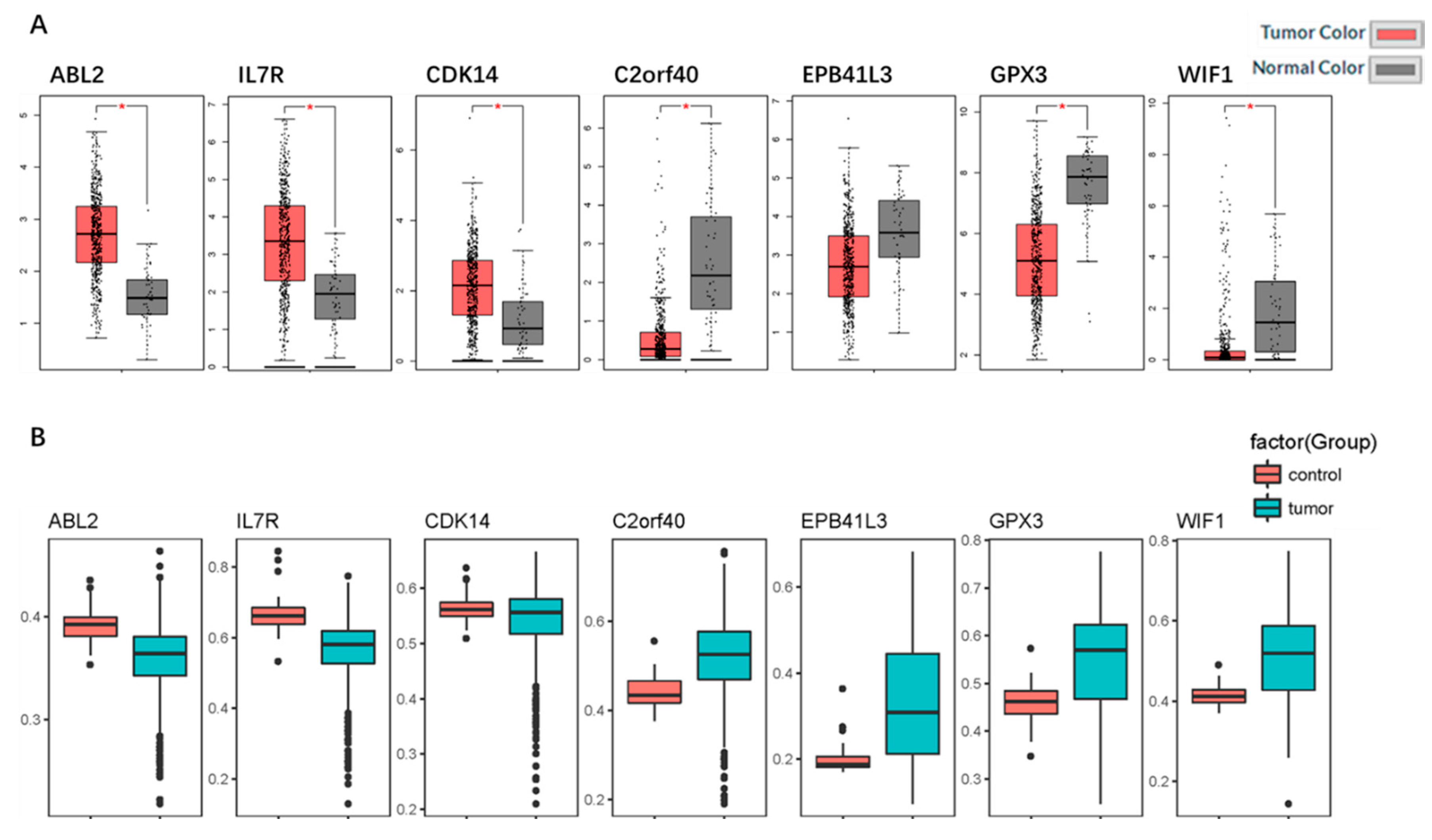
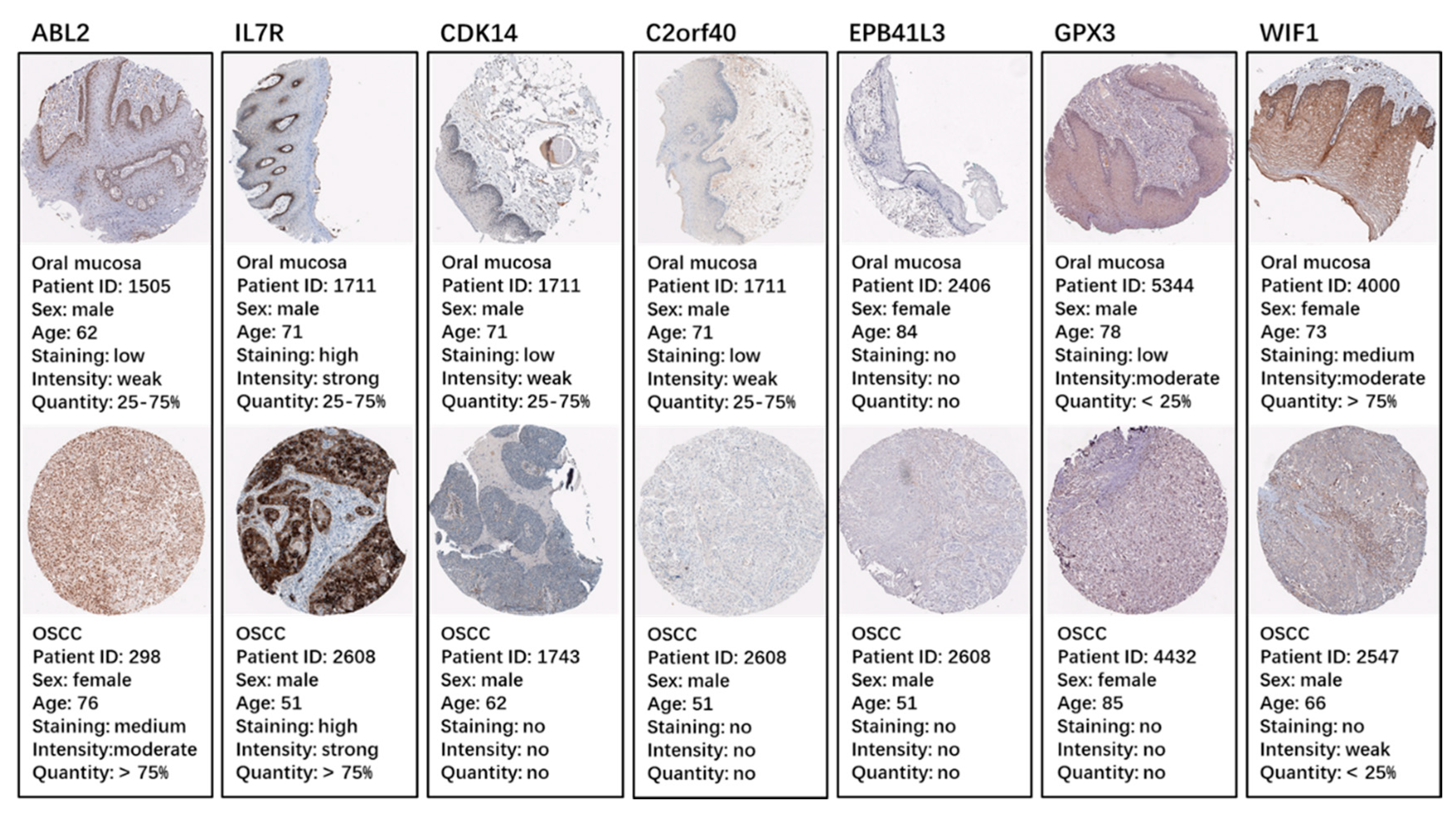
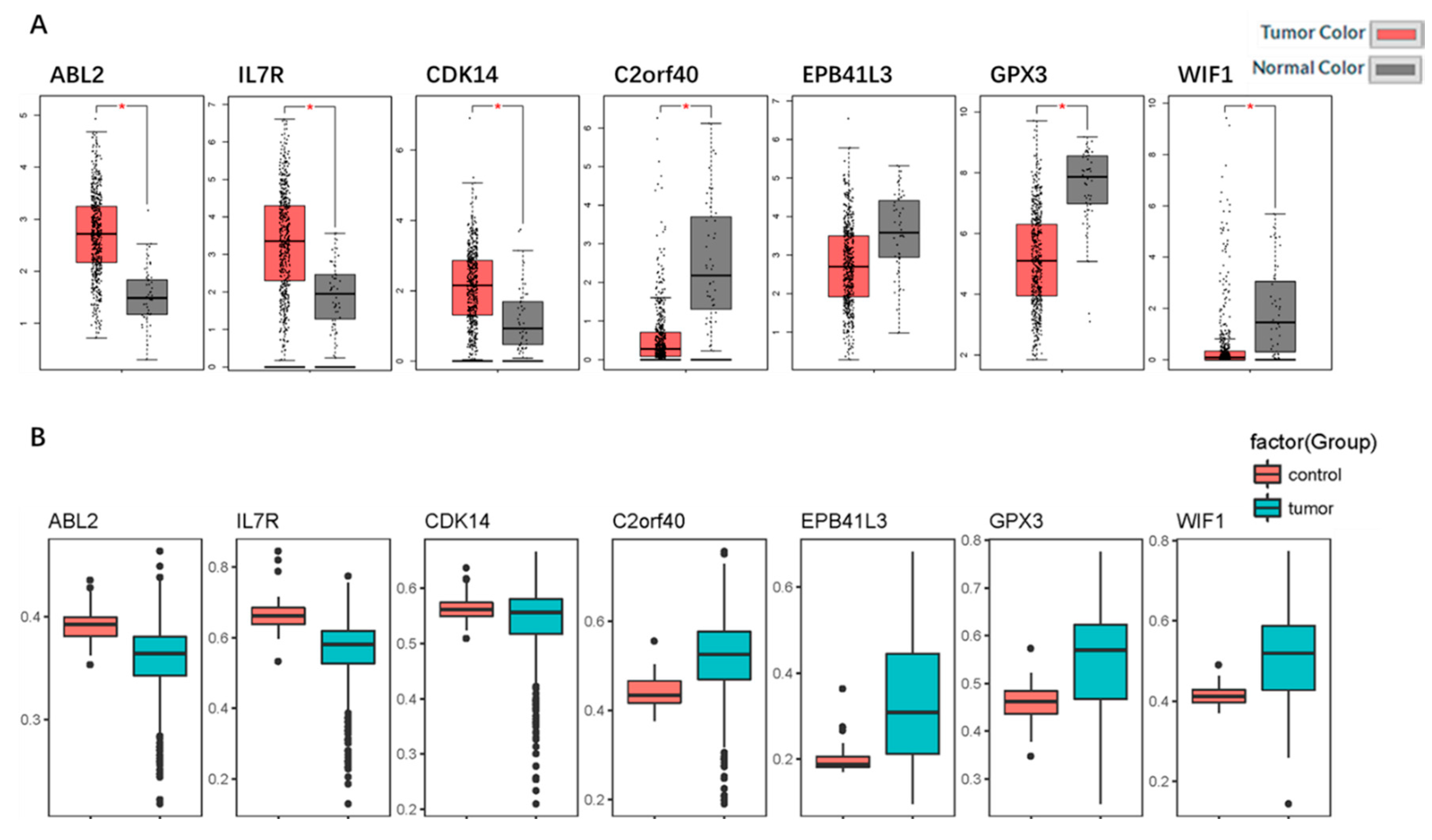
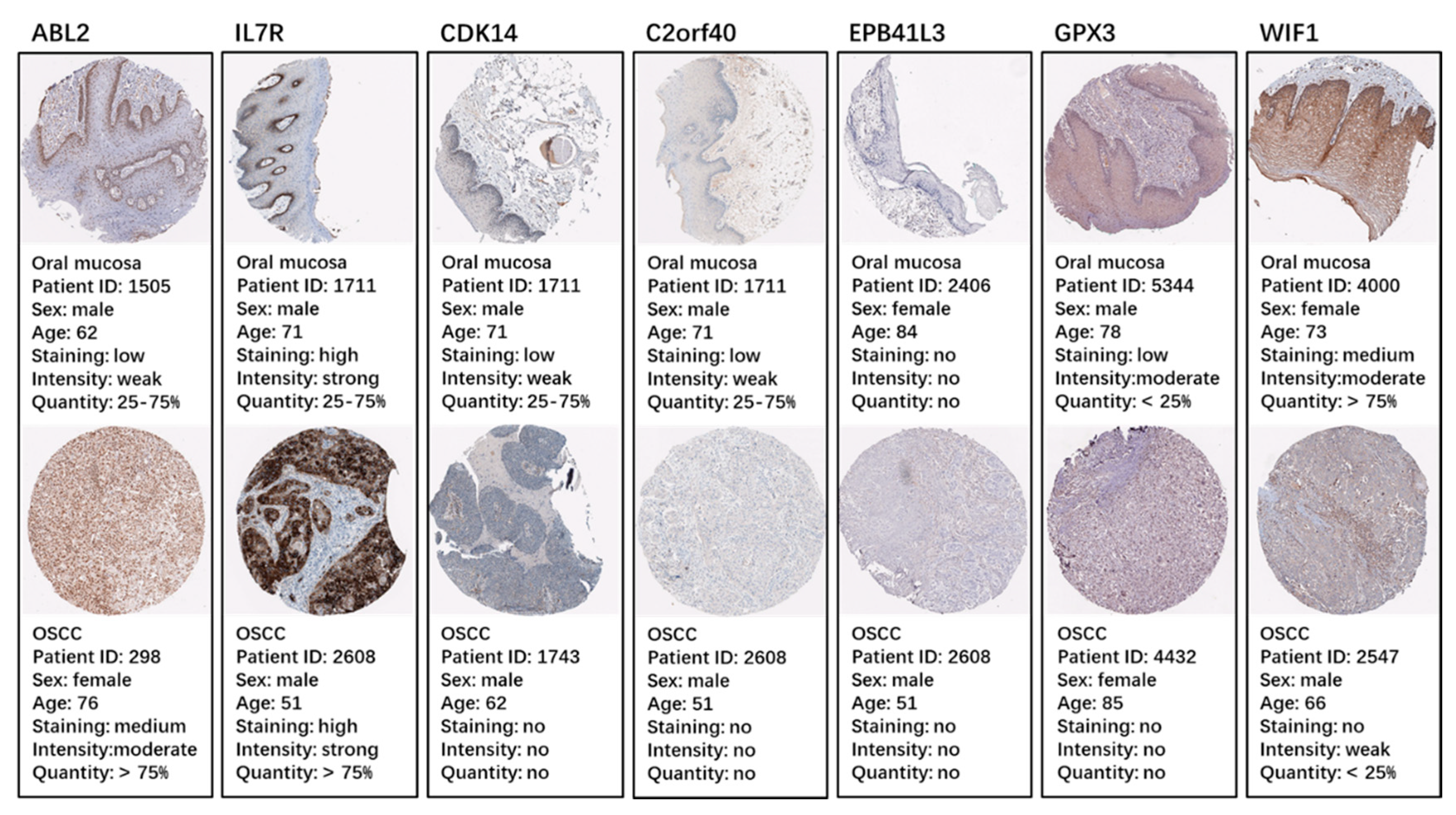
2.5. Identification and Validation of the Seven Selected Genes
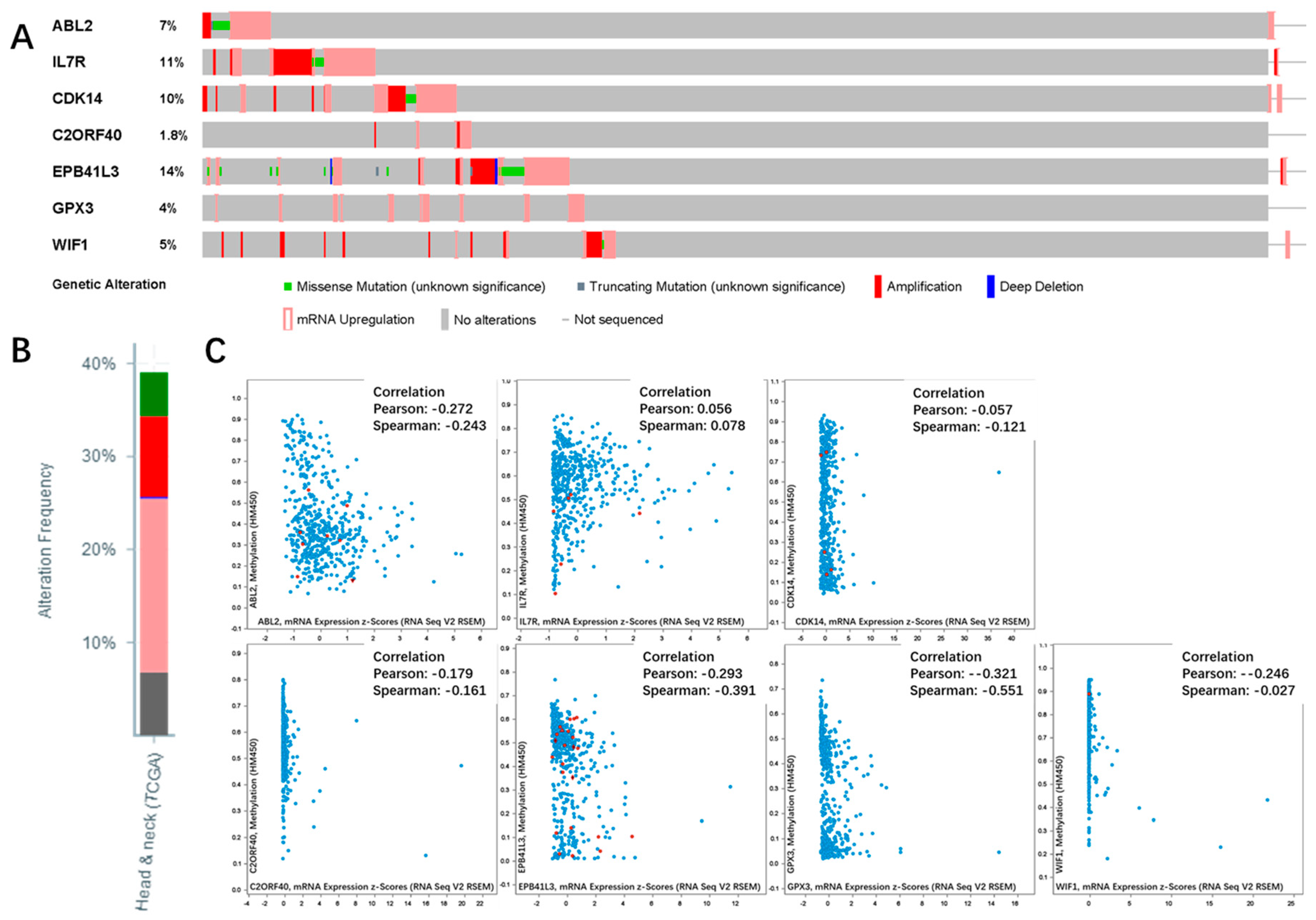
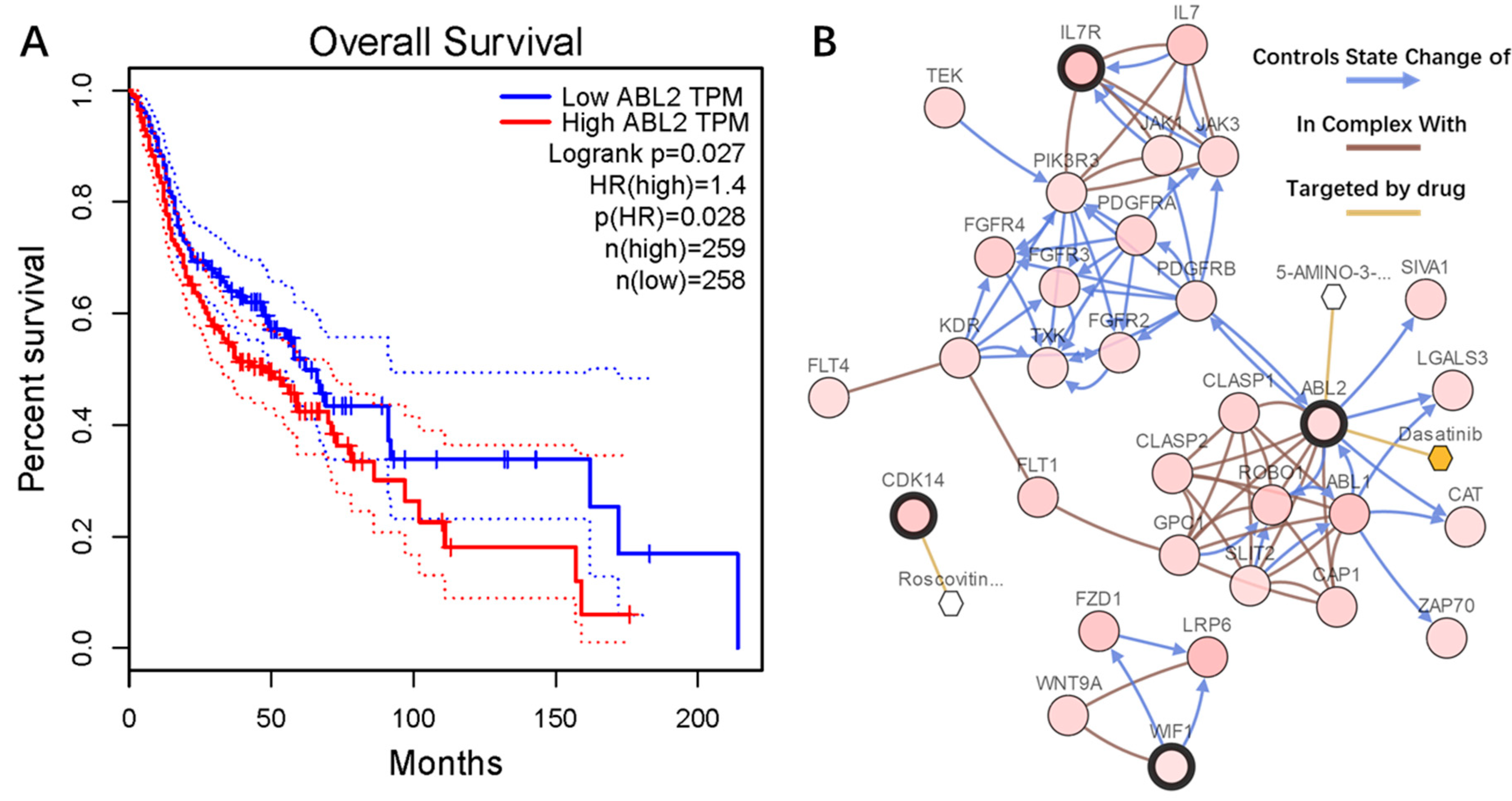
2.6. Genetic Information of the Seven Genes
3. Discussion
4. Materials and Methods
4.1. Microarray Data Information
4.2. Data Processing for the Identification of DEGs and DMGs
4.3. Gene Ontology and Pathway Enrichment Analysis
4.4. Protein–Protein Interaction (PPI) Network Construction and Module Analysis
4.5. Validation of the Seven Genes
5. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| HNSC | Head and neck squamous cell carcinoma |
| DEGs | Differentially methylated genes |
| DMGS | Differentially methylated genes |
| PPI | Protein–protein interaction |
| GO | Gene ontology |
| BP | Biological process |
| CC | Cell component |
| MF | Molecular function |
References
- Mignogna, M.D.; Fedele, S.; Lo Russo, L. The World Cancer Report and the burden of oral cancer. Eur. J. Cancer Prev. 2004, 13, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Eleftheriadou, A.; Chalastras, T.; Ferekidou, E.; Yiotakis, I.; Kyriou, L.; Tzagarakis, M.; Ferekidis, E.; Kandiloros, D. Association between squamous cell carcinoma of the head and neck and serum folate and homocysteine. Anticancer Res. 2006, 26, 2345–2348. [Google Scholar] [PubMed]
- Lo, A.K.F.; Lo, K.W.; Tsao, S.W.; Wong, H.L.; Hui, J.W.Y.; To, K.F.; Hayward, S.D.; Chui, Y.L.; Lau, Y.L.; Takada, K.; et al. Epstein-Barr virus infection alters cellular signal cascades in human nasopharyngeal epithelial cells. Neoplasia 2006, 8, 173–180. [Google Scholar] [PubMed]
- Hennessey, P.T.; Westra, W.H.; Califano, J.A. Human papillomavirus and head and neck squamous cell Carcinoma: Recent evidence and clinical implications. J. Dent. Res. 2009, 88, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.M.; Rubenstein, L.M.; Haugen, T.H.; Pawlita, M.; Turek, L.P. Complex etiology underlies risk and survival in head and neck cancer human papillomavirus, tobacco, and Alcohol: A case for multifactor disease. J. Oncol. 2012, 2012, 571862. [Google Scholar] [CrossRef] [PubMed]
- Maruya, S.; Issa, J.P.; Weber, R.S.; Rosenthal, D.I.; Haviland, J.C.; Lotan, R.; El-Naggar, A.K. Differential methylation status of tumor-associated genes in head and neck squamous Carcinoma: Incidence and potential implications. Clin. Cancer Res. 2004, 10, 3825–3830. [Google Scholar] [CrossRef] [PubMed]
- Towle, R.; Truong, D.; Hogg, K.; Robinson, W.P.; Poh, C.F.; Garnis, C. Global analysis of DNA methylation changes during progression of oral cancer. Oral Oncol. 2013, 49, 1033–1042. [Google Scholar] [CrossRef] [PubMed]
- Farkas, S.A.; Milutin-Gasperov, N.; Grce, M.; Nilsson, T.K. Genome-wide DNA methylation assay reveals novel candidate biomarker genes in cervical cancer. Epigenetics 2013, 8, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatu, A.; Sartore-Bianchi, A.; Moutinho, C.; Belotti, A.; Bencardino, K.; Chirico, G.; Cassingena, A.; Rusconi, F.; Esposito, A.; Nichelatti, M.; et al. Promoter CpG island hypermethylation of the DNA repair enzyme MGMT predicts clinical response to dacarbazine in a phase II study for metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 2265–2272. [Google Scholar] [CrossRef] [PubMed]
- Draht, M.X.; Riedl, R.R.; Niessen, H.; Carvalho, B.; Meijer, G.A.; Herman, J.G.; van Engeland, M.; Melotte, V.; Smits, K.M. Promoter CpG island methylation markers in colorectal Cancer: The road ahead. Epigenomics 2012, 4, 179–194. [Google Scholar] [CrossRef] [PubMed]
- Kulasingam, V.; Diamandis, E.P. Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat. Clin. Pract. Oncol. 2008, 5, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Demokan, S.; Chuang, A.Y.; Chang, X.; Khan, T.; Smith, I.M.; Pattani, K.M.; Dasgupta, S.; Begum, S.; Khan, Z.; Liegeois, N.J.; et al. Identification of guanine nucleotide-binding protein γ-7 as an epigenetically silenced gene in head and neck cancer by gene expression profiling. Int. J. Oncol. 2013, 42, 1427–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poage, G.M.; Houseman, E.A.; Christensen, B.C.; Butler, R.A.; Avissar-Whiting, M.; McClean, M.D.; Waterboer, T.; Pawlita, M.; Marsit, C.J.; Kelsey, K.T. Global hypomethylation identifies Loci targeted for hypermethylation in head and neck cancer. Clin. Cancer Res. 2011, 17, 3579–3589. [Google Scholar] [CrossRef] [PubMed]
- Wimmers, F.; Aarntzen, E.H.; Duiveman-deBoer, T.; Figdor, C.G.; Jacobs, J.F.; Tel, J.; de Vries, I.J. Long-lasting multifunctional CD8+ T cell responses in end-stage melanoma patients can be induced by dendritic cell vaccination. Oncoimmunology 2016, 5, e1067745. [Google Scholar] [CrossRef] [PubMed]
- De Visser, K.E.; Eichten, A.; Coussens, L.M. Paradoxical roles of the immune system during cancer development. Nat. Rev. Cancer 2006, 6, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Steelman, L.S.; Chappell, W.H.; Abrams, S.L.; Kempf, C.R.; Long, J.; Laidler, P.; Mijatovic, S.; Maksimovic-Ivanic, D.; Stivala, F.; Mazzarino, M.C.; et al. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging 2011, 3, 192–222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Carnero, A.; Blanco-Aparicio, C.; Renner, O.; Link, W.; Leal, J.F.M. The PTEN/PI3K/AKT signalling pathway in cancer, therapeutic implications. Curr. Cancer Drug Target 2008, 8, 187–198. [Google Scholar] [CrossRef]
- Bleau, A.M.; Hambardzumyan, D.; Ozawa, T.; Fomchenko, E.I.; Huse, J.T.; Brennan, C.W.; Holland, E.C. PTEN/PI3K/Akt pathway regulates the side population phenotype and ABCG2 activity in glioma tumor stem-like cells. Cell Stem Cell 2009, 4, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Iijima, Y.; Ito, T.; Oikawa, T.; Eguchi, M.; Eguchi-Ishimae, M.; Kamada, N.; Kishi, K.; Asano, S.; Sakaki, Y.; Sato, Y. A new ETV6/TEL partner gene, ARG (ABL-related gene or ABL2), identified in an AML-M3 cell line with a t(1;12)(q25;p13) translocation. Blood 2000, 95, 2126–2131. [Google Scholar] [PubMed]
- Lin, Y.C.; Yeckel, M.F.; Koleske, A.J. Abl2/Arg controls dendritic spine and dendrite arbor stability via distinct cytoskeletal control pathways. J. Neurosci. 2013, 33, 1846–1857. [Google Scholar] [CrossRef] [PubMed]
- Xing, Q.T.; Qu, C.M.; Wang, G. Overexpression of Abl2 predicts poor prognosis in hepatocellular carcinomas and is associated with cancer cell migration and invasion. Oncotargets Ther. 2014, 7, 881–885. [Google Scholar] [CrossRef] [PubMed]
- Qiang, X.F.; Zhang, Z.W.; Liu, Q.; Sun, N.; Pan, L.L.; Shen, J.; Li, T.; Yun, C.; Li, H.; Shi, L.H. miR-20a promotes prostate cancer invasion and migration through targeting ABL2. J. Cell. Biochem. 2014, 115, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Gil-Henn, H.; Patsialou, A.; Wang, Y.; Warren, M.S.; Condeelis, J.S.; Koleske, A.J. Arg/Abl2 promotes invasion and attenuates proliferation of breast cancer in vivo. Oncogene 2013, 32, 2622–2630. [Google Scholar] [CrossRef] [PubMed]
- Gregory, S.G.; Schmidt, S.; Seth, P.; Oksenberg, J.R.; Hart, J.; Prokop, A.; Caillier, S.J.; Ban, M.; Goris, A.; Barcellos, L.F.; et al. Interleukin 7 receptor a chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007, 39, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Puel, A.; Ziegler, S.F.; Buckley, R.H.; Leonard, W.J. Defective IL7R expression in T−B+NK+ severe combined immunodeficiency. Nat. Genet. 1998, 20, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Zenatti, P.P.; Ribeiro, D.; Li, W.Q.; Zuurbier, L.; Silva, M.C.; Paganin, M.; Tritapoe, J.; Hixon, J.A.; Silveira, A.B.; Cardoso, B.A.; et al. Oncogenic IL7R gain-of-function mutations in childhood T-cell acute lymphoblastic leukemia. Nat. Genet. 2011, 43, 932. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, M.; Morimoto, M.; Baradaran-Heravi, A.; Choi, K.; Kambham, N.; Jensen, K.; Dutt, S.; Dionis-Petersen, K.Y.; Liu, L.X.; Felix, K.; et al. Lack of IL7R alpha expression in T cells is a hallmark of T-cell immunodeficiency in Schimke immuno-osseous dysplasia (SIOD). Clin. Immunol. 2015, 161, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Chen, J.Y. Identification and cellular localization of human PFTAIRE1. Gene 2001, 267, 165–172. [Google Scholar] [CrossRef]
- Ji, Q.B.; Xu, X.J.; Li, L.; Goodman, S.B.; Bi, W.Z.; Xu, M.; Xu, Y.M.; Fan, Z.Y.; Maloney, W.J.; Ye, Q.N.; et al. miR-216a inhibits osteosarcoma cell proliferation, invasion and metastasis by targeting CDK14. Cell Death Dis. 2017, 8, e3103. [Google Scholar] [CrossRef] [PubMed]
- Shia, W.; Chen, D. The allele-specific copy number imbalance in CDK14/RABGAP1L/SH3BP5L and the relation to the poor prognosis in breast cancer. Breast 2017, 32, S104–S105. [Google Scholar] [CrossRef]
- Li, L.W.; Li, X.Y.; Wang, W.Y.; Gao, T.H.; Zhou, Y.; Lu, S.X. Soluble purified recombinant C2ORF40 protein inhibits tumor cell growth in vivo by decreasing telomerase activity in esophageal squamous cell carcinoma. Oncol. Lett. 2016, 12, 2820–2824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.Y.; Li, L.W.; Wang, W.Y.; Yang, Y.; Zhou, Y.; Lu, S.X. Soluble purified recombinant C2ORF40 protein inhibits esophageal cancer cell proliferation by inducing cell cycle G1 phase block. Oncol. Lett. 2015, 10, 1593–1596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.H.; Zhang, Y.; Zhang, H.W.; Liu, X.N.; Gong, T.Q.; Li, M.B.; Sun, L.; Ji, G.; Shi, Y.Q.; Han, Z.Y.; et al. miRNA-223 promotes gastric cancer invasion and metastasis by targeting tumor suppressor EPB41L3. Mol. Cancer Res. 2011, 9, 824–833. [Google Scholar] [CrossRef] [PubMed]
- Zeng, R.; Liu, Y.; Jiang, Z.J.; Huang, J.P.; Wang, Y.; Li, X.F.; Xiong, W.B.; Wu, X.C.; Zhang, J.R.; Wang, Q.E.; et al. EPB41L3 is a potential tumor suppressor gene and prognostic indicator in esophageal squamous cell carcinoma. Int. J. Oncol. 2018, 52, 1443–1454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.Y.; Yang, N.H.; Chen, J.; Zeng, T.; Yan, S.Y.; Liu, Y.Y.; Yu, G.F.; Chen, Q.X.; Du, G.Q.; Pan, W.; et al. LINC00052 upregulates EPB41L3 to inhibit migration and invasion of hepatocellular carcinoma by binding miR-452-5p. Oncotarget 2017, 8, 63724–63737. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.F. The human glutathione-peroxidase genes Gpx2, Gpx3, and Gpx4 map to chromosome-14, chromosome-5, and chromosome-19, respectively. Cytogenet. Genome Res. 1994, 66, 96–98. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Li, J.Y.; Li, X.; Han, C.; Zhang, Y.; Zheng, L.L.; Guo, M.Z. Silencing GPX3 Expression promotes tumor metastasis in human thyroid cancer. Curr. Protein Pept. Sci. 2015, 16, 316–321. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.L.; Yang, L.P.; Zou, Q.; Yuan, Y.; Li, J.H.; Liang, L.F.; Zeng, G.X.; Chen, S.L. Positive ALDH1A3 and negative GPX3 expressions are biomarkers for poor prognosis of gallbladder cancer. Dis. Markers 2013, 35, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Vassallo, I.; Zinn, P.; Lai, M.; Rajakannu, P.; Hamou, M.F.; Hegi, M.E. WIF1 re-expression in glioblastoma inhibits migration through attenuation of non-canonical WNT signaling by downregulating the lncRNA MALAT1. Oncogene 2016, 35, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Wissmann, C.; Wild, P.J.; Kaiser, S.; Roepcke, S.; Stoehr, R.; Woenckhaus, M.; Kristiansen, G.; Hsieh, J.C.; Hofstaecter, F.; Hartmann, A.; et al. WIF1, a component of the Wnt pathway, is down-regulated in prostate, breast, lung, and bladder cancer. J. Pathol. 2003, 201, 204–212. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.X.; Simoneau, A.R.; Liao, W.X.; Yi, G.; Hope, C.; Liu, F.; Li, S.Q.; Xie, J.; Holcombe, R.F.; Jurnak, F.A.; et al. WIF1, a Wnt pathway inhibitor, regulates SKP2 and c-myc expression leading to G1 arrest and growth inhibition of human invasive urinary bladder cancer cells. Mol. Cancer Ther. 2009, 8, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Node | Count | p-Value |
|---|---|---|---|
| Upregulated and hypomethylated oncogene | |||
| 1 | Proteoglycan syndecan-mediated signaling events | 32 | p < 0.001 |
| 2 | Class I PI3K signaling events | 31 | p < 0.001 |
| 3 | Insulin Pathway | 31 | p < 0.001 |
| 4 | mTOR signaling pathway | 31 | p < 0.001 |
| 5 | Class I PI3K signaling events mediated by AKT | 31 | p < 0.001 |
| Downregulated and hypermethylated TSGs | |||
| 1 | α6β4 integrin | 7 | p < 0.001 |
| 2 | Neurotrophic factor-mediated Trk receptor signaling | 8 | p < 0.001 |
| 3 | p75(NTR)-mediated signaling | 9 | p < 0.001 |
| 4 | Regulation of nuclear SMAD2/3 signaling | 10 | p < 0.001 |
| 5 | TGF-β receptor signaling | 10 | p < 0.001 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Feng, H.; Li, D.; Liu, S.; Amizuka, N.; Li, M. Identification of Differentially Expressed Genes Induced by Aberrant Methylation in Oral Squamous Cell Carcinomas Using Integrated Bioinformatic Analysis. Int. J. Mol. Sci. 2018, 19, 1698. https://doi.org/10.3390/ijms19061698
Zhang X, Feng H, Li D, Liu S, Amizuka N, Li M. Identification of Differentially Expressed Genes Induced by Aberrant Methylation in Oral Squamous Cell Carcinomas Using Integrated Bioinformatic Analysis. International Journal of Molecular Sciences. 2018; 19(6):1698. https://doi.org/10.3390/ijms19061698
Chicago/Turabian StyleZhang, Xiaoqi, Hao Feng, Dongfang Li, Shanshan Liu, Norio Amizuka, and Minqi Li. 2018. "Identification of Differentially Expressed Genes Induced by Aberrant Methylation in Oral Squamous Cell Carcinomas Using Integrated Bioinformatic Analysis" International Journal of Molecular Sciences 19, no. 6: 1698. https://doi.org/10.3390/ijms19061698
APA StyleZhang, X., Feng, H., Li, D., Liu, S., Amizuka, N., & Li, M. (2018). Identification of Differentially Expressed Genes Induced by Aberrant Methylation in Oral Squamous Cell Carcinomas Using Integrated Bioinformatic Analysis. International Journal of Molecular Sciences, 19(6), 1698. https://doi.org/10.3390/ijms19061698