The Cholinergic Anti-Inflammatory Response and the Role of Macrophages in HIV-Induced Inflammation

Abstract
1. Introduction
2. Monocytes and Macrophages: Their Origin and Role in Immune Response
3. Macrophages, Inflammation, and HIV Infection
4. Macrophages and the Cholinergic Anti-Inflammatory Response: A Focus on α7-nAChR
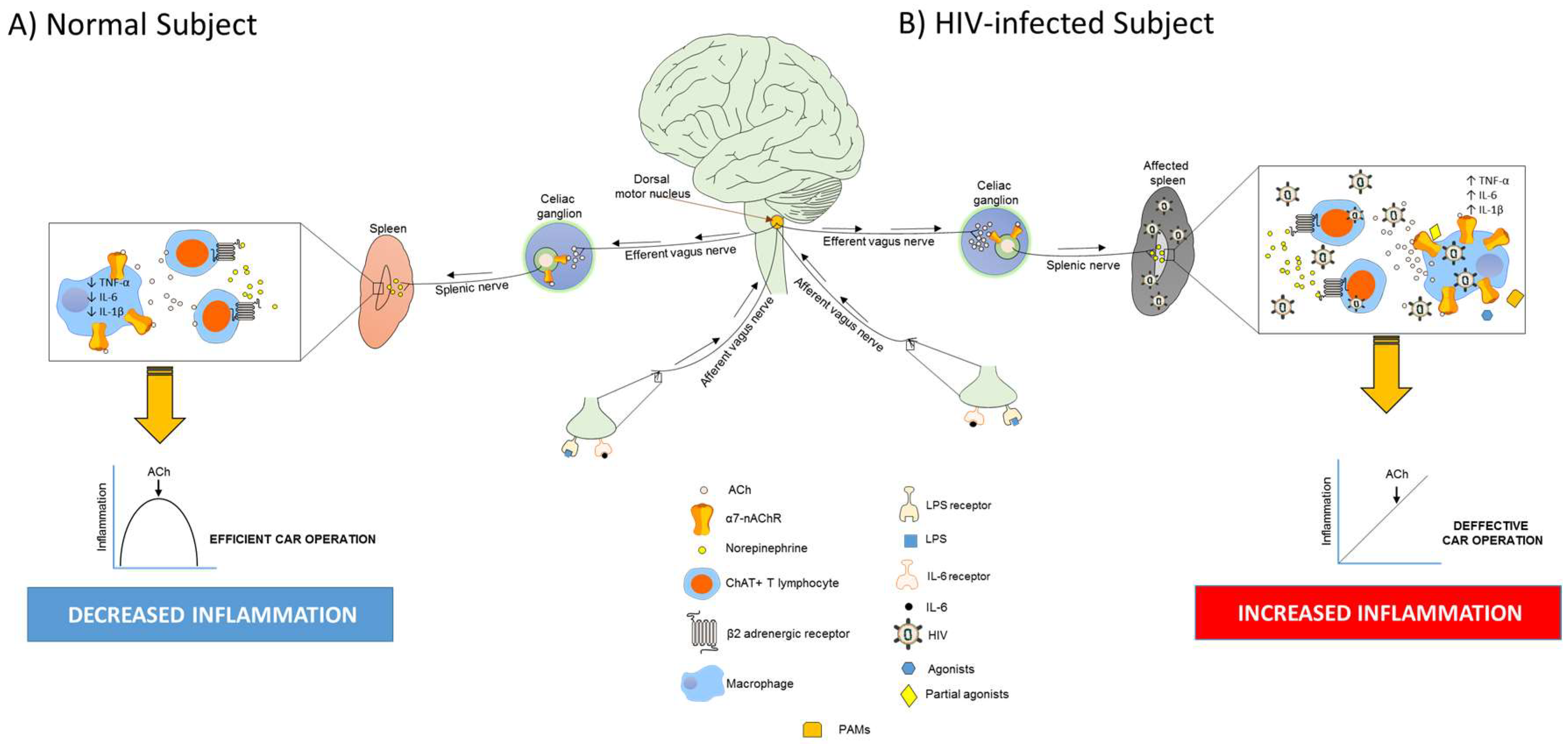
5. Macrophages, Inflammation, and the Cholinergic Anti-Inflammatory Response during HIV Infection
6. Targeting Macrophages to Reduce HIV-Induced Inflammation through α7-nAChRs
7. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Mečnikov, I.I. Leçons sur la pathologie comparée de l’inflammation: Faites à l’Institut Pasteur en avril et mai 1891/par Élie Metchnikoff; G. Masson: Paris, France, 1892. [Google Scholar]
- Highleyman, L. Inflammation, immune activation, and HIV. BETA Bull. Exp. Treat. AIDS Publ. San Franc. AIDS Found. 2010, 22, 12–26. [Google Scholar]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic Effects of Inflammation on Health during Chronic HIV Infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- Borovikova, L.V.; Ivanova, S.; Zhang, M.; Yang, H.; Botchkina, G.I.; Watkins, L.R.; Wang, H.; Abumrad, N.; Eaton, J.W.; Tracey, K.J. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 2000, 405, 458–462. [Google Scholar] [CrossRef] [PubMed]
- Famm, K.; Litt, B.; Tracey, K.J.; Boyden, E.S.; Slaoui, M. Drug discovery: A jump-start for electroceuticals. Nature 2013, 496, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Torres-Rosas, R.; Yehia, G.; Peña, G.; Mishra, P.; del Rocio Thompson-Bonilla, M.; Moreno-Eutimio, M.A.; Arriaga-Pizano, L.A.; Isibasi, A.; Ulloa, L. Dopamine mediates vagal modulation of the immune system by electroacupuncture. Nat. Med. 2014, 20, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Ulloa, L.; Quiroz-Gonzalez, S.; Torres-Rosas, R. Nerve Stimulation: Immunomodulation and Control of Inflammation. Trends Mol. Med. 2017, 23, 1103–1120. [Google Scholar] [CrossRef] [PubMed]
- Appay, V.; Sauce, D. Immune activation and inflammation in HIV-1 infection: Causes and consequences. J. Pathol. 2008, 214, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Hunt, P.W. HIV and inflammation: Mechanisms and consequences. Curr. HIV/AIDS Rep. 2012, 9, 139–147. [Google Scholar] [CrossRef] [PubMed]
- Hileman, C.O.; Funderburg, N.T. Inflammation, Immune Activation, and Antiretroviral Therapy in HIV. Curr. HIV/AIDS Rep. 2017, 14, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Hove-Skovsgaard, M.; Gaardbo, J.C.; Kolte, L.; Winding, K.; Seljeflot, I.; Svardal, A.; Berge, R.K.; Gerstoft, J.; Ullum, H.; Trøseid, M.; Nielsen, S.D. HIV-infected persons with type 2 diabetes show evidence of endothelial dysfunction and increased inflammation. BMC Infect. Dis. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Van Furth, R.; Cohn, Z.A.; Hirsch, J.G.; Humphrey, J.H.; Spector, W.G.; Langevoort, H.L. Mononuclear phagocytic system: New classification of macrophages, monocytes and of their cell line. Bull. World Health Organ. 1972, 47, 651–658. [Google Scholar] [PubMed]
- Yona, S.; Gordon, S. From the Reticuloendothelial to Mononuclear Phagocyte System—The Unaccounted Years. Front. Immunol. 2015, 6, 328. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L.; Ancuta, P.; Crowe, S.; Dalod, M.; Grau, V.; Hart, D.N.; Leenen, P.J.M.; Liu, Y.-J.; MacPherson, G.; Randolph, G.J.; et al. Nomenclature of monocytes and dendritic cells in blood. Blood 2010, 116, e74–e80. [Google Scholar] [CrossRef] [PubMed]
- Ziegler-Heitbrock, L. The CD14+ CD16+ blood monocytes: Their role in infection and inflammation. J. Leukoc. Biol. 2007, 81, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef] [PubMed]
- Passlick, B.; Flieger, D.; Ziegler-Heitbrock, H.W. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood 1989, 74, 2527–2534. [Google Scholar] [PubMed]
- Wong, K.L.; Tai, J.J.-Y.; Wong, W.-C.; Han, H.; Sem, X.; Yeap, W.-H.; Kourilsky, P.; Wong, S.-C. Gene expression profiling reveals the defining features of the classical, intermediate, and nonclassical human monocyte subsets. Blood 2011, 118, e16–e31. [Google Scholar] [CrossRef] [PubMed]
- Monie, T.P. Section 1—A Snapshot of the Innate Immune System. In The Innate Immune System; Academic Press: Cambridge, MA, USA, 2017; pp. 1–40. ISBN 978-0-12-804464-3. [Google Scholar]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [PubMed]
- Schulz, C.; Gomez Perdiguero, E.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.-W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Günther, P.; Crozet, L.; Jacome-Galarza, C.E.; Händler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Phagocytosis: The Legacy of Metchnikoff. Cell 2016, 166, 1065–1068. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14. [Google Scholar] [CrossRef] [PubMed]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17. [Google Scholar] [CrossRef] [PubMed]
- Sieweke, M.H.; Allen, J.E. Beyond stem cells: Self-renewal of differentiated macrophages. Science 2013, 342, 1242974. [Google Scholar] [CrossRef] [PubMed]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages and dendritic cells. Science 2010, 327, 656–661. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Chawla, A.; Pollard, J.W. Origins and Hallmarks of Macrophages: Development, Homeostasis, and Disease. Nature 2013, 496, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Italiani, P.; Boraschi, D. From Monocytes to M1/M2 Macrophages: Phenotypical vs. Functional Differentiation. Front. Immunol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Available online: https://www.hindawi.com/journals/mi/2015/816460/ (accessed on 6 April 2018).
- Wang, Q.; Ni, H.; Lan, L.; Wei, X.; Xiang, R.; Wang, Y. Fra-1 protooncogene regulates IL-6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010, 20, 701–712. [Google Scholar] [CrossRef] [PubMed]
- Ferrante, C.J.; Pinhal-Enfield, G.; Elson, G.; Cronstein, B.N.; Hasko, G.; Outram, S.; Leibovich, S.J. The adenosine-dependent angiogenic switch of macrophages to an M2-like phenotype is independent of interleukin-4 receptor alpha (IL-4Rα) signaling. Inflammation 2013, 36, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Charles A Janeway, J.; Travers, P.; Walport, M.; Shlomchik, M.J. Receptors of the innate immune system. In Immunobiology: The Immune System in Health and Disease, 5th ed.; NCBI: Bethesda, MD, USA, 2001. [Google Scholar]
- Lim, J.J.; Grinstein, S.; Roth, Z. Diversity and Versatility of Phagocytosis: Roles in Innate Immunity, Tissue Remodeling, and Homeostasis. Front. Cell. Infect. Microbiol. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Fact Sheet—Latest Statistics on the Status of the AIDS Epidemic. Available online: http://www.unaids.org/en/resources/fact-sheet (accessed on 27 November 2017).
- UNAIDS DATA 2017. Available online: http://www.unaids.org/en/resources/documents/2017/2017_data_book (accessed on 22 November 2017).
- Alkhatib, G.; Combadiere, C.; Broder, C.C.; Feng, Y.; Kennedy, P.E.; Murphy, P.M.; Berger, E.A. CC CKR5: A RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science 1996, 272, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Choe, H.; Farzan, M.; Sun, Y.; Sullivan, N.; Rollins, B.; Ponath, P.D.; Wu, L.; Mackay, C.R.; LaRosa, G.; Newman, W.; et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell 1996, 85, 1135–1148. [Google Scholar] [CrossRef]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Liu, R.; Ellmeier, W.; Choe, S.; Unutmaz, D.; Burkhart, M.; Di Marzio, P.; Marmon, S.; Sutton, R.E.; Hill, C.M.; et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature 1996, 381, 661–666. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Broder, C.C.; Kennedy, P.E.; Berger, E.A. HIV-1 entry cofactor: Functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science 1996, 272, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Schmidtmayerova, H.; Sherry, B.; Bukrinsky, M. Chemokines and HIV replication. Nature 1996, 382, 767. [Google Scholar] [CrossRef] [PubMed]
- Langford, S.E.; Ananworanich, J.; Cooper, D.A. Predictors of disease progression in HIV infection: A review. AIDS Res. Ther. 2007, 4, 11. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A.; Chattopadhyay, P.K.; Brodie, T.M.; Qin, J.; Gu, W.; Mascola, J.R.; Michael, N.L.; Follmann, D.A.; Roederer, M. Immunological and Virological Events in Early HIV Infection Predict Subsequent Rate of Progression. J. Infect. Dis. 2010, 201, 272–284. [Google Scholar] [CrossRef] [PubMed]
- Lackner, A.A.; Mohan, M.; Veazey, R.S. The Gastrointestinal Tract and AIDS Pathogenesis. Gastroenterology 2009, 136, 1965–1978. [Google Scholar] [CrossRef] [PubMed]
- Brenchley, J.M.; Douek, D.C. HIV infection and the gastrointestinal immune system. Mucosal Immunol. 2008, 1, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Kitchen, C.M.R.; Liu, L.; Guo, H.; Gascon, R.; Narváez, A.B.; Hunt, P.; Martin, J.N.; Kahn, J.O.; Levy, J.; et al. Immune activation set point during early HIV infection predicts subsequent CD4+ T-cell changes independent of viral load. Blood 2004, 104, 942–947. [Google Scholar] [CrossRef] [PubMed]
- Hazenberg, M.D.; Otto, S.A.; van Benthem, B.H.B.; Roos, M.T.L.; Coutinho, R.A.; Lange, J.M.A.; Hamann, D.; Prins, M.; Miedema, F. Persistent immune activation in HIV-1 infection is associated with progression to AIDS. AIDS Lond. Engl. 2003, 17, 1881–1888. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar] [CrossRef] [PubMed]
- Dillon, S.M.; Frank, D.N.; Wilson, C.C. The gut microbiome and HIV-1 pathogenesis: A two-way street. AIDS Lond. Engl. 2016, 30, 2737–2751. [Google Scholar] [CrossRef] [PubMed]
- Dubourg, G.; Surenaud, M.; Lévy, Y.; Hüe, S.; Raoult, D. Microbiome of HIV-infected people. Microb. Pathog. 2017, 106, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Zilberman-Schapira, G.; Zmora, N.; Itav, S.; Bashiardes, S.; Elinav, H.; Elinav, E. The gut microbiome in human immunodeficiency virus infection. BMC Med. 2016, 14. [Google Scholar] [CrossRef] [PubMed]
- Douek, D. HIV disease progression: Immune activation, microbes, and a leaky gut. Top. HIV Med. Publ. Int. AIDS Soc. USA 2007, 15, 114–117. [Google Scholar]
- Wallet, M.A.; Rodriguez, C.A.; Yin, L.; Saporta, S.; Chinratanapisit, S.; Hou, W.; Sleasman, J.W.; Goodenow, M.M. Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. AIDS Lond. Engl. 2010, 24, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Kelesidis, T.; Kendall, M.A.; Yang, O.O.; Hodis, H.N.; Currier, J.S. Biomarkers of Microbial Translocation and Macrophage Activation: Association With Progression of Subclinical Atherosclerosis in HIV-1 Infection. J. Infect. Dis. 2012, 206, 1558–1567. [Google Scholar] [CrossRef] [PubMed]
- Stacey, A.R.; Norris, P.J.; Qin, L.; Haygreen, E.A.; Taylor, E.; Heitman, J.; Lebedeva, M.; DeCamp, A.; Li, D.; Grove, D.; et al. Induction of a striking systemic cytokine cascade prior to peak viremia in acute human immunodeficiency virus type 1 infection, in contrast to more modest and delayed responses in acute hepatitis B and C virus infections. J. Virol. 2009, 83, 3719–3733. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.; Passmore, J.-A.S.; Williamson, C.; Little, F.; Bebell, L.M.; Mlisana, K.; Burgers, W.A.; van Loggerenberg, F.; Walzl, G.; Djoba Siawaya, J.F.; et al. Plasma cytokine levels during acute HIV-1 infection predict HIV disease progression. AIDS Lond. Engl. 2010, 24, 819–831. [Google Scholar] [CrossRef] [PubMed]
- Mugwe, J.N.; Gicheru, M.M.; Mwatha, J. Plasma Cytokine Profiles as Predictive Biomarkers of HIV and Aids Progression among HIV Patients Attending Nakuru Provincial General Hospital, Kenya. Am. J. Med. Biol. Res. Am. J. Med. Biol. Res. 2016, 4, 20–25. [Google Scholar] [CrossRef]
- Shebl, F.M.; Yu, K.; Landgren, O.; Goedert, J.J.; Rabkin, C.S. Increased Levels of Circulating Cytokines with HIV-Related Immunosuppression. AIDS Res. Hum. Retroviruses 2012, 28, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Tien, P.C.; Choi, A.I.; Zolopa, A.R.; Benson, C.; Tracy, R.; Scherzer, R.; Bacchetti, P.; Shlipak, M.; Grunfeld, C. Inflammation and mortality in HIV-infected adults: Analysis of the FRAM study cohort. J. Acquir. Immune Defic. Syndr. 1999 2010, 55, 316–322. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.V.; Neuhaus, J.; Duprez, D.; Kuller, L.H.; Tracy, R.; Belloso, W.H.; De Wit, S.; Drummond, F.; Lane, H.C.; Ledergerber, B.; et al. Changes in inflammatory and coagulation biomarkers: A randomized comparison of immediate versus deferred antiretroviral therapy in patients with HIV infection. J. Acquir. Immune Defic. Syndr. 1999 2011, 56, 36–43. [Google Scholar] [CrossRef] [PubMed]
- McDonald, B.; Moyo, S.; Gabaitiri, L.; Gaseitsiwe, S.; Bussmann, H.; Koethe, J.R.; Musonda, R.; Makhema, J.; Novitsky, V.; Marlink, R.G.; et al. Persistently elevated serum interleukin-6 predicts mortality among adults receiving combination antiretroviral therapy in Botswana: Results from a clinical trial. AIDS Res. Hum. Retroviruses 2013, 29, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Caruana, G.; Vidili, G.; Serra, P.A.; Bagella, P.; Spanu, A.; Fiore, V.; Calvisi, D.F.; Manetti, R.; Rocchitta, G.; Nuvoli, S.; et al. The burden of HIV-associated neurocognitive disorder (HAND) in post-HAART era: A multidisciplinary review of the literature. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2290–2301. [Google Scholar] [PubMed]
- Phair, J.; Palella, F. Renal disease in HIV infected Individuals. Curr. Opin. HIV AIDS 2011, 6, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Compston, J. HIV infection and bone disease. J. Intern. Med. 2016, 280, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Farahani, M.; Mulinder, H.; Farahani, A.; Marlink, R. Prevalence and distribution of non-AIDS causes of death among HIV-infected individuals receiving antiretroviral therapy: A systematic review and meta-analysis. Int. J. STD AIDS 2017, 28, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.; Gange, S.J.; Moore, R.D.; Justice, A.C.; Buchacz, K.; Abraham, A.G.; Rebeiro, P.F.; Koethe, J.R.; Martin, J.N.; Horberg, M.A.; et al. Multimorbidity among Persons Living with HIV in the U.S. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2017. [Google Scholar] [CrossRef]
- Ruan, A.; Tobin, N.H.; Mulligan, K.; Rollie, A.; Li, F.; Sleasman, J.; Aldrovandi, G.M. Brief Report: Macrophage Activation in HIV-Infected Adolescent Males Contributes to Differential Bone Loss by Sex Adolescent Trials Network Study 021. JAIDS J. Acquir. Immune Defic. Syndr. 2016, 72, 372. [Google Scholar] [CrossRef] [PubMed]
- Kearns, A.; Gordon, J.; Burdo, T.H.; Qin, X. HIV-1-Associated Atherosclerosis: Unraveling the Missing Link. J. Am. Coll. Cardiol. 2017, 69, 3084–3098. [Google Scholar] [CrossRef] [PubMed]
- Hanna, D.B.; Lin, J.; Post, W.S.; Hodis, H.N.; Xue, X.; Anastos, K.; Cohen, M.H.; Gange, S.J.; Haberlen, S.A.; Heath, S.L.; et al. Association of Macrophage Inflammation Biomarkers With Progression of Subclinical Carotid Artery Atherosclerosis in HIV-Infected Women and Men. J. Infect. Dis. 2017, 215, 1352–1361. [Google Scholar] [CrossRef] [PubMed]
- Shaked, I.; Hanna, D.B.; Gleißner, C.; Marsh, B.; Plants, J.; Tracy, D.; Anastos, K.; Cohen, M.; Golub, E.T.; Karim, R.; et al. Macrophage inflammatory markers are associated with subclinical carotid artery disease in women with human immunodeficiency virus or hepatitis C virus infection. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1085–1092. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Badiee, J.; Vaida, F.; Letendre, S.; Heaton, R.K.; Clifford, D.; Collier, A.C.; Gelman, B.; McArthur, J.; Morgello, S.; et al. CD4 nadir is a predictor of HIV neurocognitive impairment in the era of combination antiretroviral therapy. AIDS Lond. Engl. 2011, 25, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Fischer-Smith, T.; Croul, S.; Sverstiuk, A.E.; Capini, C.; L’Heureux, D.; Régulier, E.G.; Richardson, M.W.; Amini, S.; Morgello, S.; Khalili, K.; Rappaport, J. CNS invasion by CD14+/CD16+ peripheral blood-derived monocytes in HIV dementia: Perivascular accumulation and reservoir of HIV infection. J. Neurovirol. 2001, 7, 528–541. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.L.; Uno, H.; Ancuta, P.; Kamat, A.; Moore, D.J.; Singer, E.J.; Morgello, S.; Gabuzda, D. Plasma sCD14 is a biomarker associated with impaired neurocognitive test performance in attention and learning domains in HIV infection. J. Acquir. Immune Defic. Syndr. 1999 2011, 57, 371–379. [Google Scholar] [CrossRef] [PubMed]
- McCombe, J.A.; Vivithanaporn, P.; Gill, M.J.; Power, C. Predictors of symptomatic HIV-associated neurocognitive disorders in universal health care. HIV Med. 2013, 14, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, J.; Volsky, D.J. Role of the Macrophage in HIV-Associated Neurocognitive Disorders and Other Comorbidities in Patients on Effective Antiretroviral Treatment. J. Neurovirol. 2015, 21, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Hassan, J.; Browne, K.; De Gascun, C. HIV-1 in Monocytes and Macrophages: An Overlooked Reservoir? Viral Immunol. 2016, 29, 532–533. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, S.K.; Lennox, J.; Caliendo, A.M.; Brown, L.A.; Guidot, D.M. Healthy HIV-1-infected individuals on highly active antiretroviral therapy harbor HIV-1 in their alveolar macrophages. AIDS Res. Hum. Retroviruses 2015, 31, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Ray, P.E.; Liu, X.H.; Henry, D.; Dye, L.; Xu, L.; Orenstein, J.M.; Schuztbank, T.E. Infection of human primary renal epithelial cells with HIV-1 from children with HIV-associated nephropathy. Kidney Int. 1998, 53, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- O’Donnell, M.P.; Chao, C.C.; Gekker, G.; Modi, K.S.; Kasiske, B.L.; Keane, W.F. Renal cell cytokine production stimulates HIV-1 expression in chronically HIV-1-infected monocytes. Kidney Int. 1998, 53, 593–597. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.; Ma, Y.; Scherzer, R.; Price, J.C.; French, A.L.; Plankey, M.W.; Grunfeld, C.; Tien, P.C. Higher CD163 levels are associated with insulin resistance in hepatitis C virus-infected and HIV-infected adults. AIDS Lond. Engl. 2017, 31, 385–393. [Google Scholar] [CrossRef]
- Macrophages linked to lymphoma. AIDS Patient Care STDs 2010, 24, 395. [CrossRef]
- Huysentruyt, L.C.; McGrath, M.S. The role of macrophages in the development and progression of AIDS-related non-Hodgkin lymphoma. J. Leukoc. Biol. 2010, 87, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Hosoi, T.; Okuma, Y.; Matsuda, T.; Nomura, Y. Novel pathway for LPS-induced afferent vagus nerve activation: Possible role of nodose ganglion. Auton. Neurosci. Basic Clin. 2005, 120, 104–107. [Google Scholar] [CrossRef] [PubMed]
- Ek, M.; Kurosawa, M.; Lundeberg, T.; Ericsson, A. Activation of vagal afferents after intravenous injection of interleukin-1beta: Role of endogenous prostaglandins. J. Neurosci. 1998, 18, 9471–9479. [Google Scholar] [CrossRef] [PubMed]
- Van der Kleij, H.; Charles, N.; Karimi, K.; Mao, Y.-K.; Foster, J.; Janssen, L.; Yang, P.C.; Kunze, W.; Rivera, J.; Bienenstock, J. Evidence for neuronal expression of functional Fc (ε and γ) receptors. J. Allergy Clin. Immunol. 2010, 125, 757–760. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; O’Donnell, T.A.; Blackshaw, L.A. P2X purinoceptor-induced sensitization of ferret vagal mechanoreceptors in oesophageal inflammation. J. Physiol. 2000, 523, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Niijima, A. The afferent discharges from sensors for interleukin 1 beta in the hepatoportal system in the anesthetized rat. J. Auton. Nerv. Syst. 1996, 61, 287–291. [Google Scholar] [CrossRef]
- Inoue, T.; Tanaka, S.; Okusa, M.D. Neuroimmune Interactions in Inflammation and Acute Kidney Injury. Front. Immunol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Lang, P.M.; Tracey, D.J.; Irnich, D.; Sippel, W.; Grafe, P. Activation of adenosine and P2Y receptors by ATP in human peripheral nerve. Naunyn Schmiedebergs Arch. Pharmacol. 2002, 366, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Ochani, M.; Gallowitsch-Puerta, M.; Ochani, K.; Huston, J.M.; Czura, C.J.; Al-Abed, Y.; Tracey, K.J. Central muscarinic cholinergic regulation of the systemic inflammatory response during endotoxemia. Proc. Natl. Acad. Sci. USA 2006, 103, 5219–5223. [Google Scholar] [CrossRef] [PubMed]
- Pavlov, V.A.; Parrish, W.R.; Rosas-Ballina, M.; Ochani, M.; Puerta, M.; Ochani, K.; Chavan, S.; Al-Abed, Y.; Tracey, K.J. Brain acetylcholinesterase activity controls systemic cytokine levels through the cholinergic anti-inflammatory pathway. Brain Behav. Immun. 2009, 23, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Langley, R.J.; Kalra, R.; Mishra, N.C.; Sopori, M.L. Central but not the peripheral action of cholinergic compounds suppresses the immune system. J. Neuroimmunol. 2004, 148, 140–145. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Ballina, M.; Olofsson, P.S.; Ochani, M.; Valdés-Ferrer, S.I.; Levine, Y.A.; Reardon, C.; Tusche, M.W.; Pavlov, V.A.; Andersson, U.; Chavan, S.; et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science 2011, 334, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Chernyavsky, A.I.; Arredondo, J.; Skok, M.; Grando, S.A. Auto/paracrine control of inflammatory cytokines by acetylcholine in macrophage-like U937 cells through nicotinic receptors. Int. Immunopharmacol. 2010, 10, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Galvis, G.; Lips, K.S.; Kummer, W. Expression of nicotinic acetylcholine receptors on murine alveolar macrophages. J. Mol. Neurosci. 2006, 30, 107–108. [Google Scholar] [CrossRef]
- Kim, S.-S.; Ye, C.; Kumar, P.; Chiu, I.; Subramanya, S.; Wu, H.; Shankar, P.; Manjunath, N. Targeted Delivery of siRNA to Macrophages for Anti-inflammatory Treatment. Mol. Ther. 2010, 18, 993–1001. [Google Scholar] [CrossRef] [PubMed]
- Shytle, R.D.; Mori, T.; Townsend, K.; Vendrame, M.; Sun, N.; Zeng, J.; Ehrhart, J.; Silver, A.A.; Sanberg, P.R.; Tan, J. Cholinergic modulation of microglial activation by alpha 7 nicotinic receptors. J. Neurochem. 2004, 89, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Mikulski, Z.; Hartmann, P.; Jositsch, G.; Zasłona, Z.; Lips, K.S.; Pfeil, U.; Kurzen, H.; Lohmeyer, J.; Clauss, W.G.; Grau, V.; et al. Nicotinic receptors on rat alveolar macrophages dampen ATP-induced increase in cytosolic calcium concentration. Respir. Res. 2010, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Van der Zanden, E.P.; Snoek, S.A.; Heinsbroek, S.E.; Stanisor, O.I.; Verseijden, C.; Boeckxstaens, G.E.; Peppelenbosch, M.P.; Greaves, D.R.; Gordon, S.; De Jonge, W.J. Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor α4β2. Gastroenterology 2009, 137, 1029–1039. [Google Scholar] [CrossRef] [PubMed]
- Nemethova, A.; Michel, K.; Gomez-Pinilla, P.J.; Boeckxstaens, G.E.; Schemann, M. Nicotine Attenuates Activation of Tissue Resident Macrophages in the Mouse Stomach through the β2 Nicotinic Acetylcholine Receptor. PLoS ONE 2013, 8, e79264. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Vélez, M.; Báez-Pagán, C.A.; Gerena, Y.; Quesada, O.; Santiago-Pérez, L.I.; Capó-Vélez, C.M.; Wojna, V.; Meléndez, L.; León-Rivera, R.; Silva, W.; Lasalde-Dominicci, J.A. The α7-nicotinic receptor is upregulated in immune cells from HIV-seropositive women: Consequences to the cholinergic anti-inflammatory response. Clin. Transl. Immunol. 2015, 4, e53. [Google Scholar] [CrossRef] [PubMed]
- Paiardini, M.; Müller-Trutwin, M. HIV-associated chronic immune activation. Immunol. Rev. 2013, 254, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Estes, J.D.; Schlievert, P.M.; Duan, L.; Brosnahan, A.J.; Southern, P.J.; Reilly, C.S.; Peterson, M.L.; Schultz-Darken, N.; Brunner, K.G.; et al. Glycerol monolaurate prevents mucosal SIV transmission. Nature 2009, 458, 1034–1038. [Google Scholar] [CrossRef] [PubMed]
- Manches, O.; Frleta, D.; Bhardwaj, N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 2014, 35, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Funderburg, N.T. Markers of coagulation and inflammation often remain elevated in ART-treated HIV-infected patients. Curr. Opin. HIV AIDS 2014, 9, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Neri, P.; Bracci, L.; Rustici, M.; Santucci, A. Sequence homology between HIV gp120, rabies virus glycoprotein, and snake venom neurotoxins. Is the nicotinic acetylcholine receptor an HIV receptor? Arch. Virol. 1990, 114, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Sönnerborg, A.; Johansson, B. The neurotoxin-like sequence of human immunodeficiency virus gp120: A comparison of sequence data from patients with and without neurological symptoms. Virus Genes 1993, 7, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Bracci, L.; Lozzi, L.; Rustici, M.; Neri, P. Binding of HIV-1 gp120 to the nicotinic receptor. FEBS Lett. 1992, 311, 115–118. [Google Scholar] [CrossRef]
- Bracci, L.; Ballas, S.K.; Spreafico, A.; Neri, P. Molecular mimicry between the rabies virus glycoprotein and human immunodeficiency virus-1 GP120: Cross-reacting antibodies induced by rabies vaccination. Blood 1997, 90, 3623–3628. [Google Scholar] [PubMed]
- Yoshikawa, H.; Kurokawa, M.; Ozaki, N.; Nara, K.; Atou, K.; Takada, E.; Kamochi, H.; Suzuki, N. Nicotine inhibits the production of proinflammatory mediators in human monocytes by suppression of I-kappaB phosphorylation and nuclear factor-kappaB transcriptional activity through nicotinic acetylcholine receptor alpha7. Clin. Exp. Immunol. 2006, 146, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Ciardo, A.; Meldolesi, J. Effects of the HIV-1 envelope glycoprotein gp120 in cerebellar cultures. [Ca2+]i increases in a glial cell subpopulation. Eur. J. Neurosci. 1993, 5, 1711–1718. [Google Scholar] [CrossRef] [PubMed]
- Hesselgesser, J.; Taub, D.; Baskar, P.; Greenberg, M.; Hoxie, J.; Kolson, D.L.; Horuk, R. Neuronal apoptosis induced by HIV-1 gp120 and the chemokine SDF-1α is mediated by the chemokine receptor CXCR4. Curr. Biol. CB 1998, 8, 595–598. [Google Scholar] [CrossRef]
- Feligioni, M.; Raiteri, L.; Pattarini, R.; Grilli, M.; Bruzzone, S.; Cavazzani, P.; Raiteri, M.; Pittaluga, A. The human immunodeficiency virus-1 protein Tat and its discrete fragments evoke selective release of acetylcholine from human and rat cerebrocortical terminals through species-specific mechanisms. J. Neurosci. Off. J. Soc. Neurosci. 2003, 23, 6810–6818. [Google Scholar] [CrossRef]
- Ballester, L.Y.; Capó-Vélez, C.M.; García-Beltrán, W.F.; Ramos, F.M.; Vázquez-Rosa, E.; Ríos, R.; Mercado, J.R.; Meléndez, R.I.; Lasalde-Dominicci, J.A. Up-regulation of the neuronal nicotinic receptor α7 by HIV glycoprotein 120: Potential implications for HIV-associated neurocognitive disorder. J. Biol. Chem. 2012, 287, 3079–3086. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Wang, S.; Wang, J.; Cui, W.; Nesil, T.; Vigorito, M.; Chang, S.L.; Li, M.D. RNA deep sequencing analysis reveals that nicotine restores impaired gene expression by viral proteins in the brains of HIV-1 transgenic rats. PLoS ONE 2013, 8, e68517. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yu, J.-Y.; Liu, L.-Q.; Peng, L.; Chi, F.; Wu, C.-H.; Jong, A.; Wang, S.-F.; Cao, H.; Huang, S.-H. Alpha7 nicotinic acetylcholine receptor is required for blood-brain barrier injury-related CNS disorders caused by Cryptococcus neoformans and HIV-1 associated comorbidity factors. BMC Infect. Dis. 2015, 15, 352. [Google Scholar] [CrossRef] [PubMed]
- Ramos, F.M.; Delgado-Vélez, M.; Ortiz, Á.L.; Báez-Pagán, C.A.; Quesada, O.; Lasalde-Dominicci, J.A. Expression of CHRFAM7A and CHRNA7 in neuronal cells and postmortem brain of HIV-infected patients: Considerations for HIV-associated neurocognitive disorder. J. Neurovirol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yu, J.; Li, L.; Zhang, B.; Liu, L.; Wu, C.-H.; Jong, A.; Mao, D.-A.; Huang, S.-H. Alpha7 nicotinic acetylcholine receptor is required for amyloid pathology in brain endothelial cells induced by Glycoprotein 120, methamphetamine and nicotine. Sci. Rep. 2017, 7, 40467. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Mathews, P.; Saito, E.K.; Diaz, N.; Naylor, D.; Chung, J.; McMurtray, A.M. α7-Nicotinic acetylcholine receptor inhibition by indinavir: Implications for cognitive dysfunction in treated HIV disease. AIDS Lond. Engl. 2017, 31, 1083–1089. [Google Scholar] [CrossRef] [PubMed]
- Capó-Vélez, C.M.; Morales-Vargas, B.; García-González, A.; Grajales-Reyes, J.G.; Delgado-Vélez, M.; Madera, B.; Báez-Pagán, C.A.; Quesada, O.; Lasalde-Dominicci, J.A. The alpha7-nicotinic receptor contributes to gp120-induced neurotoxicity: Implications in HIV-associated neurocognitive disorders. Sci. Rep. 2018, 8, 1829. [Google Scholar] [CrossRef] [PubMed]
- Gundavarapu, S.; Mishra, N.C.; Singh, S.P.; Langley, R.J.; Saeed, A.I.; Feghali-Bostwick, C.A.; McIntosh, J.M.; Hutt, J.; Hegde, R.; Buch, S.; et al. HIV gp120 induces mucus formation in human bronchial epithelial cells through CXCR4/α7-nicotinic acetylcholine receptors. PLoS ONE 2013, 8, e77160. [Google Scholar] [CrossRef] [PubMed]
- Catani, M.V.; Corasaniti, M.T.; Navarra, M.; Nisticò, G.; Finazzi-Agrò, A.; Melino, G. gp120 induces cell death in human neuroblastoma cells through the CXCR4 and CCR5 chemokine receptors. J. Neurochem. 2000, 74, 2373–2379. [Google Scholar] [CrossRef] [PubMed]
- Giunta, B.; Ehrhart, J.; Townsend, K.; Sun, N.; Vendrame, M.; Shytle, D.; Tan, J.; Fernandez, F. Galantamine and nicotine have a synergistic effect on inhibition of microglial activation induced by HIV-1 gp120. Brain Res. Bull. 2004, 64, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Rock, R.B.; Gekker, G.; Aravalli, R.N.; Hu, S.; Sheng, W.S.; Peterson, P.K. Potentiation of HIV-1 expression in microglial cells by nicotine: Involvement of transforming growth factor-beta 1. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2008, 3, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Abbud, R.A.; Finegan, C.K.; Guay, L.A.; Rich, E.A. Enhanced production of human immunodeficiency virus type 1 by in vitro-infected alveolar macrophages from otherwise healthy cigarette smokers. J. Infect. Dis. 1995, 172, 859–863. [Google Scholar] [CrossRef] [PubMed]
- Loram, L.C.; Harrison, J.A.; Chao, L.; Taylor, F.R.; Reddy, A.; Travis, C.L.; Giffard, R.; Al-Abed, Y.; Tracey, K.; Maier, S.F.; et al. Intrathecal injection of an alpha seven nicotinic acetylcholine receptor agonist attenuates gp120-induced mechanical allodynia and spinal pro-inflammatory cytokine profiles in rats. Brain Behav. Immun. 2010, 24, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Alfano, M.; Crotti, A.; Vicenzi, E.; Poli, G. New players in cytokine control of HIV infection. Curr. HIV/AIDS Rep. 2008, 5, 27–32. [Google Scholar] [CrossRef] [PubMed]
- Zaritsky, L.A.; Dery, A.; Leong, W.Y.; Gama, L.; Clements, J.E. Tissue-specific interferon alpha subtype response to SIV infection in brain, spleen, and lung. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2013, 33, 24–33. [Google Scholar] [CrossRef] [PubMed]
- Ward, J.M.; O’Leary, T.J.; Baskin, G.B.; Benveniste, R.; Harris, C.A.; Nara, P.L.; Rhodes, R.H. Immunohistochemical localization of human and simian immunodeficiency viral antigens in fixed tissue sections. Am. J. Pathol. 1987, 127, 199–205. [Google Scholar] [PubMed]
- Williams, D.W.; Engle, E.L.; Shirk, E.N.; Queen, S.E.; Gama, L.; Mankowski, J.L.; Zink, M.C.; Clements, J.E. Splenic Damage during SIV Infection. Am. J. Pathol. 2016, 186, 2068–2087. [Google Scholar] [CrossRef] [PubMed]
- McIlroy, D.; Autran, B.; Cheynier, R.; Wain-Hobson, S.; Clauvel, J.P.; Oksenhendler, E.; Debré, P.; Hosmalin, A. Infection frequency of dendritic cells and CD4+ T lymphocytes in spleens of human immunodeficiency virus-positive patients. J. Virol. 1995, 69, 4737–4745. [Google Scholar] [PubMed]
- Falk, S.; Müller, H.; Stutte, H.J. The spleen in acquired immunodeficiency syndrome (AIDS). Pathol. Res. Pract. 1988, 183, 425–433. [Google Scholar] [CrossRef]
- Ansovini, R.; Barbolini, G.; Migaldi, M.; Botticelli, L.; De Rienzo, B. AIDS splenomegaly and related iron problems. Pathologica 1998, 90, 133–139. [Google Scholar] [PubMed]
- Falk, S.; Stutte, H.J. The spleen in HIV infection—Morphological evidence of HIV-associated macrophage dysfunction. Res. Virol. 1990, 141, 161–169. [Google Scholar] [CrossRef]
- Kelley, S.P.; Moynihan, J.A.; Stevens, S.Y.; Grota, L.J.; Felten, D.L. Sympathetic nerve destruction in spleen in murine AIDS. Brain Behav. Immun. 2003, 17, 94–109. [Google Scholar] [CrossRef]
- Skok, M.V. Editorial: To channel or not to channel? Functioning of nicotinic acetylcholine receptors in leukocytes. J. Leukoc. Biol. 2009, 86, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Villiger, Y.; Szanto, I.; Jaconi, S.; Blanchet, C.; Buisson, B.; Krause, K.-H.; Bertrand, D.; Romand, J.-A. Expression of an alpha7 duplicate nicotinic acetylcholine receptor-related protein in human leukocytes. J. Neuroimmunol. 2002, 126, 86–98. [Google Scholar] [CrossRef]
- Báez-Pagán, C.A.; Delgado-Vélez, M.; Lasalde-Dominicci, J.A. Activation of the Macrophage α7 Nicotinic Acetylcholine Receptor and Control of Inflammation. J. Neuroimmune Pharmacol. Off. J. Soc. NeuroImmune Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; Brucker, W.J.; Hawrot, E. Proteomic analysis of an alpha7 nicotinic acetylcholine receptor interactome. J. Proteome Res. 2009, 8, 1849–1858. [Google Scholar] [CrossRef] [PubMed]
- Nordman, J.C.; Kabbani, N. An interaction between α7 nicotinic receptors and a G-protein pathway complex regulates neurite growth in neural cells. J. Cell Sci. 2012, 125, 5502–5513. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Nordman, J.C.; Bridges, S.P.; Lin, M.-K.; Kabbani, N. Identification and Characterization of a G Protein-binding Cluster in α7 Nicotinic Acetylcholine Receptors. J. Biol. Chem. 2015, 290, 20060–20070. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Kabbani, N. Alpha 7 nicotinic receptor coupling to heterotrimeric G proteins modulates RhoA activation, cytoskeletal motility, and structural growth. J. Neurochem. 2016, 138, 532–545. [Google Scholar] [CrossRef] [PubMed]
- King, J.R.; Gillevet, T.C.; Kabbani, N. A G protein-coupled α7 nicotinic receptor regulates signaling and TNF-α release in microglia. FEBS Open Bio 2017, 7, 1350–1361. [Google Scholar] [CrossRef] [PubMed]
- Benfante, R.; Antonini, R.A.; De Pizzol, M.; Gotti, C.; Clementi, F.; Locati, M.; Fornasari, D. Expression of the α7 nAChR subunit duplicate form (CHRFAM7A) is down-regulated in the monocytic cell line THP-1 on treatment with LPS. J. Neuroimmunol. 2011, 230, 74–84. [Google Scholar] [CrossRef] [PubMed]
- De Lucas-Cerrillo, A.M.; Maldifassi, M.C.; Arnalich, F.; Renart, J.; Atienza, G.; Serantes, R.; Cruces, J.; Sánchez-Pacheco, A.; Andrés-Mateos, E.; Montiel, C. Function of partially duplicated human α77 nicotinic receptor subunit CHRFAM7A gene: Potential implications for the cholinergic anti-inflammatory response. J. Biol. Chem. 2011, 286, 594–606. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Xiao, T.; Sun, Q.; Wang, K. The current agonists and positive allosteric modulators of α7 nAChR for CNS indications in clinical trials. Acta Pharm. Sin. B 2017, 7, 611–622. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.; Bonneau, R.H.; Chinchilli, V.M.; Tracey, K.J.; Cockroft, K.M. A novel inhibitor of inflammatory cytokine production (CNI-1493) reduces rodent post-hemorrhagic vasospasm. Neurocrit. Care 2006, 5, 222–229. [Google Scholar] [CrossRef]
- van Westerloo, D.J.; Giebelen, I.A.; Florquin, S.; Bruno, M.J.; Larosa, G.J.; Ulloa, L.; Tracey, K.J.; van der Poll, T. The vagus nerve and nicotinic receptors modulate experimental pancreatitis severity in mice. Gastroenterology 2006, 130, 1822–1830. [Google Scholar] [CrossRef] [PubMed]
- Giebelen, I.A.J.; van Westerloo, D.J.; LaRosa, G.J.; de Vos, A.F.; van der Poll, T. Stimulation of alpha 7 cholinergic receptors inhibits lipopolysaccharide-induced neutrophil recruitment by a tumor necrosis factor alpha-independent mechanism. Shock Augusta Ga 2007, 27, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Giebelen, I.A.J.; van Westerloo, D.J.; LaRosa, G.J.; de Vos, A.F.; van der Poll, T. Local stimulation of alpha7 cholinergic receptors inhibits LPS-induced TNF-alpha release in the mouse lung. Shock Augusta Ga 2007, 28, 700–703. [Google Scholar] [CrossRef]
- Rosas-Ballina, M.; Goldstein, R.S.; Gallowitsch-Puerta, M.; Yang, L.; Valdés-Ferrer, S.I.; Patel, N.B.; Chavan, S.; Al-Abed, Y.; Yang, H.; Tracey, K.J. The Selective α7 Agonist GTS-21 Attenuates Cytokine Production in Human Whole Blood and Human Monocytes Activated by Ligands for TLR2, TLR3, TLR4, TLR9, and RAGE. Mol. Med. 2009, 15, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Sitapara, R.A.; Antoine, D.J.; Sharma, L.; Patel, V.S.; Ashby, C.R.; Gorasiya, S.; Yang, H.; Zur, M.; Mantell, L.L. The α7 nicotinic acetylcholine receptor agonist GTS-21 improves bacterial clearance in mice by restoring hyperoxia-compromised macrophage function. Mol. Med. Camb. Mass. 2014, 20, 238–247. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Liu, R.; Cheng, W.; Hu, Y.; Li, J.; Pan, X.; Peng, J.; Zhang, P. GTS-21 attenuates lipopolysaccharide-induced inflammatory cytokine production in vitro by modulating the Akt and NF-κB signaling pathway through the α7 nicotinic acetylcholine receptor. Int. Immunopharmacol. 2015, 29, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Van Maanen, M.A.; Lebre, M.C.; van der Poll, T.; LaRosa, G.J.; Elbaum, D.; Vervoordeldonk, M.J.; Tak, P.P. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009, 60, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Ek, C.J.; Mallard, C.; Johansson, M.E. Perinatal hypoxia-ischemia reduces α 7 nicotinic receptor expression and selective α 7 nicotinic receptor stimulation suppresses inflammation and promotes microglial Mox phenotype. BioMed Res. Int. 2014, 2014, 718769. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, A.; Ichiki, T.; Kojima, H.; Takahara, Y.; Hurt-Camejo, E.; Michaëlsson, E.; Sankoda, C.; Ikeda, J.; Inoue, E.; Tokunou, T.; et al. Suppression of abdominal aortic aneurysm formation by AR-R17779, an agonist for the α7 nicotinic acetylcholine receptor. Atherosclerosis 2016, 244, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Targeting of α7 Nicotinic Acetylcholine Receptors in the Treatment of Schizophrenia and the Use of Auditory Sensory Gating as a Translational Biomarker. Curr. Pharm. Des. 2015, 21, 3797–3806. [Google Scholar] [CrossRef] [PubMed]
- Maehara, T.; Matsumoto, K.; Horiguchi, K.; Kondo, M.; Iino, S.; Horie, S.; Murata, T.; Tsubone, H.; Shimada, S.; Ozaki, H.; et al. Therapeutic action of 5-HT3 receptor antagonists targeting peritoneal macrophages in post-operative ileus. Br. J. Pharmacol. 2015, 172, 1136–1147. [Google Scholar] [CrossRef] [PubMed]
- Tasaka, Y.; Yasunaga, D.; Kiyoi, T.; Tanaka, M.; Tanaka, A.; Suemaru, K.; Araki, H. Involvement of stimulation of α7 nicotinic acetylcholine receptors in the suppressive effect of tropisetron on dextran sulfate sodium-induced colitis in mice. J. Pharmacol. Sci. 2015, 127, 275–283. [Google Scholar] [CrossRef] [PubMed]
- Fiebich, B.L.; Akundi, R.S.; Lieb, K.; Candelario-Jalil, E.; Gmeiner, D.; Haus, U.; Müller, W.; Stratz, T.; Muñoz, E. Antiinflammatory effects of 5-HT3 receptor antagonists in lipopolysaccharide-stimulated primary human monocytes. Scand. J. Rheumatol. 2004, 33, 28–32. [Google Scholar] [CrossRef]
- Munro, G.; Hansen, R.; Erichsen, H.; Timmermann, D.; Christensen, J.; Hansen, H. The α7 nicotinic ACh receptor agonist compound B and positive allosteric modulator PNU-120596 both alleviate inflammatory hyperalgesia and cytokine release in the rat. Br. J. Pharmacol. 2012, 167, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Zhang, Z.-Z.; Zhan, J.; He, X.-H.; Song, X.-M.; Wang, Y.-L. Protective effect of PNU-120596, a selective alpha7 nicotinic acetylcholine receptor-positive allosteric modulator, on myocardial ischemia-reperfusion injury in rats. J. Cardiovasc. Pharmacol. 2012, 59, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.B.; Montero-Melendez, T.; Greco, K.V.; Perretti, M. Melanocortin Receptors as Novel Effectors of Macrophage Responses in Inflammation. Front. Immunol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Thorp, E.B. Contrasting Inflammation Resolution during Atherosclerosis and Post Myocardial Infarction at the Level of Monocyte/Macrophage Phagocytic Clearance. Front. Immunol. 2012, 3. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, K.; Janssen, W.J.; Fessler, M.B.; McPhillips, K.A.; Borges, V.M.; Bowler, R.P.; Xiao, Y.-Q.; Kench, J.A.; Henson, P.M.; Vandivier, R.W. Lovastatin enhances clearance of apoptotic cells (efferocytosis) with implications for chronic obstructive pulmonary disease. J. Immunol. Baltim. Md 1950 2006, 176, 7657–7665. [Google Scholar] [CrossRef]
- Kennedy, A.; Fearon, U.; Veale, D.J.; Godson, C. Macrophages in Synovial Inflammation. Front. Immunol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, W.C.; Kempf, M.-C.; Moneyham, L.; Vance, D.E. The potential role of vagus-nerve stimulation in the treatment of HIV-associated depression: A review of literature. Neuropsychiatr. Dis. Treat. 2017, 13, 1677–1689. [Google Scholar] [CrossRef] [PubMed]
- Skaper, S.D.; Facci, L.; Zusso, M.; Giusti, P. An Inflammation-Centric View of Neurological Disease: Beyond the Neuron. Front. Cell. Neurosci. 2018, 12, 72. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Rivera, Y.; García, Y.; Toro, V.; Cappas, N.; López, P.; Yamamura, Y.; Rivera-Amill, V. Depression Correlates with Increased Plasma Levels of Inflammatory Cytokines and a Dysregulated Oxidant/Antioxidant Balance in HIV-1-Infected Subjects Undergoing Antiretroviral Therapy. J. Clin. Cell. Immunol. 2014, 5. [Google Scholar] [CrossRef]
- Norcini Pala, A.; Steca, P.; Bagrodia, R.; Helpman, L.; Colangeli, V.; Viale, P.; Wainberg, M.L. Subtypes of depressive symptoms and inflammatory biomarkers: An exploratory study on a sample of HIV-positive patients. Brain. Behav. Immun. 2016, 56, 105–113. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Rivera, Y.; Vázquez-Santiago, F.J.; Albino, E.; Sánchez, M.D.C.; Rivera-Amill, V. Impact of Depression and Inflammation on the Progression of HIV Disease. J. Clin. Cell. Immunol. 2016, 7. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Drug | Type of Drug | Effects on Inflammation | Experimental Model/Cell Type/Cell Line | Reference(s) |
|---|---|---|---|---|
| CNI-1493 | Agonist | ↓ IL-6 |
| [153] |
| GTS-21 (DMXB-A) | Agonist | ↓ IL-6 ↓ TNF-α ↓ TNF-α and IL-1β ↓ IL-1β ↓ high mobility group box-1 ↓ IL-1β, IL-6, and TNF-α |
| [132,154,155,156,157,158,159] |
| AR-R17779 | Agonist | ↓ TNF-α ↓ TNF-α ↓ IL-1β, IL-6, and MCP-1 |
| [160,161,162] |
| Tropisetron | Partial Agonist | ↓ TNF-α and IL-1β |
| [163,164,165,166] |
| PNU-120596 | PAM | ↓ TNF-α and IL-6 ↓ TNF-α and IL-6 |
| [167,168] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Vélez, M.; Lasalde-Dominicci, J.A. The Cholinergic Anti-Inflammatory Response and the Role of Macrophages in HIV-Induced Inflammation. Int. J. Mol. Sci. 2018, 19, 1473. https://doi.org/10.3390/ijms19051473
Delgado-Vélez M, Lasalde-Dominicci JA. The Cholinergic Anti-Inflammatory Response and the Role of Macrophages in HIV-Induced Inflammation. International Journal of Molecular Sciences. 2018; 19(5):1473. https://doi.org/10.3390/ijms19051473
Chicago/Turabian StyleDelgado-Vélez, Manuel, and José A. Lasalde-Dominicci. 2018. "The Cholinergic Anti-Inflammatory Response and the Role of Macrophages in HIV-Induced Inflammation" International Journal of Molecular Sciences 19, no. 5: 1473. https://doi.org/10.3390/ijms19051473
APA StyleDelgado-Vélez, M., & Lasalde-Dominicci, J. A. (2018). The Cholinergic Anti-Inflammatory Response and the Role of Macrophages in HIV-Induced Inflammation. International Journal of Molecular Sciences, 19(5), 1473. https://doi.org/10.3390/ijms19051473