Application of 3D-QSAR, Pharmacophore, and Molecular Docking in the Molecular Design of Diarylpyrimidine Derivatives as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors


Abstract
:
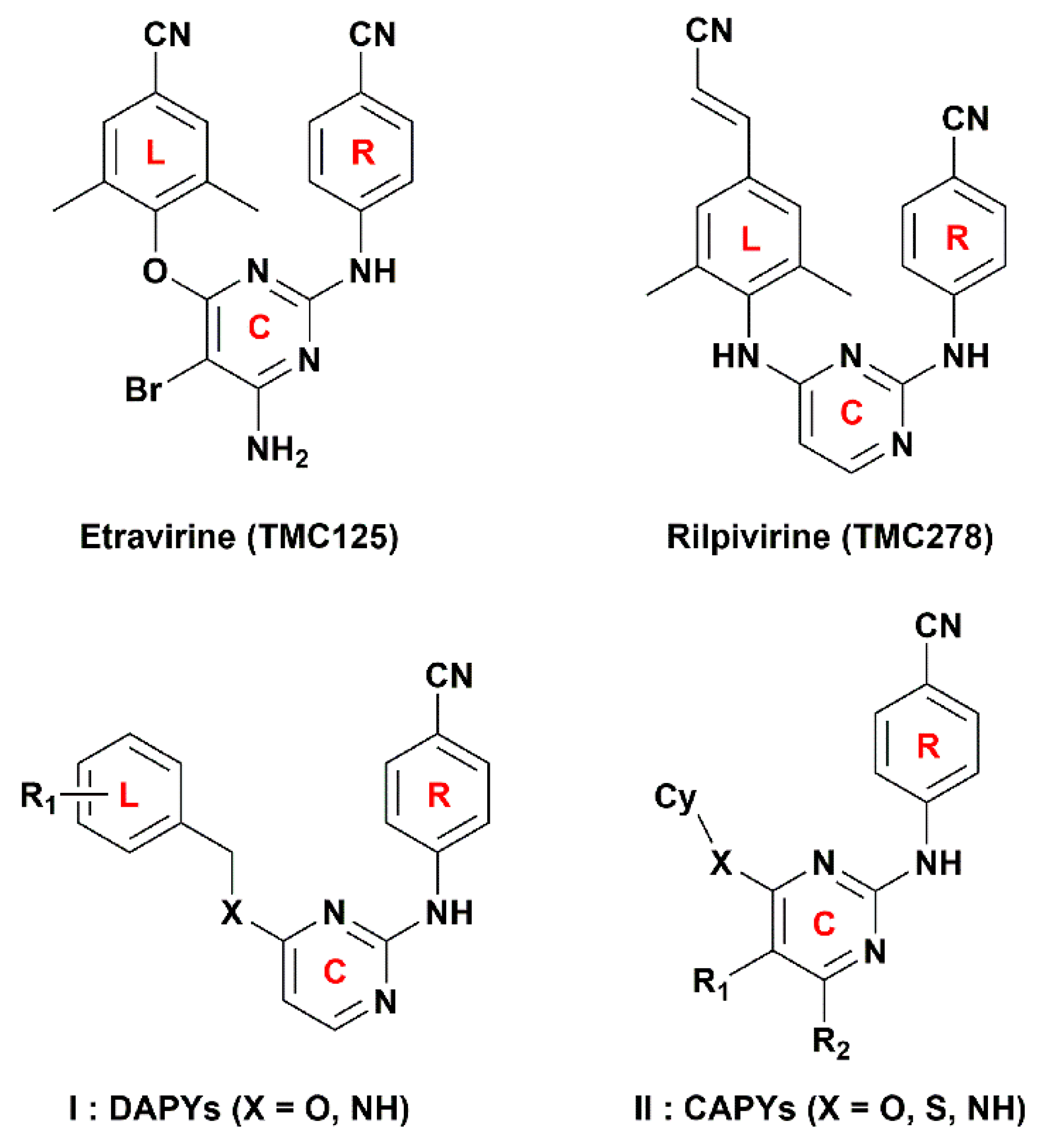
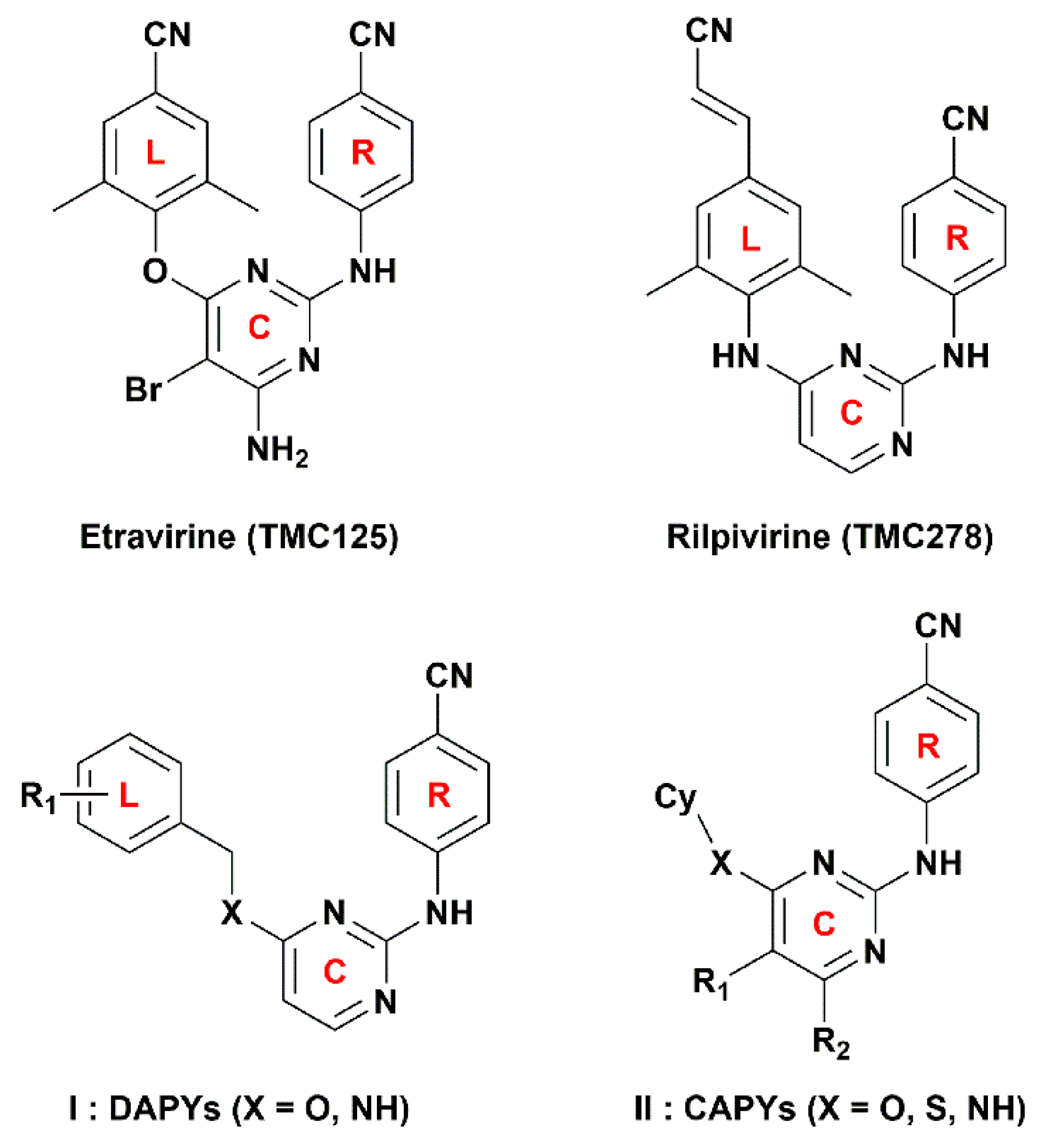
1. Introduction
2. Results and Discussion
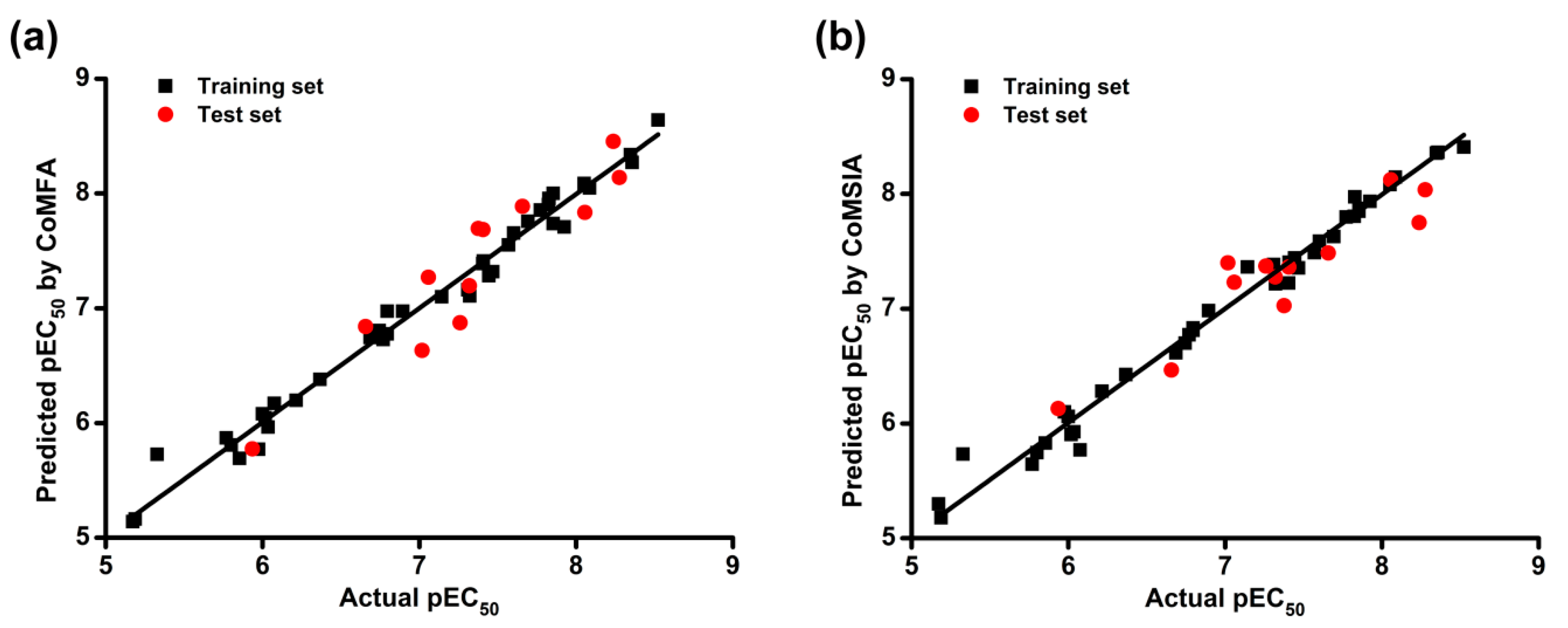
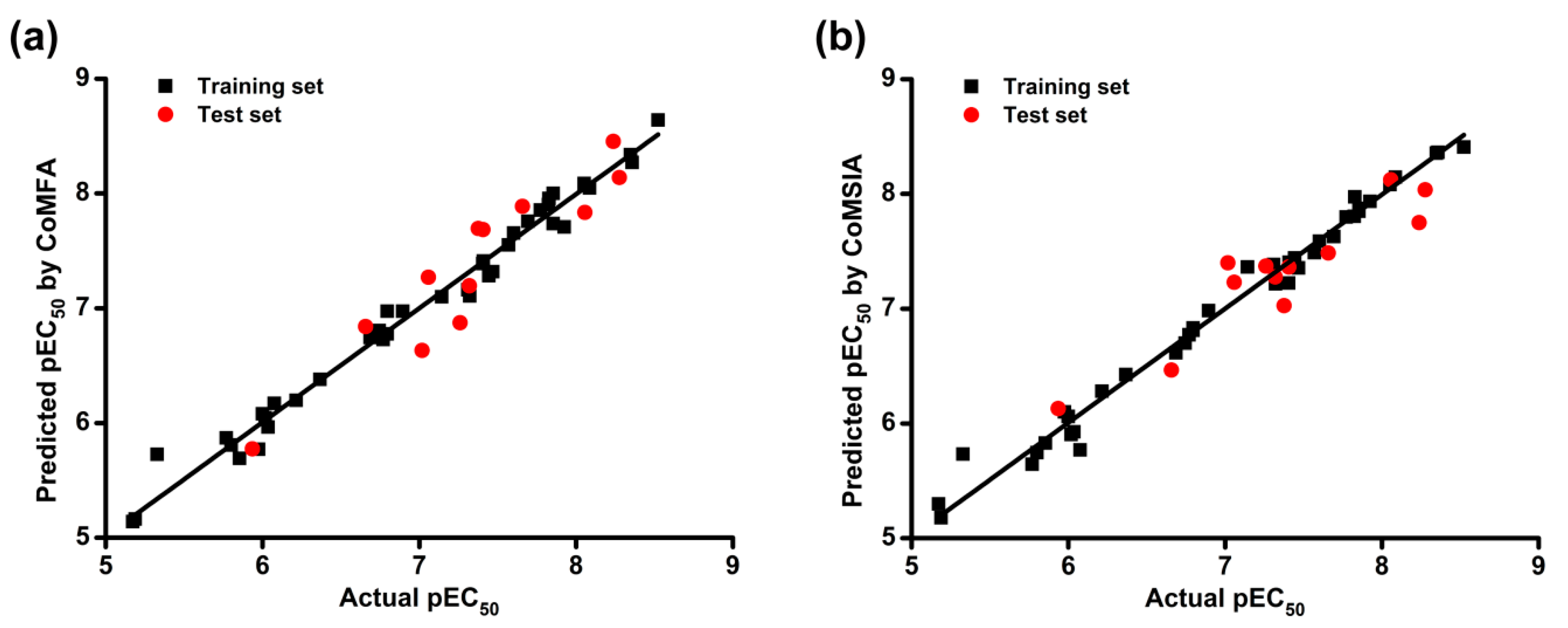
2.1. CoMFA and CoMSIA Statistical Results
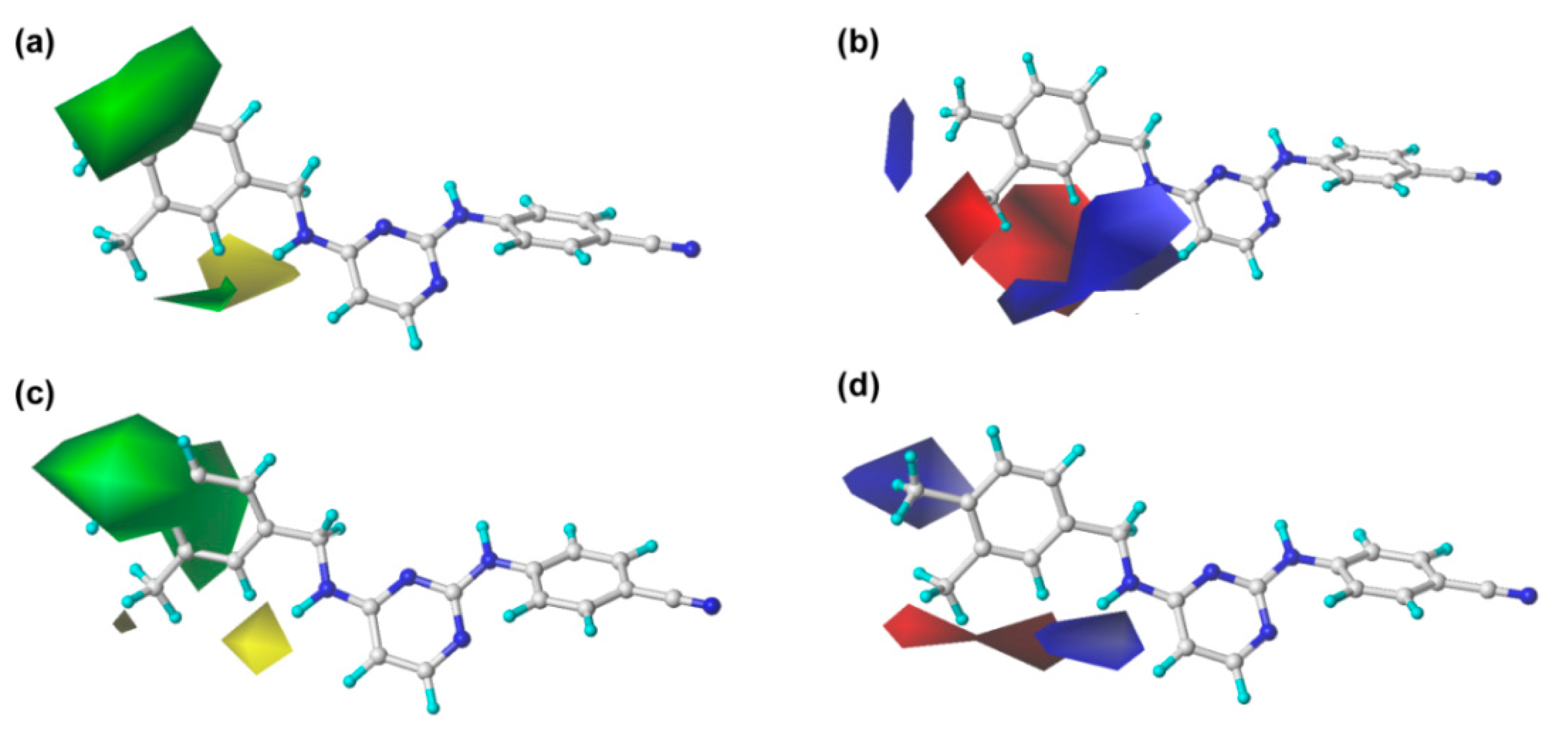
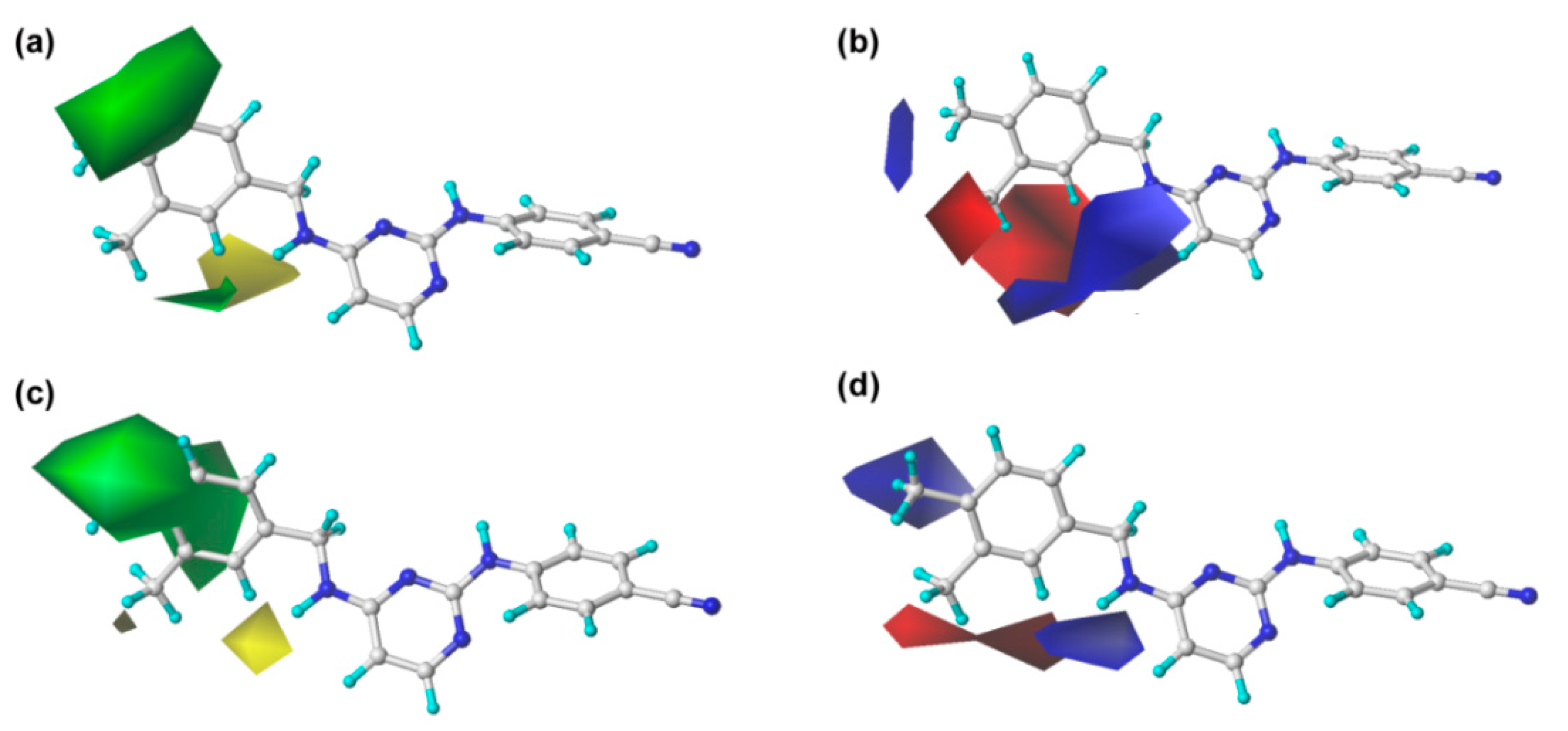
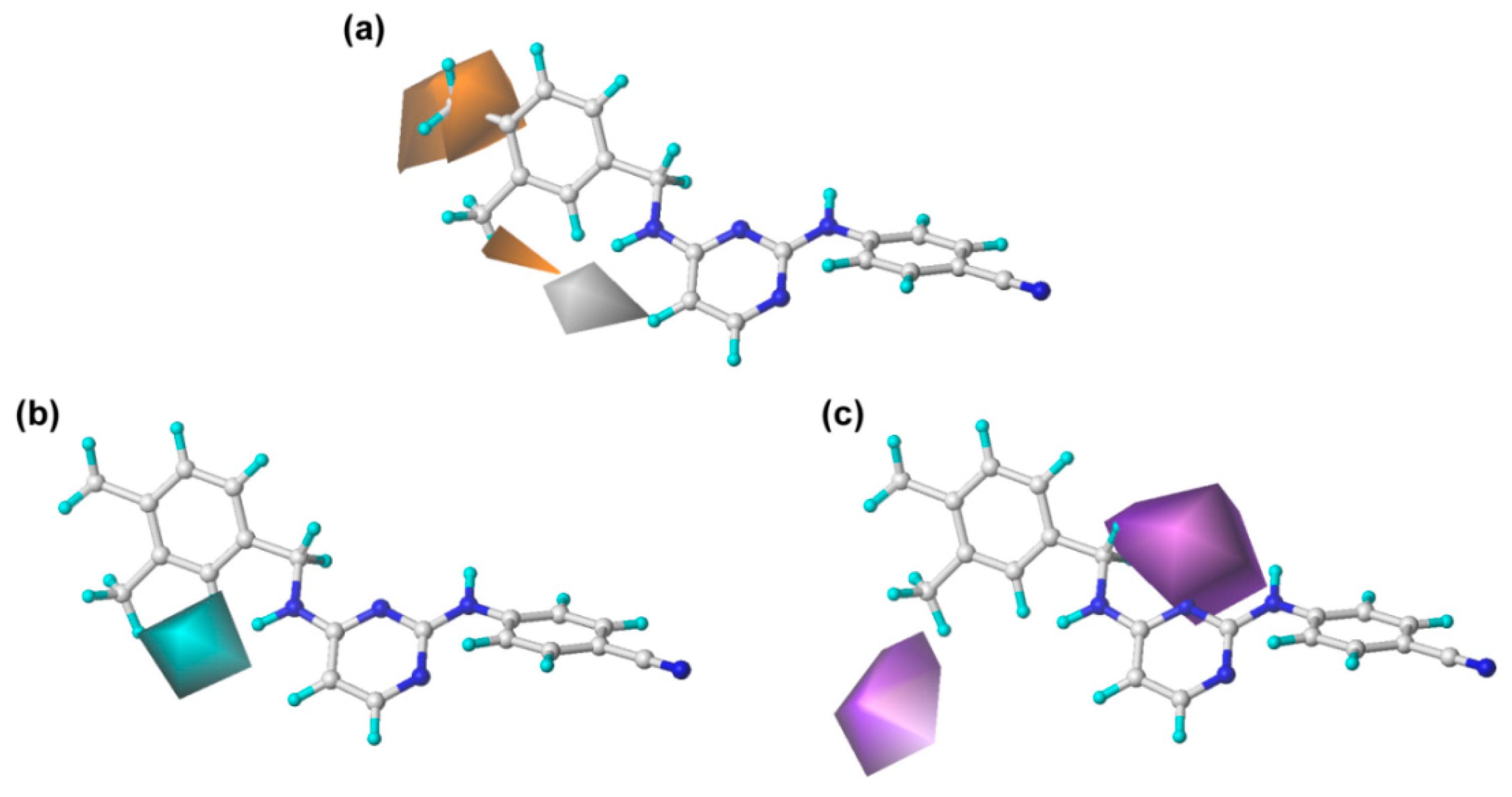
2.2. CoMFA and CoMSIA Contour Maps
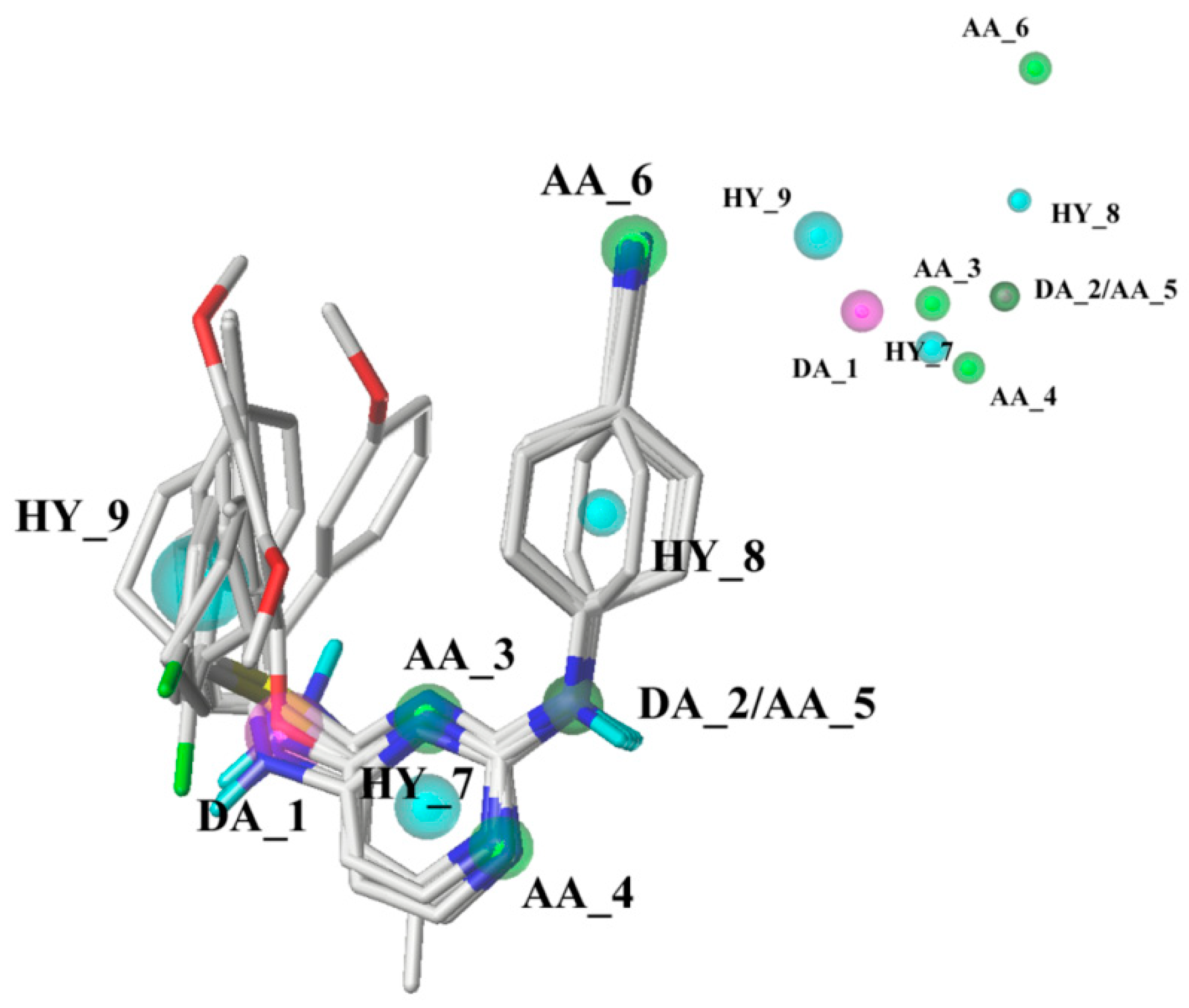
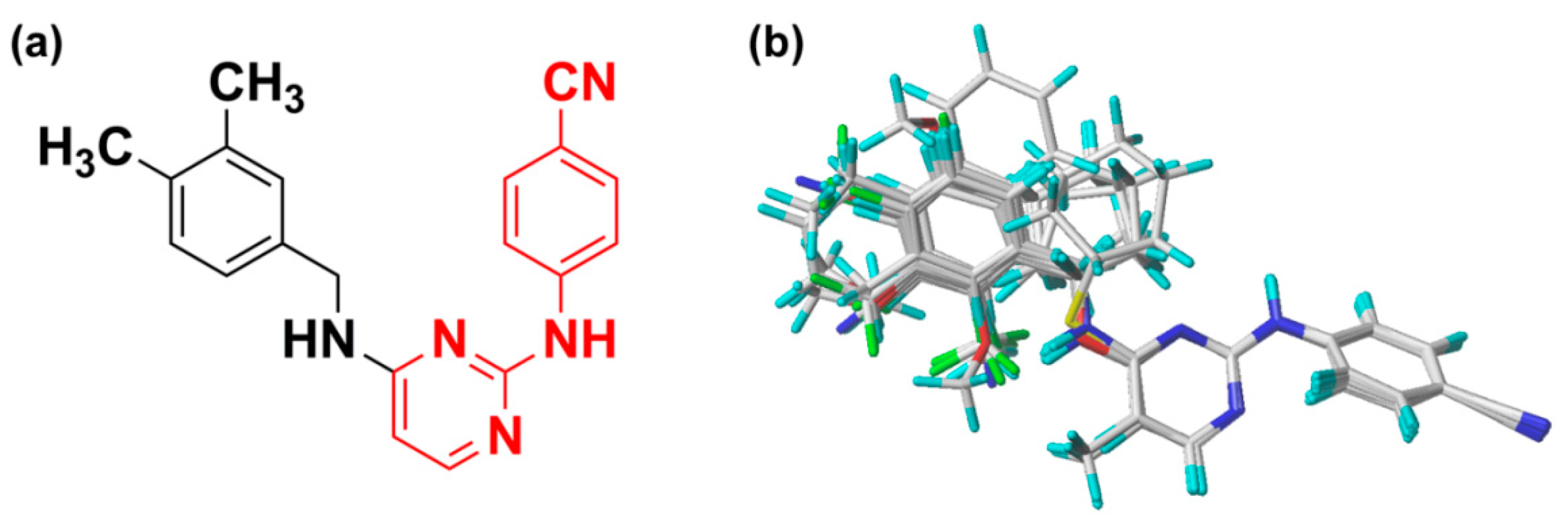
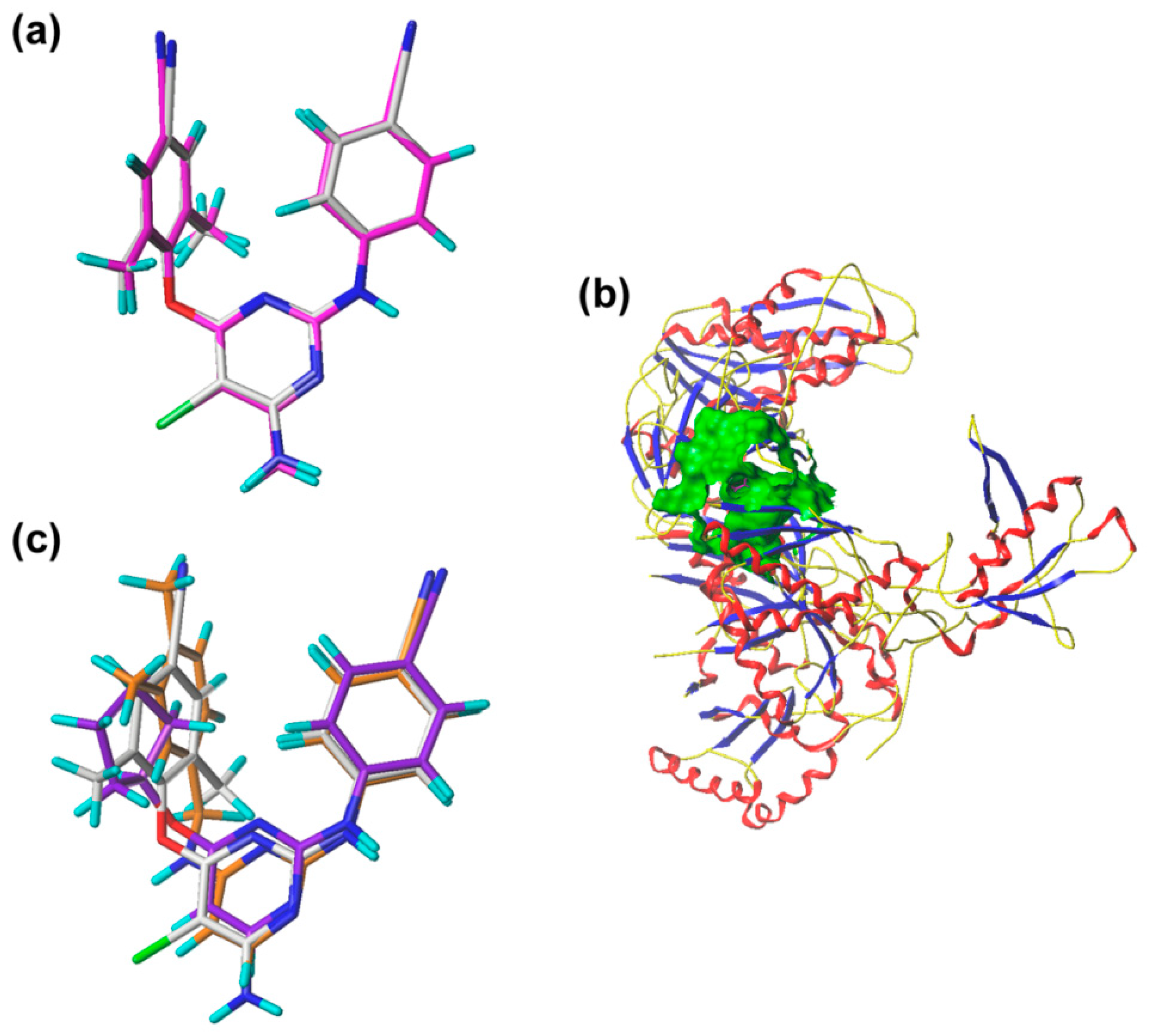
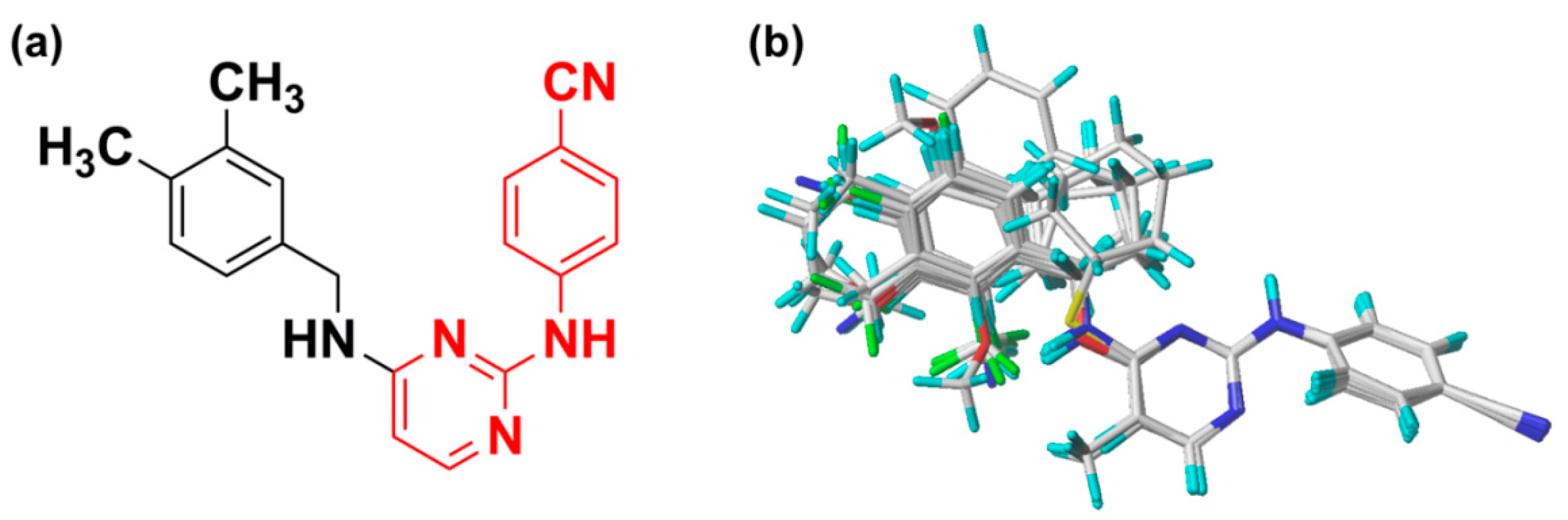
2.3. Pharmacophore Model
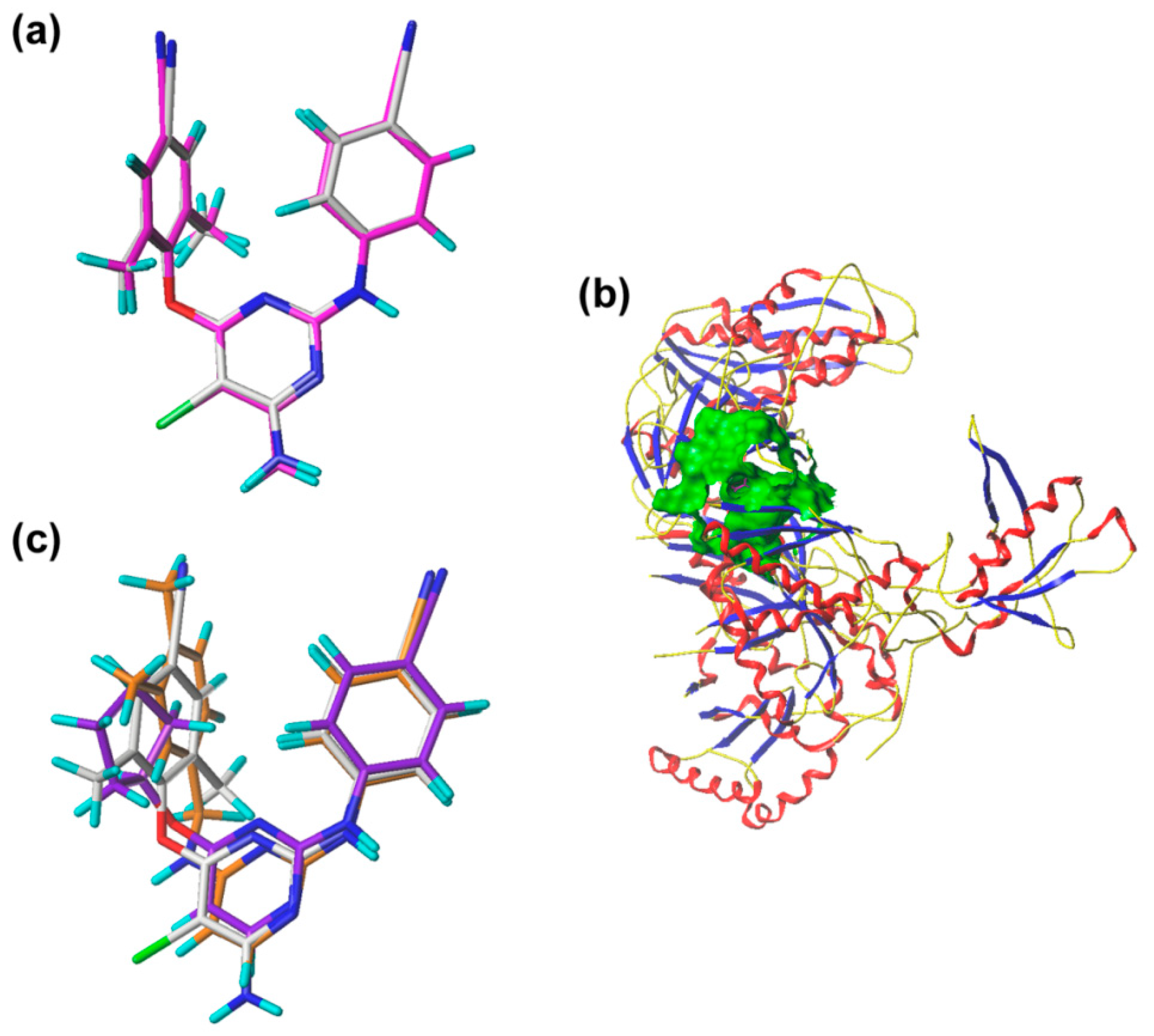
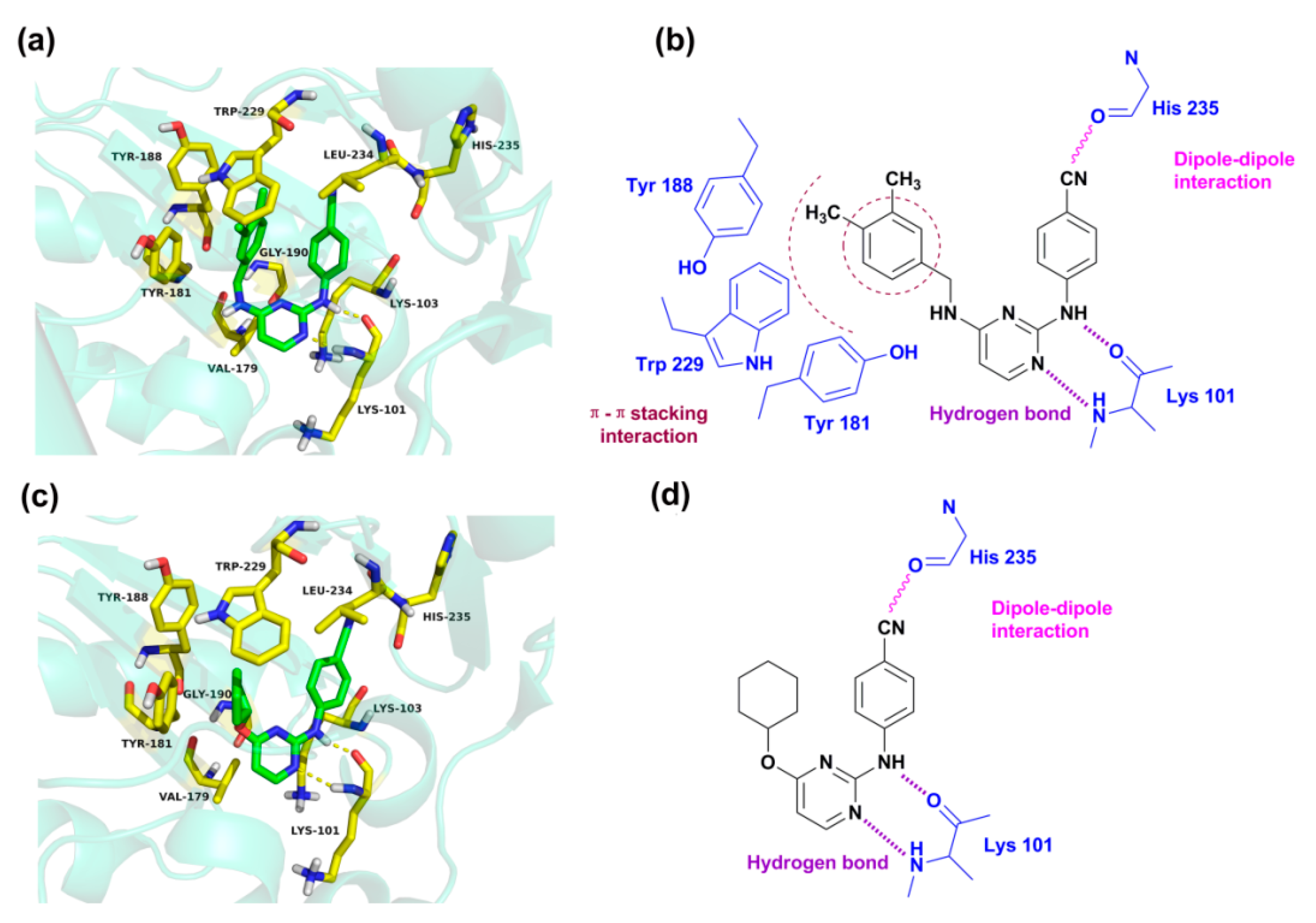
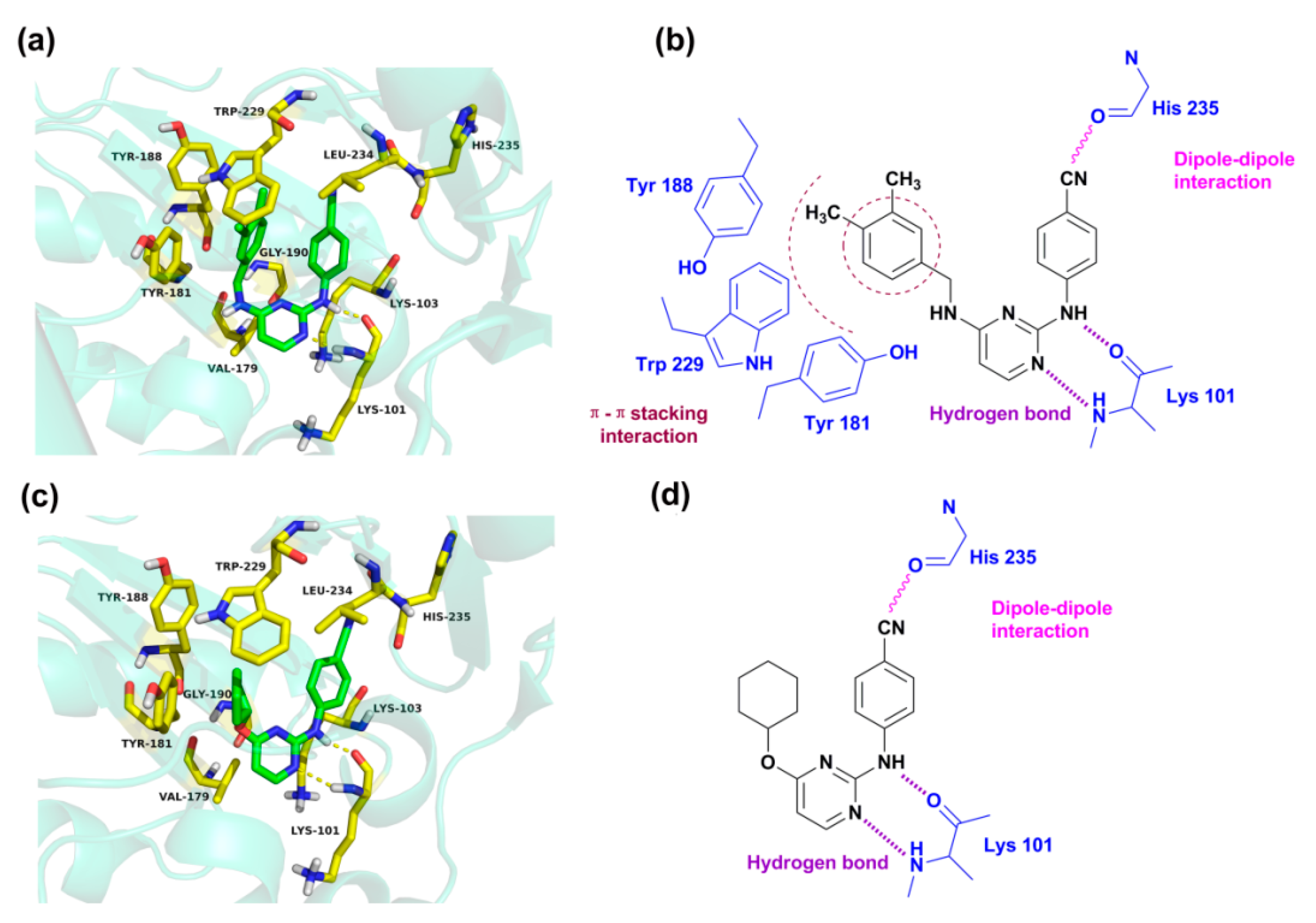
2.4. Molecular Docking Analysis
2.5. Newly Designed DAPYs
3. Materials and Methods
3.1. Dataset and Alignment
3.2. CoMFA and CoMSIA Models
3.3. Pharmacophore Model
3.4. Molecular Docking
4. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| DAPY | diarylpyrimidine |
| NNRTI | nonnucleoside reverse transcriptase inhibitor |
| AIDS | acquired immunodeficiency syndrome |
| 3D-QSAR | three-dimensional quantitative structure-activity relationship |
| CoMFA | comparative molecular field analysis |
| CoMSIA | comparative molecular similarity indices analysis |
| HIV | human immunodeficiency virus |
| HAART | highly active antiretroviral therapy |
| RT | reverse transcriptase |
| HIV-1 | HIV type 1 |
| WT | wild-type |
| CAPY | cycloalkyl arylpyrimidine |
| cross-validated correlation coefficient | |
| ONC | optimal number of components |
| non-cross-validated correlation coefficient | |
| predictive correlation coefficient | |
| SEE | standard error of estimate |
| F | F-statistic values |
| RMSE | root mean square error |
| MAE | mean absolute error |
| GALAHAD | Genetic Algorithm with Linear Assignment of Hypermolecular Alignment of Datasets |
| SE | strain energy |
| SO | steric overlap |
| PhS | pharmacophoric similarity |
| DA | hydrogen-bond donor atom |
| AA | hydrogen-bond acceptor atom |
| HY | hydrophobe |
| RMSD | root-mean-square deviation |
| LOO | leave one out |
| SDEP | standard deviation error of predictions |
| PRESS | predicted residuals sum of squares |
References
- Fauci, A.S. HIV and AIDS: 20 years of science. Nat. Med. 2003, 9, 839–843. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhan, P.; Li, D.; De Clercq, E.; Liu, X. Recent advances in DAPYs and related analogues as HIV-1 NNRTIs. Curr. Med. 2011, 18, 359–376. [Google Scholar] [CrossRef]
- Das, K.; Martinez, S.E.; Bauman, J.D.; Arnold, E. HIV-1 reverse transcriptase complex with DNA and nevirapine reveals non-nucleoside inhibition mechanism. Nat. Struct. Mol. Biol. 2012, 19, 253–259. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Available online: http://www.who.int/gho/publications/world_health_statistics/en/ (accessed on 31 July 2017).
- Gu, S.X.; Qiao, H.; Zhu, Y.Y.; Shu, Q.C.; Liu, H.; De Clercq, E.; Balzarini, J.; Pannecouque, C. A novel family of diarylpyrimidines (DAPYs) featuring a diatomic linker: Design, synthesis and anti-HIV activities. Bioorg. Med. Chem. 2015, 23, 6587–6593. [Google Scholar] [CrossRef] [PubMed]
- Unissa, A.N.; Hanna, L.E. Insights into computational analysis of HIV-1 reverse transcriptase resistance to non-nucleoside inhibitors: Efavirenz, etravirine, nevirapine and rilpivine. Int. J. Pharma Bio Sci. 2017, 8, 839–848. [Google Scholar] [CrossRef]
- De Clercq, E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antivir. Res. 1998, 38, 153–179. [Google Scholar] [CrossRef]
- Lu, H.H.; Xue, P.; Zhu, Y.Y.; Jiu, X.L.; Zhen, X.J.; Zhan, X.; Xiao, T.; Christophe, P.; Li, T.T.; Gu, S.X. Structural modifications of diarylpyrimidines (DAPYs) as HIV-1 NNRTIs: Synthesis, anti-HIV activities and SAR. Bioorg. Med. Chem. 2017, 25, 2491–2497. [Google Scholar] [CrossRef] [PubMed]
- Croxtall, J.D. Etravirine: A review of its use in the management of treatment-experienced patients with HIV-1 infection. Drugs 2012, 72, 847–869. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.J.; Short, W.R. Rilpivirine, a novel non-nucleoside reverse transcriptase inhibitor for the management of HIV-1 infection: A systematic review. Antivir. Ther. 2012, 17, 1495–1502. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Yang, S.Q.; He, Q.Q.; Ma, X.D.; Chen, F.E.; Dai, H.F.; De Clercq, E.; Jan, B.; Christophe, P. Design, synthesis and biological evaluation of cycloalkyl arylpyrimidines (CAPYs) as HIV-1 NNRTIs. Bioorg. Med. Chem. 2011, 19, 7093–7099. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.; Wu, Y.; Bai, M.; Zhan, P. 3D-QSAR and molecular docking studies on HIV protease inhibitors. J. Mol. Struct. 2016, 1129, 17–22. [Google Scholar] [CrossRef]
- Kumar, S.; Tiwari, M. Topomer-CoMFA-based predictive modelling on 2,3-diaryl-substituted-1,3-thiazolidin-4-ones as non-nucleoside reverse transcriptase inhibitors. Med. Chem. Res. 2014, 24, 245–257. [Google Scholar] [CrossRef]
- Maltarollo, V.G.; Silva, D.C.; Honório, K.M. Advanced QSAR studies on PPARδ ligands related to metabolic diseases. J. Braz. Chem. Soc. 2012, 23, 78–84. [Google Scholar] [CrossRef]
- Khan, K.M.; Wadood, A.; Ali, M.; Lodhi, M.A.; Khan, M.; Perveen, S.; Choudhary, M.I. Identification of potent urease inhibitors via ligand- and structure-based virtual screening and in vitro assays. J. Mol. Graph. Model. 2010, 28, 792–798. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhan, P.; Li, Z.; Liu, H.; De Clercq, E.; Pannecouque, C.; Liu, X. Discovery of nitropyridine derivatives as potent HIV-1 non-nucleoside reverse transcriptase inhibitors via a structure-based core refining approach. Eur. J. Med. Chem. 2014, 76, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Madrid, M.; Madura, J.D. Docking of non-nucleoside inhibitors: Neotripterifordin and its derivatives to HIV-1 reverse transcriptase. Proteins 2002, 49, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Barreiro, G.; Guimarães, C.R.; Tubert-Brohman, I.; Lyons, T.M.; Tirado-Rives, J.; Jorgensen, W.L. Search for non-nucleoside inhibitors of HIV-1 reverse transcriptase using chemical similarity, molecular docking, and MM-GB/SA scoring. J. Chem. Inf. Model. 2007, 47, 2416–2428. [Google Scholar] [CrossRef] [PubMed]
- Xue, P.; Lu, H.H.; Zhu, Y.Y.; Jiu, X.L.; Christophe, P.; Zheng, X.J.; Liu, G.Y.; Zhang, X.L.; Gu, S.X. Design and synthesis of hybrids of diarylpyrimidines and diketo acids as HIV-1 inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 1640–1643. [Google Scholar] [CrossRef] [PubMed]
- Ghaleb, A.; Aouidate, A.; Ghamali, M.; Sbai, A.; Bouachrine, M.; Lakhlifi, T. 3D-QSAR modeling and molecular docking studies on a series of 2,5 disubstituted 1,3,4-oxadiazoles. J. Mol. Struct. 2017, 1145, 278–284. [Google Scholar] [CrossRef]
- Fatemi, M.H.; Heidari, A.; Gharaghani, S. QSAR prediction of HIV-1 protease inhibitory activities using docking derived molecular descriptors. J. Theor. Biol. 2015, 369, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Li, S.R.; Fan, J.L.; Peng, C.K.; Chang, Y.Q.; Guo, L.X.; Hou, J.S.; Huang, M.Q.; Wu, B.Y.; Zheng, J.X.; Lin, L.X.; et al. New molecular insights into the tyrosyl-tRNA synthase inhibitors: CoMFA, CoMSIA analyses and molecular docking studies. Sci. Rep. 2017, 7, 11525. [Google Scholar] [CrossRef] [PubMed]
- Ojha, P.K.; Mitra, I.; Das, R.N.; Roy, K. Further exploring metrics for validation of QSPR models. Chemom. Intell. Lab. Syst. 2011, 107, 194–205. [Google Scholar] [CrossRef]
- Elidrissi, B.; Ousaa, A.; Aouidate, A.; Zaki, H.; Ajana, M.; Lakhlif, T.; Bouachrine, M. 3D-QSAR studies of isatin derivatives with anti-cancer in vitro: Advanced CoMFA, CoMSIA and docking methods. Chem. Sci. J. 2017, 8, 158. [Google Scholar] [CrossRef]
- Roy, K.; Das, R.N.; Ambure, P.; Aher, R.B. Be aware of error measures. Further studies on validation of predictive QSAR models. Chemom. Intell. Lab. Syst. 2016, 152, 18–33. [Google Scholar] [CrossRef]
- Golbraikh, A.; Tropsha, A. Beware of q2! J. Mol. Graph. Model. 2002, 20, 269–276. [Google Scholar] [CrossRef]
- Caballero, J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model. 2010, 29, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.D.; Yang, S.Q.; Gu, S.X.; He, Q.Q.; Chen, F.E.; De Clercq, E.; Jan, B.; Christophe, P. Synthesis and anti-HIV activity of aryl-2-[(4-cyanophenyl)amino]-4-pyrimidinone hydrazones as potent non-nucleoside reverse transcriptase inhibitors. ChemMedChem 2011, 6, 2225–2232. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.D.; Dixit, S.R.; Kirankumar, M.N.; Aminabhavi, T.M.; Raju, K.V.; Narayan, R.; Lherbet, C.; Yang, K.S. Synthesis, antimycobacterial screening and ligand-based molecular docking studies on novel pyrrole derivatives bearing pyrazoline, isoxazole and phenyl thiourea moieties. Eur. J. Med. Chem. 2016, 107, 133–152. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | ONC | SEE | F | Filed Contribution (%) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| E | H | S | D | A | ||||||||
| CoMFA | E + S | 0.679 | 8 | 0.983 | 0.884 | 0.136 | 229.756 | 53.70 | — | 46.30 | — | — |
| CoMSIA | E + H + S | 0.721 | 9 | 0.972 | 0.734 | 0.180 | 114.912 | 52.40 | 32.40 | 15.20 | — | — |
| E + S + D + A | 0.705 | 9 | 0.947 | 0.743 | 0.247 | 59.278 | 58.80 | — | 14.90 | 14.00 | 12.20 | |
| E + H + D + A | 0.695 | 11 | 0.987 | 0.826 | 0.125 | 198.216 | 45.10 | 34.00 | — | 12.60 | 8.20 | |
| E + H + S + A | 0.74 | 9 | 0.984 | 0.827 | 0.135 | 206.773 | 44.80 | 29.70 | 13.60 | — | 11.90 | |
| E + H + S + D | 0.703 | 9 | 0.972 | 0.698 | 0.178 | 117.713 | 48.70 | 29.70 | 14.30 | 7.30 | — | |
| E + H + S + D + A | 0.734 | 9 | 0.985 | 0.891 | 0.132 | 215.609 | 41.40 | 27.60 | 12.30 | 11.20 | 7.50 | |
| Statistics | CoMFA (E + S) | CoMSIA (E + H + S + D + A) | ||
|---|---|---|---|---|
| Training Set | Test Set | Training Set | Test Set | |
| RMSE | 0.160 | 0.155 | ||
| MAE | 0.124 | 0.108 | ||
| 0.9834 | 0.8764 | 0.9848 | 0.8657 | |
| 0.9830 | 0.8454 | 0.9848 | 0.8613 | |
| 0.9831 | 0.8750 | 0.9845 | 0.8144 | |
| k | 1.0003 | 0.8408 | 1.0000 | 1.0848 |
| k’ | 0.9831 | 0.9982 | 0.9997 | 0.9917 |
| 0.9637 | 0.7221 | 0.9848 | 0.8083 | |
| 0.9664 | 0.8436 | 0.9677 | 0.6696 | |
| Δ | 0.0026 | 0.1215 | 0.0171 | 0.1387 |
| 0.9690 | 0.7829 | 0.9763 | 0.7389 | |
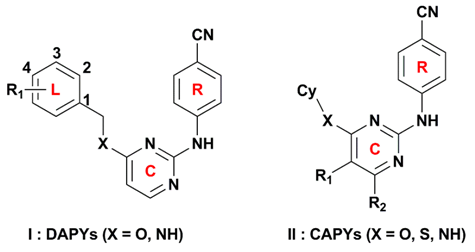
| No. | Type | X | R1 | R2 | Cy | Actual pEC50 | CoMFA | CoMSIA | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Predicted | Residuals | Predicted | Residuals | |||||||
| 1 | I | O | 2-Cl | — | — | 7.469 | 7.318 | −0.151 | 7.355 | −0.114 |
| 2 | I | O | 3-Cl | — | — | 7.310 | 7.161 | −0.149 | 7.384 | 0.074 |
| 3 | I | O | 4-Cl | — | — | 6.076 | 6.172 | 0.096 | 5.769 | −0.307 |
| 4 | I | O | 2-Br | — | — | 7.444 | 7.283 | −0.161 | 7.442 | −0.002 |
| 5 * | I | O | 3-Br | — | — | 7.319 | 7.196 | −0.123 | 7.277 | −0.042 |
| 6 | I | O | 4-Br | — | — | 5.328 | 5.727 | 0.399 | 5.733 | 0.405 |
| 7 * | I | O | 2-F | — | — | 8.237 | 8.454 | 0.217 | 7.750 | −0.487 |
| 8 | I | O | 3-F | — | — | 7.854 | 7.737 | −0.117 | 7.848 | −0.006 |
| 9 | I | O | 4-F | — | — | 6.036 | 5.964 | −0.072 | 5.927 | −0.109 |
| 10 | I | O | 2-CH3 | — | — | 6.796 | 6.974 | 0.178 | 6.831 | 0.035 |
| 11 * | I | O | 3-CH3 | — | — | 8.056 | 7.834 | −0.222 | 8.124 | 0.068 |
| 12 | I | O | 4-CH3 | — | — | 6.796 | 6.776 | −0.020 | 6.811 | 0.015 |
| 13 | I | O | 2-OCH3 | — | — | 7.569 | 7.551 | −0.018 | 7.488 | −0.081 |
| 14 | I | O | 3-OCH3 | — | — | 7.602 | 7.654 | 0.052 | 7.587 | −0.015 |
| 15 | I | O | 4-OCH3 | — | — | 6.000 | 6.080 | 0.080 | 6.061 | 0.061 |
| 16 | I | O | 2-CF3 | — | — | 5.770 | 5.870 | 0.100 | 5.644 | −0.126 |
| 17 | I | O | 3-CF3 | — | — | 6.215 | 6.197 | −0.018 | 6.281 | 0.066 |
| 18 | I | O | 3,5-diMeO | — | — | 8.051 | 8.086 | 0.035 | 8.080 | 0.029 |
| 19 | I | O | 5-F-2-Br | — | — | 7.409 | 7.411 | 0.002 | 7.404 | −0.005 |
| 20 * | I | O | 2-F-4-Br | — | — | 6.658 | 6.841 | 0.183 | 6.465 | −0.193 |
| 21 | I | O | 2-CN | — | — | 7.854 | 8.000 | 0.146 | 7.872 | 0.018 |
| 22 | I | O | 3-CN | — | — | 6.745 | 6.805 | 0.060 | 6.700 | −0.045 |
| 23 | I | O | 4-CN | — | — | 5.854 | 5.691 | −0.163 | 5.829 | −0.025 |
| 24 * | I | O | H | — | — | 7.658 | 7.887 | 0.229 | 7.485 | −0.173 |
| 25 | I | O | 1-NaPh | — | — | 7.824 | 7.901 | 0.077 | 7.804 | −0.020 |
| 26 | I | O | 2-NaPh | — | — | 6.770 | 6.726 | −0.044 | 6.772 | 0.002 |
| 27 * | I | NH | H | — | — | 8.276 | 8.139 | −0.137 | 8.036 | −0.240 |
| 28 | I | NH | 2-Cl | — | — | 7.827 | 7.956 | 0.129 | 7.973 | 0.146 |
| 29 | I | NH | 3-Cl | — | — | 7.924 | 7.707 | −0.217 | 7.935 | 0.011 |
| 30 | I | NH | 4-Cl | — | — | 7.322 | 7.106 | −0.216 | 7.216 | −0.106 |
| 31 * | I | NH | 2-Br | — | — | 7.376 | 7.694 | 0.318 | 7.026 | −0.350 |
| 32 | I | NH | 3-Br | — | — | 7.693 | 7.757 | 0.064 | 7.628 | −0.065 |
| 33 | I | NH | 4-Br | — | — | 7.405 | 7.389 | −0.016 | 7.223 | −0.182 |
| 34 | I | NH | 3-F | — | — | 8.357 | 8.271 | −0.086 | 8.362 | 0.005 |
| 35 | I | NH | 4-F | — | — | 7.143 | 7.099 | −0.044 | 7.363 | 0.220 |
| 36 | I | NH | 4-CH3 | — | — | 8.086 | 8.047 | −0.039 | 8.146 | 0.060 |
| 37 * | I | NH | 2-OCH3 | — | — | 7.058 | 7.270 | 0.212 | 7.230 | 0.172 |
| 38 | I | NH | 3-OCH3 | — | — | 8.347 | 8.338 | −0.009 | 8.355 | 0.008 |
| 39 * | I | NH | 4-OCH3 | — | — | 7.406 | 7.685 | 0.279 | 7.363 | −0.043 |
| 40 | I | NH | 2-CF3 | — | — | 5.976 | 5.771 | −0.205 | 6.101 | 0.125 |
| 41 | I | NH | 3-CF3 | — | — | 6.687 | 6.744 | 0.057 | 6.615 | −0.072 |
| 42 | I | NH | 4-CF3 | — | — | 6.895 | 6.973 | 0.078 | 6.983 | 0.088 |
| 43 | I | NH | 3,4-diMe | — | — | 8.523 | 8.640 | 0.117 | 8.408 | −0.115 |
| 44 | I | NH | 2,4-diF | — | — | 7.772 | 7.854 | 0.082 | 7.798 | 0.026 |
| 45 * | II | O | H | H | cyclohexyl | 5.936 | 5.775 | −0.161 | 6.130 | 0.194 |
| 46 | II | O | H | H | cyclopentyl | 5.173 | 5.140 | −0.033 | 5.297 | 0.124 |
| 47 | II | O | H | CH3 | cyclohexyl | 5.188 | 5.162 | −0.026 | 5.178 | −0.010 |
| 48 | II | NH | H | H | cyclohexyl | 6.018 | 6.046 | 0.028 | 5.903 | −0.115 |
| 49 | II | NH | H | H | cyclopentyl | 5.801 | 5.808 | 0.007 | 5.746 | −0.055 |
| 50 * | II | S | H | H | cyclohexyl | 7.260 | 6.874 | −0.386 | 7.371 | 0.111 |
| 51 | II | S | H | CH3 | cyclohexyl | 6.367 | 6.380 | 0.013 | 6.425 | 0.058 |
| 52 * | II | S | CH3 | H | cyclohexyl | 7.018 | 6.633 | −0.385 | 7.399 | 0.381 |
| Receptor | Ligand | Hydrogen-Bond Receptor | Hydrogen-Bond Donor | Distance (Å) | Angle (°) |
|---|---|---|---|---|---|
| 3MEC | 43 | –O (Lys101 –C=O) | –N (–NH) | 1.725 | 151.02 |
| –N (pyrimidine) | –N (Lys101 –NH2) | 2.302 | 168.45 | ||
| 46 | –O (Lys101 –C=O) | –N (–NH) | 1.843 | 157.47 | |
| –N (pyrimidine) | –N (Lys101 –NH2) | 2.477 | 163.30 |
| No. | R1 | X | Predicted pEC50 | |
|---|---|---|---|---|
| CoMFA | CoMSIA | |||
| 53 | 4-CH2CH3 | NH | 8.655 | 9.508 |
| 54 | 4-CH(CH3)2 | NH | 8.765 | 10.212 |
| 55 | 4-C(CH3)3 | NH | 8.276 | 8.959 |
| 56 | 4-NH2 | NH | 8.695 | 9.050 |
| 57 | 3-CN | NH | 8.334 | 8.584 |
| 58 | 3-NO2 | NH | 8.306 | 8.281 |
| 59 | 3-OOCCH3 | NH | 8.227 | 8.873 |
| 60 | 3-OH | NH | 9.020 | 9.026 |
| 61 | 2-OH | NH | 7.845 | 8.690 |
| 62 | 2-F, 4-CH3 | NH | 8.675 | 9.388 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, G.; Wang, W.; Wan, Y.; Ju, X.; Gu, S. Application of 3D-QSAR, Pharmacophore, and Molecular Docking in the Molecular Design of Diarylpyrimidine Derivatives as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors. Int. J. Mol. Sci. 2018, 19, 1436. https://doi.org/10.3390/ijms19051436
Liu G, Wang W, Wan Y, Ju X, Gu S. Application of 3D-QSAR, Pharmacophore, and Molecular Docking in the Molecular Design of Diarylpyrimidine Derivatives as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors. International Journal of Molecular Sciences. 2018; 19(5):1436. https://doi.org/10.3390/ijms19051436
Chicago/Turabian StyleLiu, Genyan, Wenjie Wang, Youlan Wan, Xiulian Ju, and Shuangxi Gu. 2018. "Application of 3D-QSAR, Pharmacophore, and Molecular Docking in the Molecular Design of Diarylpyrimidine Derivatives as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors" International Journal of Molecular Sciences 19, no. 5: 1436. https://doi.org/10.3390/ijms19051436
APA StyleLiu, G., Wang, W., Wan, Y., Ju, X., & Gu, S. (2018). Application of 3D-QSAR, Pharmacophore, and Molecular Docking in the Molecular Design of Diarylpyrimidine Derivatives as HIV-1 Nonnucleoside Reverse Transcriptase Inhibitors. International Journal of Molecular Sciences, 19(5), 1436. https://doi.org/10.3390/ijms19051436