A Rationally Designed Hsp70 Variant Rescues the Aggregation-Associated Toxicity of Human IAPP in Cultured Pancreatic Islet β-Cells



Abstract
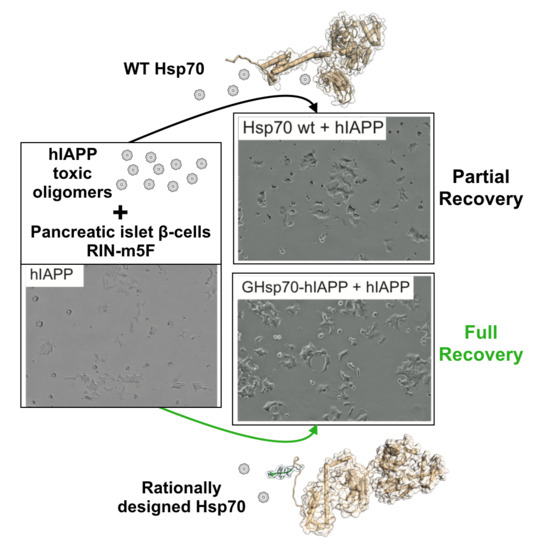
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
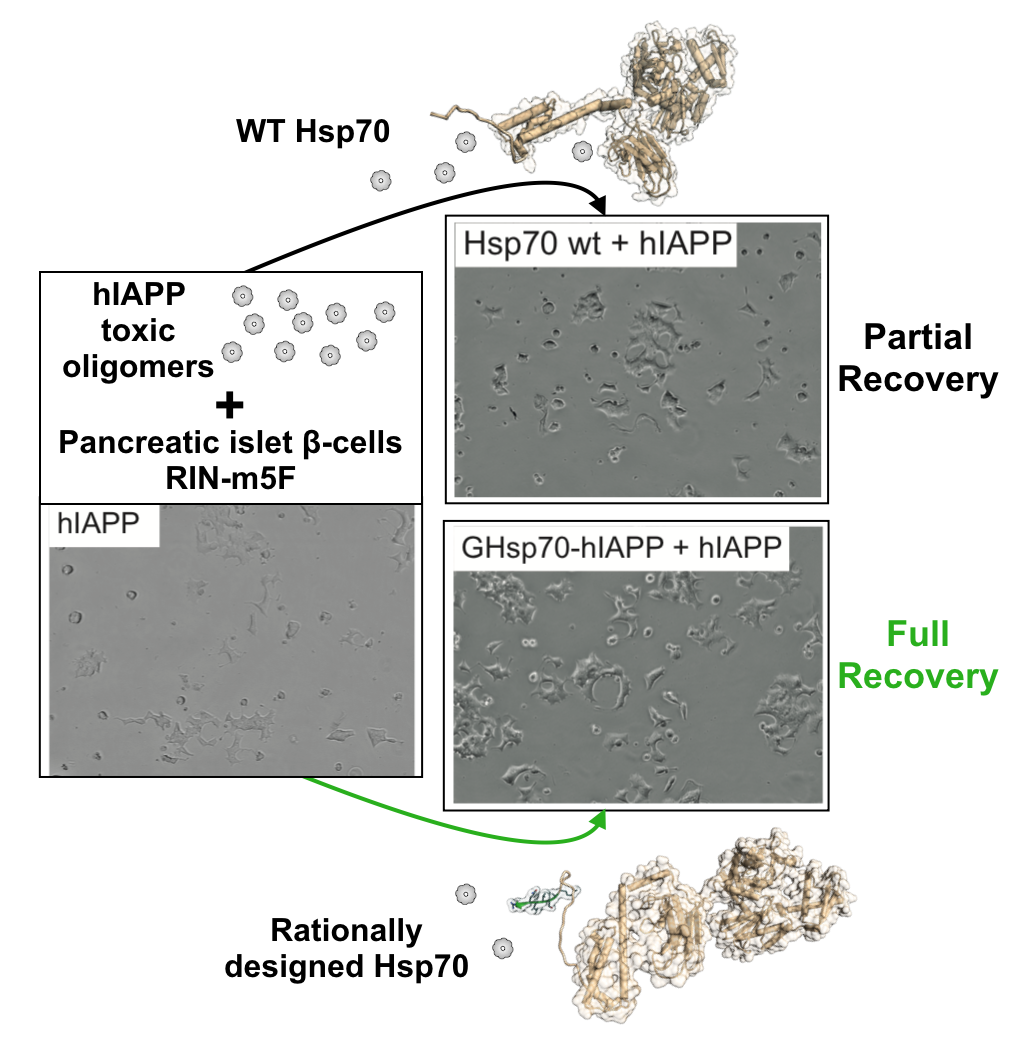
2.1. Rational Design of an Hsp70 Variant against hIAPP
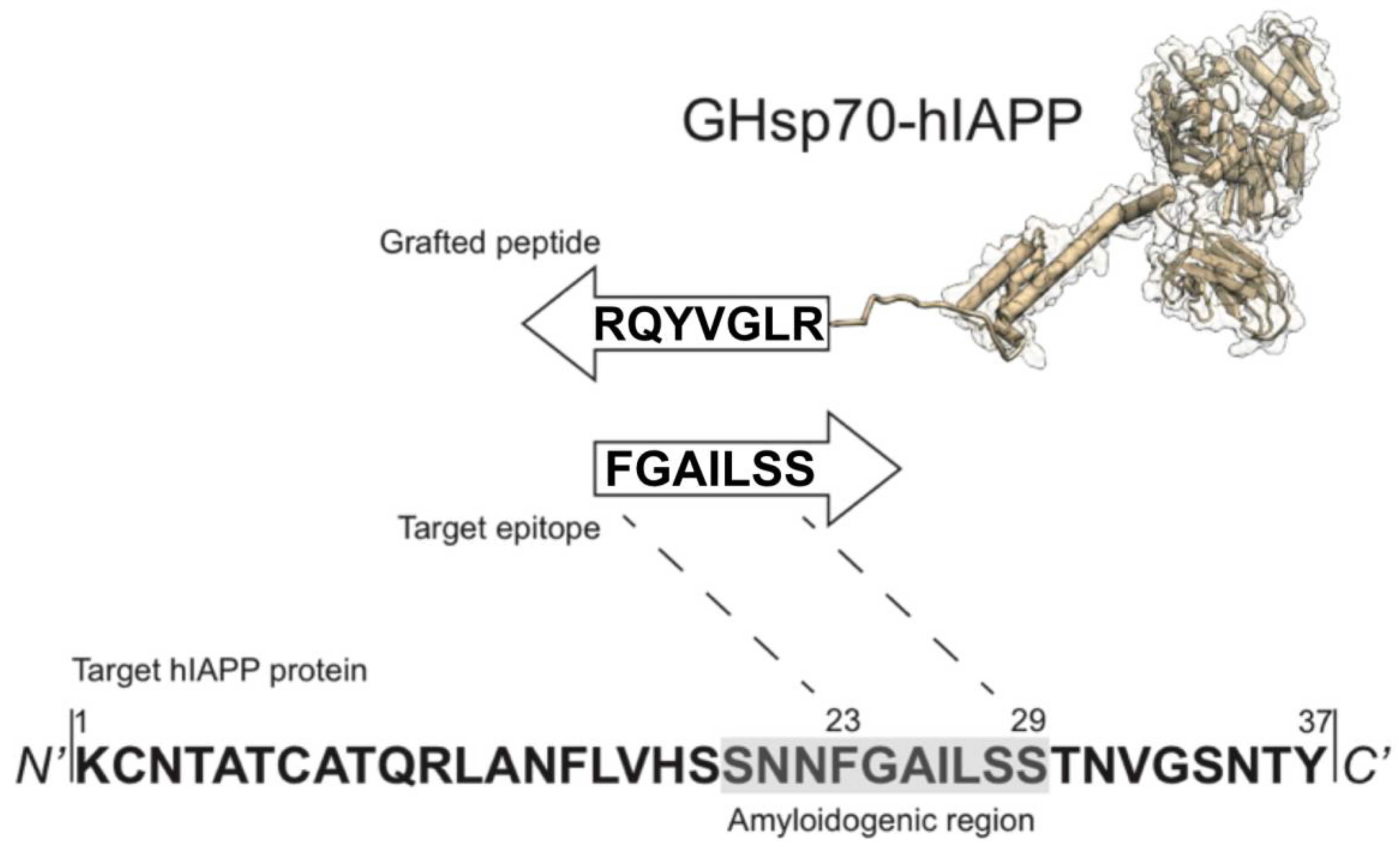
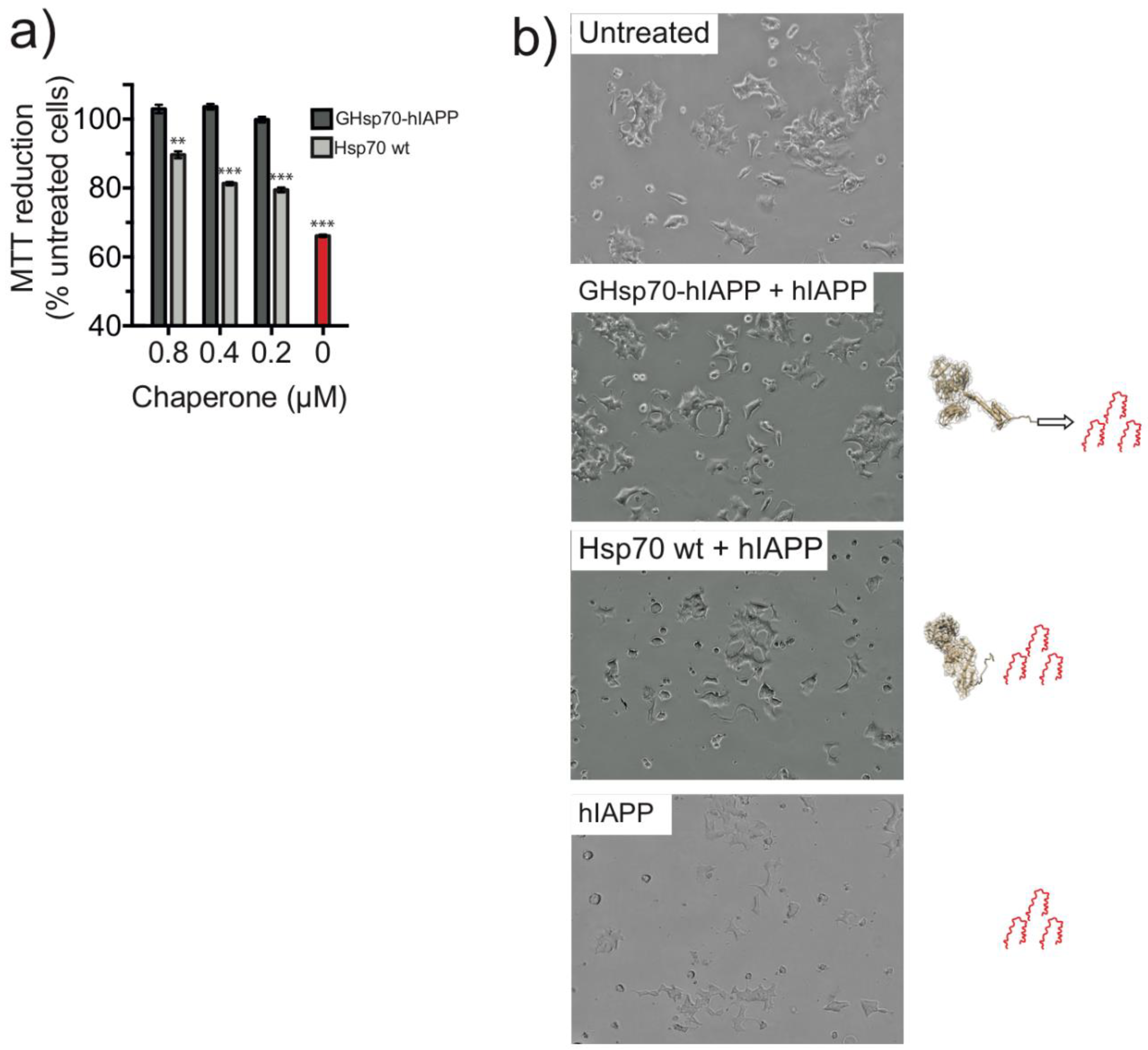
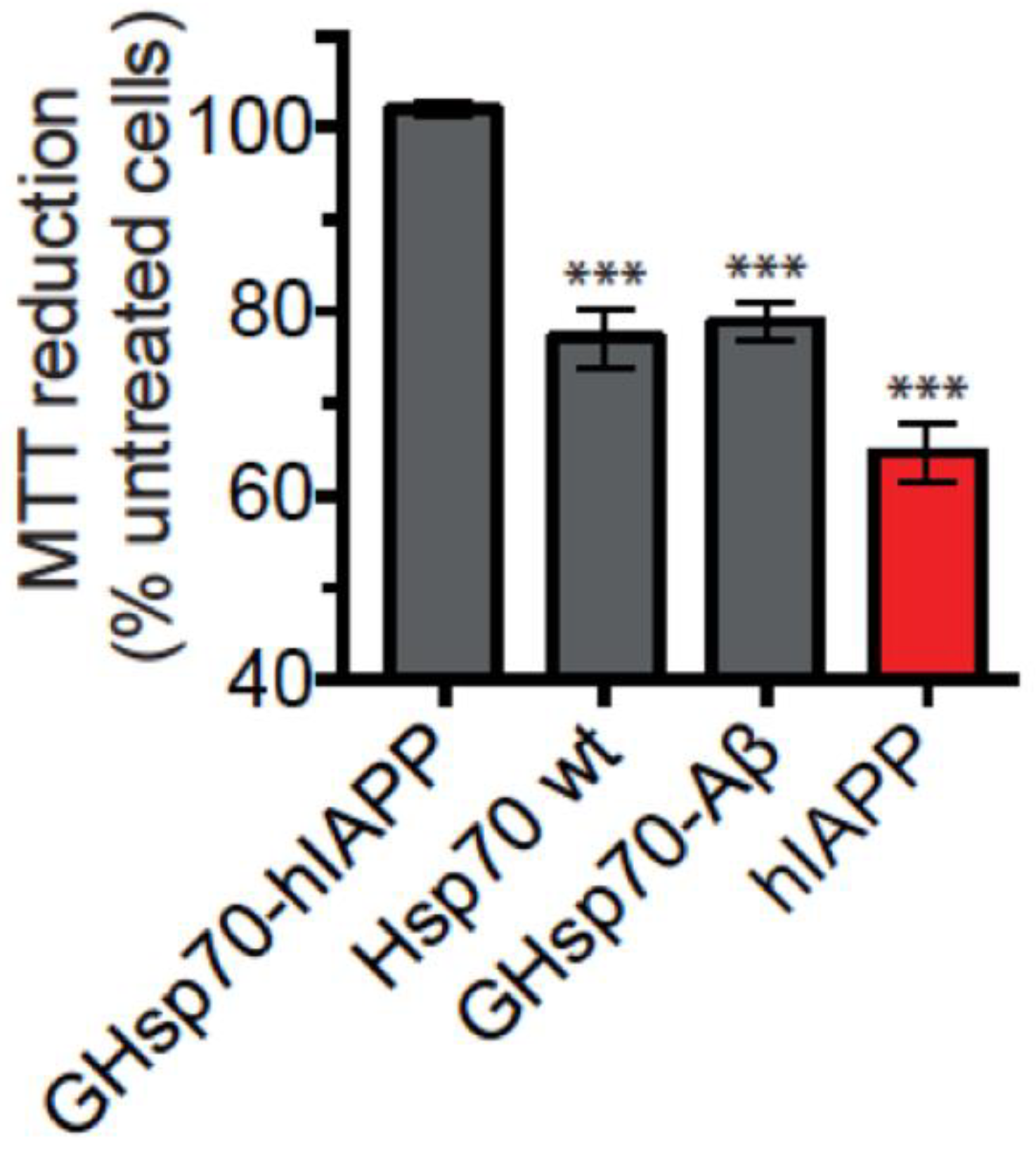
2.2. The Designed GHsp70-hIAPP Variant Increases Pancreatic Islet β-Cell Viability in Cell Culture Experiments
2.3. The Designed GHsp70-Aβ Variant Does Not Increase Pancreatic Islet β-Cell Viability
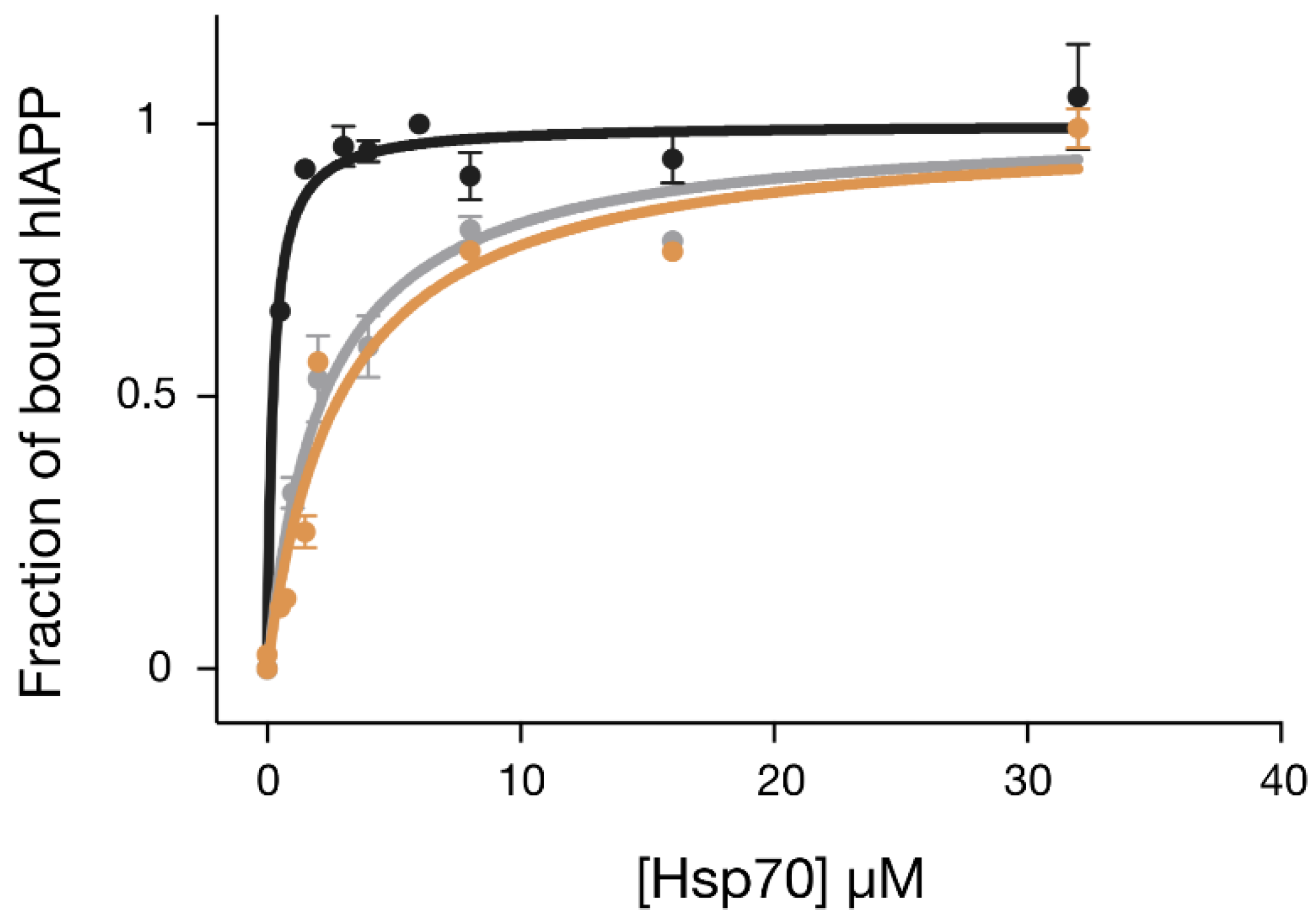
2.4. The Designed GHsp70-hIAPP Variant Binds hIAPP with Higher Affinity than Wild-Type Hsp70
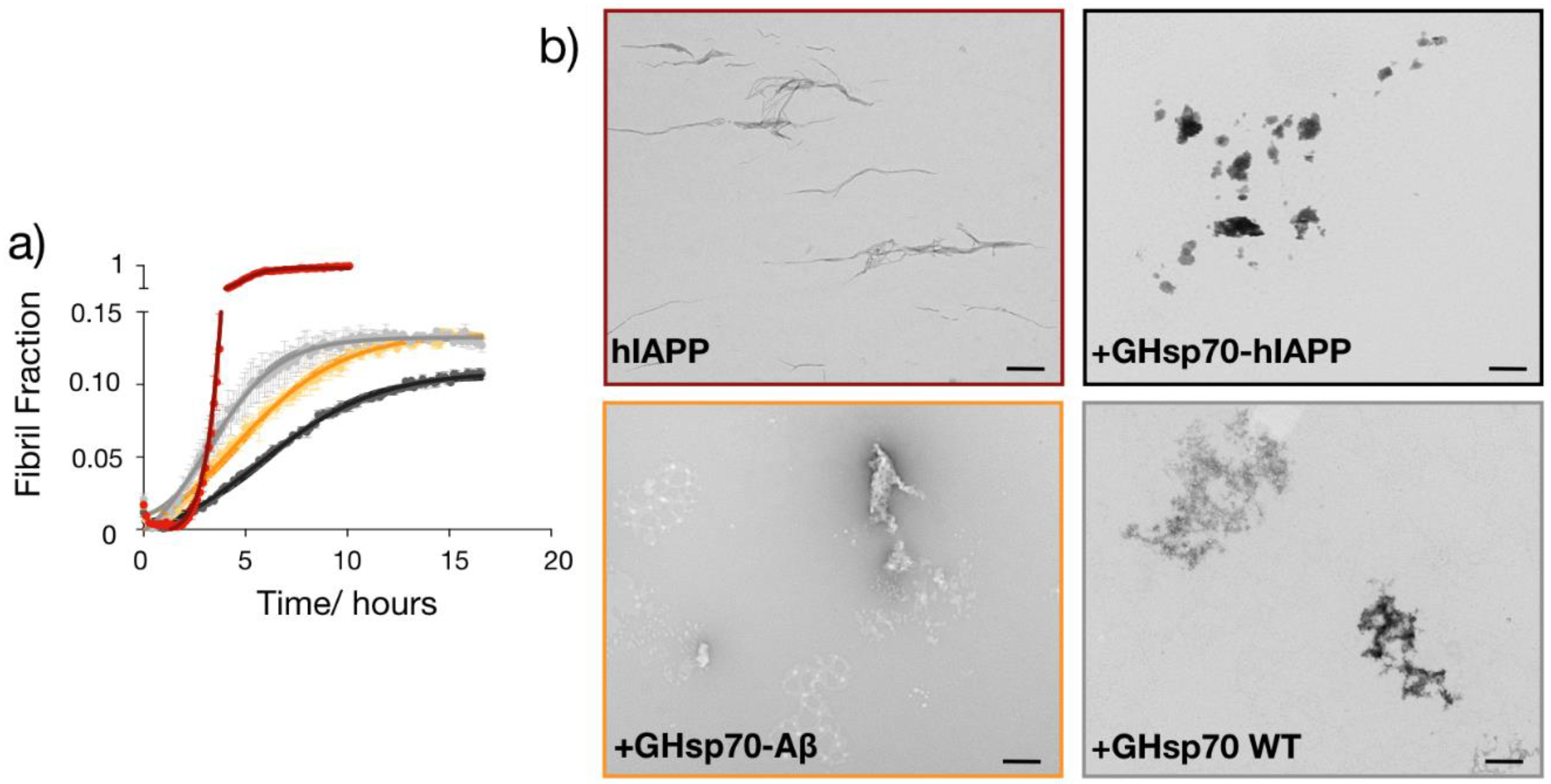
2.5. The Designed GHsp70-hIAPP Variant Inhibits hIAPP Aggregation Better Than Wild-Type Hsp70 In Vitro
3. Discussion
4. Materials and Methods
4.1. Hsp70 Variant Constructs
4.2. Preparation of hIAPP for Cell Viability Tests
4.3. Cell Culture
4.4. Cell Viability Tests
4.5. MTT Reduction Assay
4.6. ELISA Binding Assay
4.7. Fluorescence Titration Assay
4.8. Protein Aggregation Assay
4.9. Transmission Electron Microscopy (TEM)
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| Hsp70 | Heat shock protein 70 kDa |
| GHsp70 | Grafted Heat shock protein 70 kDa |
| hIAPP | Human islet amyloid polypeptide |
| MTT | 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide |
| ThT | Thioflavin-T |
References
- Hartl, F.U. Molecular chaperones in cellular protein folding. Nature 1996, 381, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Bukau, B.; Weissman, J.; Horwich, A. Molecular Chaperones and Protein Quality Control. Cell 2006, 125, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting Proteostasis for Disease Intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, S.L.; Kelly, J.W. Chemical and Biological Approaches for Adapting Proteostasis to Ameliorate Protein Misfolding and Aggregation Diseases–Progress and Prognosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a004507. [Google Scholar] [CrossRef] [PubMed]
- Hartl, F.U.; Bracher, A.; Hayer-Hartl, M. Molecular chaperones in protein folding and proteostasis. Nature 2011, 475, 324–332. [Google Scholar] [CrossRef] [PubMed]
- Brehme, M.; Voisine, C.; Rolland, T.; Wachi, S.; Soper, J.H.; Zhu, Y.; Orton, K.; Villella, A.; Garza, D.; Vidal, M.; et al. A Chaperome Subnetwork Safeguards Proteostasis in Aging and Neurodegenerative Disease. Cell Rep. 2014, 9, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Arosio, P.; Michaels, T.C.; Linse, S.; Månsson, C.; Emanuelsson, C.; Presto, J.; Johansson, J.; Vendruscolo, M.; Dobson, C.M.; Knowles, T.P. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat. Comm. 2016, 7, 10948. [Google Scholar] [CrossRef] [PubMed]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef] [PubMed]
- Gestwicki, J.E.; Crabtree, G.R.; Graef, I.A. Harnessing chaperones to generate small-molecule inhibitors of amyloid β aggregation. Science 2004, 306, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Muchowski, P.J.; Wacker, J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005, 6, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kampinga, H.H.; Bergink, S. Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol. 2016, 15, 748–759. [Google Scholar] [CrossRef]
- Pratt, W.B.; Gestwicki, J.E.; Osawa, Y.; Lieberman, A.P. Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 353–371. [Google Scholar] [CrossRef] [PubMed]
- Shorter, J. Designer protein disaggregases to counter neurodegenerative disease. Curr. Opin. Genet. Dev. 2017, 44, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Protein folding and misfolding. Nature 2003, 426, 884–890. [Google Scholar] [CrossRef] [PubMed]
- Hipp, M.S.; Park, S.-H.; Hartl, F.U. Proteostasis impairment in protein-misfolding and -aggregation diseases. Trends Cell Biol. 2014, 24, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Aguzzi, A.; O’Connor, T. Protein aggregation diseases: Pathogenicity and therapeutic perspectives. Nat. Rev. Drug Discov. 2010, 9, 237–248. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Selkoe, D.J. Soluble protein oligomers in neurodegeneration: Lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007, 8, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Knowles, T.P.J.; Vendruscolo, M.; Dobson, C.M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S101. [Google Scholar] [CrossRef] [PubMed]
- Tyedmers, J.; Mogk, A.; Bukau, B. Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol. 2010, 11, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Caughey, B.; Lansbury, P.T. Protofibrils, pores, fibrils, and neurodegeneration: Separating the responsible protein aggregates from the innocent bystanders. Annu. Rev. Neurosci. 2003, 26, 267–298. [Google Scholar] [CrossRef] [PubMed]
- Eisenberg, D.; Jucker, M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. [Google Scholar] [CrossRef] [PubMed]
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef] [PubMed]
- Dedmon, M.M.; Christodoulou, J.; Wilson, M.R.; Dobson, C.M. Heat Shock Protein 70 Inhibits α-Synuclein Fibril Formation via Preferential Binding to Prefibrillar Species. J. Biol. Chem. 2005, 280, 14733–14740. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.G.; Wisén, S.; Gestwicki, J.E. Heat Shock Proteins 70 and 90 Inhibit Early Stages of Amyloid β-(1–42) Aggregation in Vitro. J. Biol. Chem. 2006, 281, 33182–33191. [Google Scholar] [CrossRef] [PubMed]
- Kumita, J.R.; Poon, S.; Caddy, G.L.; Hagan, C.L.; Dumoulin, M.; Yerbury, J.J.; Stewart, E.M.; Robinson, C.V.; Wilson, M.R.; Dobson, C.M. The Extracellular Chaperone Clusterin Potently Inhibits Human Lysozyme Amyloid Formation by Interacting with Prefibrillar Species. J. Mol. Biol. 2007, 369, 157–167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Roodveldt, C.; Bertoncini, C.W.; Andersson, A.; van der Goot, A.T.; Hsu, S.-T.; Fernández-Montesinos, R.; de Jong, J.; van Ham, T.J.; Nollen, E.A.; Pozo, D.; et al. Chaperone proteostasis in Parkinson’s disease: Stabilization of the Hsp70/α-synuclein complex by Hip. EMBO J. 2009, 28, 3758–3770. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.Z.; Quan, Y.; Feng, Z.-P. Multifaceted Role of Heat Shock Protein 70 in Neurons. Mol. Neurobiol. 2010, 42, 114–123. [Google Scholar] [CrossRef] [PubMed]
- Mayer, M.P.; Bukau, B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell. Mol. Life Sci. 2005, 62, 670–684. [Google Scholar] [CrossRef] [PubMed]
- Auluck, P.K.; Chan, H.Y.E.; Trojanowski, J.Q.; Lee, V.M.Y.; Bonini, N.M. Chaperone Suppression of α-Synuclein Toxicity in a Drosophila Model for Parkinson’s Disease. Science 2002, 295, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Cummings, C.J.; Sun, Y.; Opal, P.; Antalffy, B.; Mestril, R.; Orr, H.T.; Dillmann, W.H.; Zoghbi, H.Y. Over-expression of inducible HSP70 chaperone suppresses neuropathology and improves motor function in SCA1 mice. Hum. Mol. Genet. 2001, 10, 1511–1518. [Google Scholar] [CrossRef] [PubMed]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 Reduces α-Synuclein Aggregation and Toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [PubMed]
- Ran, R.; Lu, A.; Zhang, L.; Tang, Y.; Zhu, H.; Xu, H.; Feng, Y.; Han, C.; Zhou, G.; Rigby, A.C.; et al. Hsp70 promotes TNF-mediated apoptosis by binding IKKγ and impairing NF-κB survival signaling. Genes Dev. 2004, 18, 1466–1481. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, G.; Valax, P. Expression of correctly folded proteins in Escherichia coli. Curr. Opin. Biotech. 1996, 7, 190–197. [Google Scholar] [CrossRef]
- Ebrahimi-Fakhari, D.; Saidi, L.-J.; Wahlster, L. Molecular chaperones and protein folding as therapeutic targets in Parkinson’s disease and other synucleinopathies. Acta Neuropathol. Commun. 2013, 1, 79. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Koren, J.; O’Leary, J.C.; Jones, J.R.; Abisambra, J.F.; Dickey, C.A. Hsp70 ATPase Modulators as Therapeutics for Alzheimer’s and other Neurodegenerative Diseases. Mol. Cell. Pharmacol. 2010, 2, 43–46. [Google Scholar] [PubMed]
- Jones, D.R.; Moussaud, S.; McLean, P. Targeting heat shock proteins to modulate α-synuclein toxicity. Ther. Adv. Neurol. Disord. 2014, 7, 33–51. [Google Scholar] [CrossRef] [PubMed]
- Kalia, S.K.; Kalia, L.V.; McLean, P.J. Molecular chaperones as rational drug targets for Parkinson’s disease therapeutics. CNS Neurol. Disord. Drug Targets 2010, 9, 741–753. [Google Scholar] [CrossRef] [PubMed]
- Montgomery, D.L.; Morimoto, R.I.; Gierasch, L.M. Mutations in the substrate binding domain of the Escherichia coli 70 kda molecular chaperone, DnaK, which alter substrate affinity or interdomain coupling1. J. Mol. Biol. 1999, 286, 915–932. [Google Scholar] [CrossRef] [PubMed]
- Vogel, M.; Bukau, B.; Mayer, M.P. Allosteric Regulation of Hsp70 Chaperones by a Proline Switch. Mol. Cell 2006, 21, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Nylandsted, J.; Brand, K.; Jäättelä, M. Heat Shock Protein 70 Is Required for the Survival of Cancer Cells. Ann. N. Y. Acad. Sci. 2000, 926, 122–125. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.Y.; Gabai, V.L. Hsp70 in cancer: Back to the future. Oncogene 2015, 34, 4153–4161. [Google Scholar] [CrossRef] [PubMed]
- Whitesell, L.; Lindquist, S.L. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 2005, 5, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Vendruscolo, M. A Rational Design Strategy for the Selective Activity Enhancement of a Molecular Chaperone toward a Target Substrate. Biochemistry 2015, 54, 5103–5112. [Google Scholar] [CrossRef] [PubMed]
- Westermark, P.; Engström, U.; Johnson, K.H.; Westermark, G.T.; Betsholtz, C. Islet amyloid polypeptide: Pinpointing amino acid residues linked to amyloid fibril formation. Proc. Natl. Acad. Sci. USA 1990, 87, 5036–5040. [Google Scholar] [CrossRef] [PubMed]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic islet cell toxicity of amylin associated with type-2 diabetes mellitus. Nature 1994, 368, 756. [Google Scholar] [CrossRef] [PubMed]
- Engel, M.F.M.; Khemtémourian, L.; Kleijer, C.C.; Meeldijk, H.J.D.; Jacobs, J.; Verkleij, A.J.; Kruijff, B.D.; Killian, J.A.; Höppener, J.W.M. Membrane damage by human islet amyloid polypeptide through fibril growth at the membrane. Proc. Natl. Acad. Sci. USA 2008, 105, 6033–6038. [Google Scholar] [CrossRef] [PubMed]
- Lopez, L.C.; Varea, O.; Navarro, S.; Carrodeguas, J.A.; Sanchez de Groot, N.; Ventura, S.; Sancho, J. Benzbromarone, Quercetin, and Folic Acid Inhibit Amylin Aggregation. Intl. J. Mol. Sci. 2016, 17, 964. [Google Scholar] [CrossRef] [PubMed]
- Mishra, R.; Bulic, B.; Sellin, D.; Jha, S.; Waldmann, H.; Winter, R. Small-Molecule Inhibitors of Islet Amyloid Polypeptide Fibril Formation. Angew. Chem. Int. Ed. 2008, 47, 4679–4682. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Abedini, A.; Plesner, A.; Verchere, C.B.; Raleigh, D.P. The flavanol (−)-epigallocatechin 3-gallate inhibits amyloid formation by islet amyloid polypeptide, disaggregates amyloid fibrils, and protects cultured cells against IAPP-induced toxicity. Biochemistry 2010, 49, 8127–8133. [Google Scholar] [CrossRef] [PubMed]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [PubMed]
- Chien, V.; Aitken, J.F.; Zhang, S.; Buchanan, C.M.; Hickey, A.; Brittain, T.; Cooper, G.J.S.; Loomes, K.M. The chaperone proteins HSP70, HSP40/DnaJ and GRP78/BiP suppress misfolding and formation of β-sheet-containing aggregates by human amylin: A potential role for defective chaperone biology in Type 2 diabetes. Biochem. J. 2010, 432, 113–121. [Google Scholar] [CrossRef] [PubMed]
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; Maio, A.D. Hsp70 Translocates into the Plasma Membrane after Stress and Is Released into the Extracellular Environment in a Membrane-Associated Form that Activates Macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Liu, J.; Saafi, E.L.; Cooper, G.J.S. Induction of apoptosis by human amylin in RINm5F islet β-cells is associated with enhanced expression of p53 and p21WAF1/CIP1. FEBS Lett. 1999, 455, 315–320. [Google Scholar] [CrossRef]
- Trikha, S.; Jeremic, A.M. Clustering and Internalization of Toxic Amylin Oligomers in Pancreatic Cells Require Plasma Membrane Cholesterol. J. Biol. Chem. 2011, 286, 36086–36097. [Google Scholar] [CrossRef] [PubMed]
- Trikha, S.; Jeremic, A.M. Distinct internalization pathways of human amylin monomers and its cytotoxic oligomers in pancreatic cells. PLoS ONE 2013, 8, e73080. [Google Scholar] [CrossRef] [PubMed]
- Sormanni, P.; Aprile, F.A.; Vendruscolo, M. Rational design of antibodies targeting specific epitopes within intrinsically disordered proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 9902–9907. [Google Scholar] [CrossRef] [PubMed]
- Pillay, K.; Govender, P.; Pillay, K.; Govender, P. Amylin Uncovered: A Review on the Polypeptide Responsible for Type II Diabetes. BioMed. Res. Int. 2013, e826706. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.J.; Day, A.J.; Willis, A.C.; Roberts, A.N.; Reid, K.B.; Leighton, B. Amylin and the amylin gene: Structure, function and relationship to islet amyloid and to diabetes mellitus. Biochim. Biophys. Acta 1989, 1014, 247–258. [Google Scholar] [CrossRef]
- Saafi, E.L.; Konarkowska, B.; Zhang, S.; Kistler, J.; Cooper, G.J. Ultrastructural evidence that apoptosis is the mechanism by which human amylin evokes death in RINm5F pancreatic islet β-cells. Cell Biol. Int. 2001, 25, 339–350. [Google Scholar] [CrossRef] [PubMed]
- Goldsbury, C.; Kistler, J.; Aebi, U.; Arvinte, T.; Cooper, G.J.S. Watching amyloid fibrils grow by time-lapse atomic force microscopy1. J. Mol. Biol. 1999, 285, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Goldsbury, C.; Goldie, K.; Pellaud, J.; Seelig, J.; Frey, P.; Müller, S.A.; Kistler, J.; Cooper, G.J.S.; Aebi, U. Amyloid Fibril Formation from Full-Length and Fragments of Amylin. J. Struct. Biol. 2000, 130, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.J.; Kayed, R.; Lin, C.-Y.; Gurlo, T.; Haataja, L.; Jayasinghe, S.; Langen, R.; Glabe, C.G.; Butler, P.C. Inhibition of human IAPP fibril formation does not prevent β -cell death: Evidence for distinct actions of oligomers and fibrils of human IAPP. Am. J. Physiol. Endocrinol. MeTable 2006, 291, E1317–E1324. [Google Scholar] [CrossRef] [PubMed]
- Magzoub, M.; Miranker, A.D. Concentration-dependent transitions govern the subcellular localization of islet amyloid polypeptide. FASEB J. 2012, 26, 1228–1238. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bongiovanni, M.N.; Aprile, F.A.; Sormanni, P.; Vendruscolo, M. A Rationally Designed Hsp70 Variant Rescues the Aggregation-Associated Toxicity of Human IAPP in Cultured Pancreatic Islet β-Cells. Int. J. Mol. Sci. 2018, 19, 1443. https://doi.org/10.3390/ijms19051443
Bongiovanni MN, Aprile FA, Sormanni P, Vendruscolo M. A Rationally Designed Hsp70 Variant Rescues the Aggregation-Associated Toxicity of Human IAPP in Cultured Pancreatic Islet β-Cells. International Journal of Molecular Sciences. 2018; 19(5):1443. https://doi.org/10.3390/ijms19051443
Chicago/Turabian StyleBongiovanni, Marie Nicole, Francesco Antonio Aprile, Pietro Sormanni, and Michele Vendruscolo. 2018. "A Rationally Designed Hsp70 Variant Rescues the Aggregation-Associated Toxicity of Human IAPP in Cultured Pancreatic Islet β-Cells" International Journal of Molecular Sciences 19, no. 5: 1443. https://doi.org/10.3390/ijms19051443
APA StyleBongiovanni, M. N., Aprile, F. A., Sormanni, P., & Vendruscolo, M. (2018). A Rationally Designed Hsp70 Variant Rescues the Aggregation-Associated Toxicity of Human IAPP in Cultured Pancreatic Islet β-Cells. International Journal of Molecular Sciences, 19(5), 1443. https://doi.org/10.3390/ijms19051443