Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death



Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
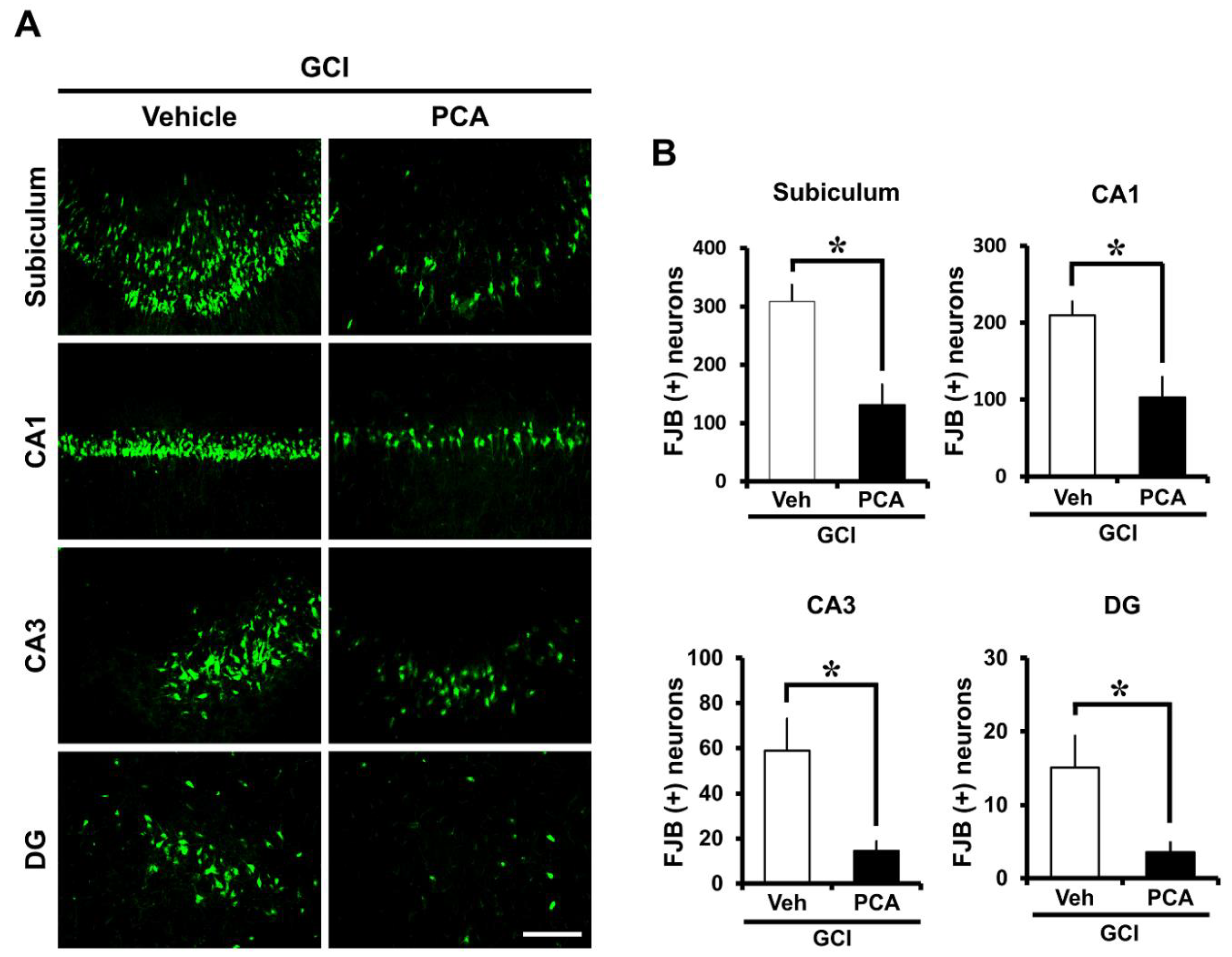
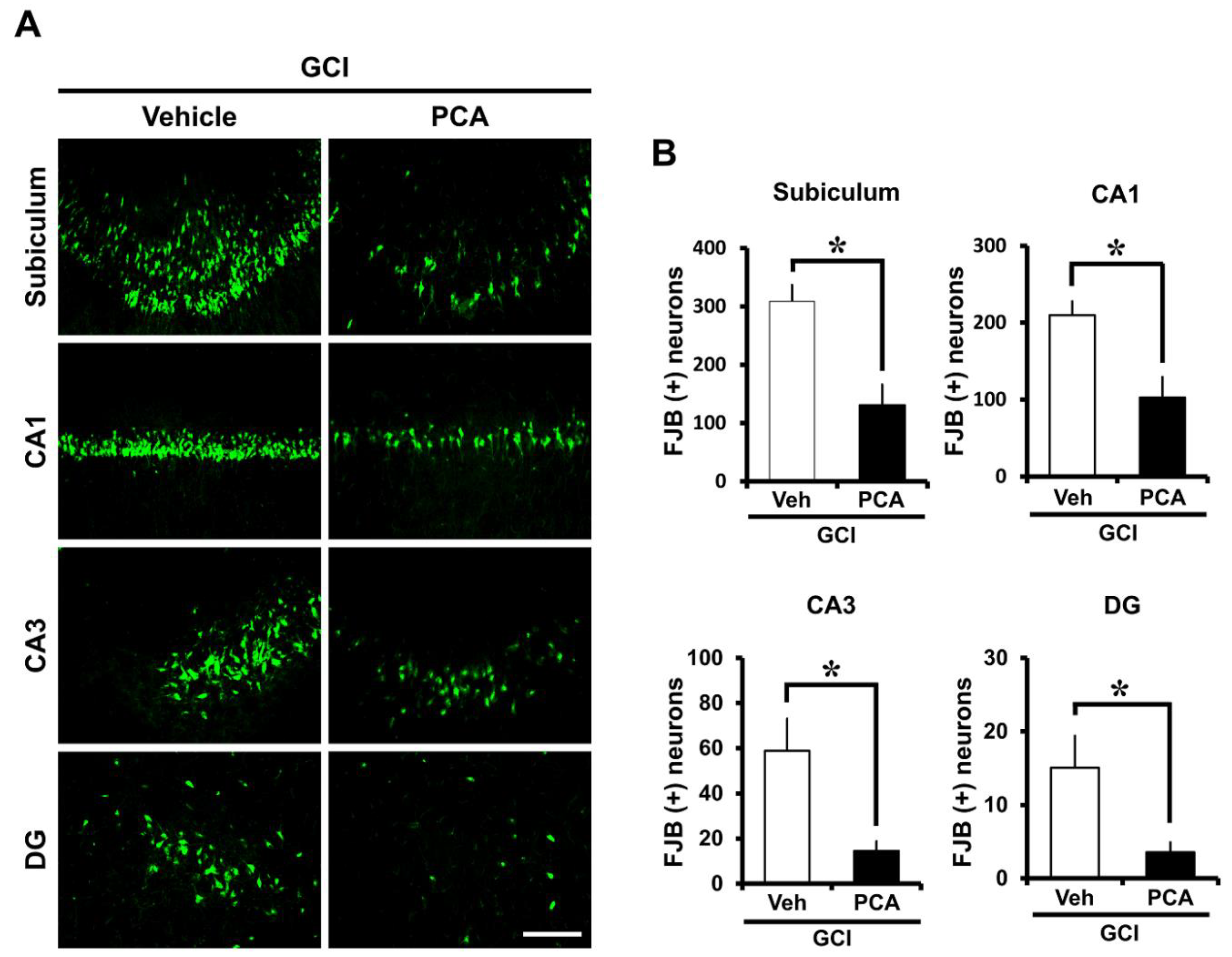
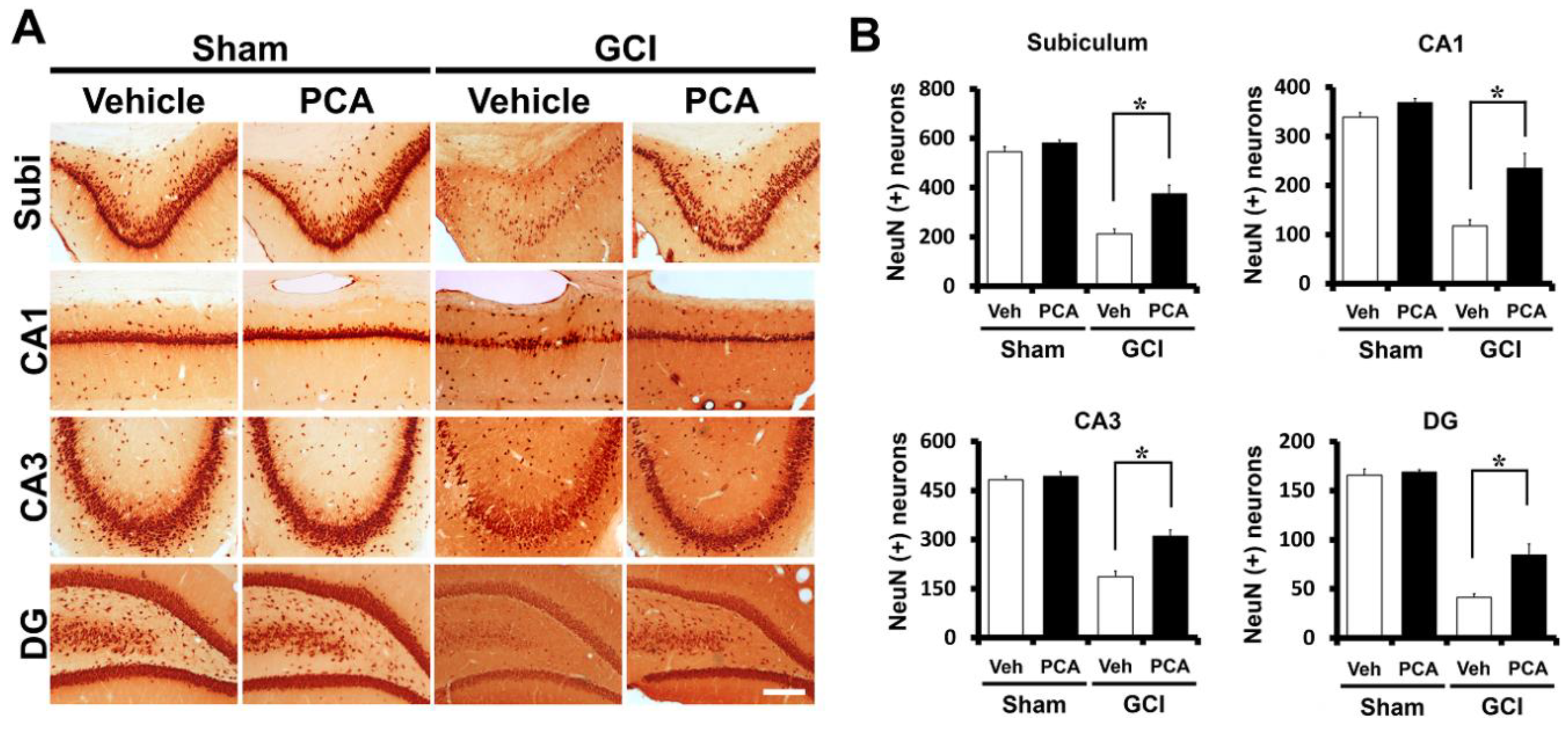
2.1. PCA Reduces Neuronal Death after Global Ischemia
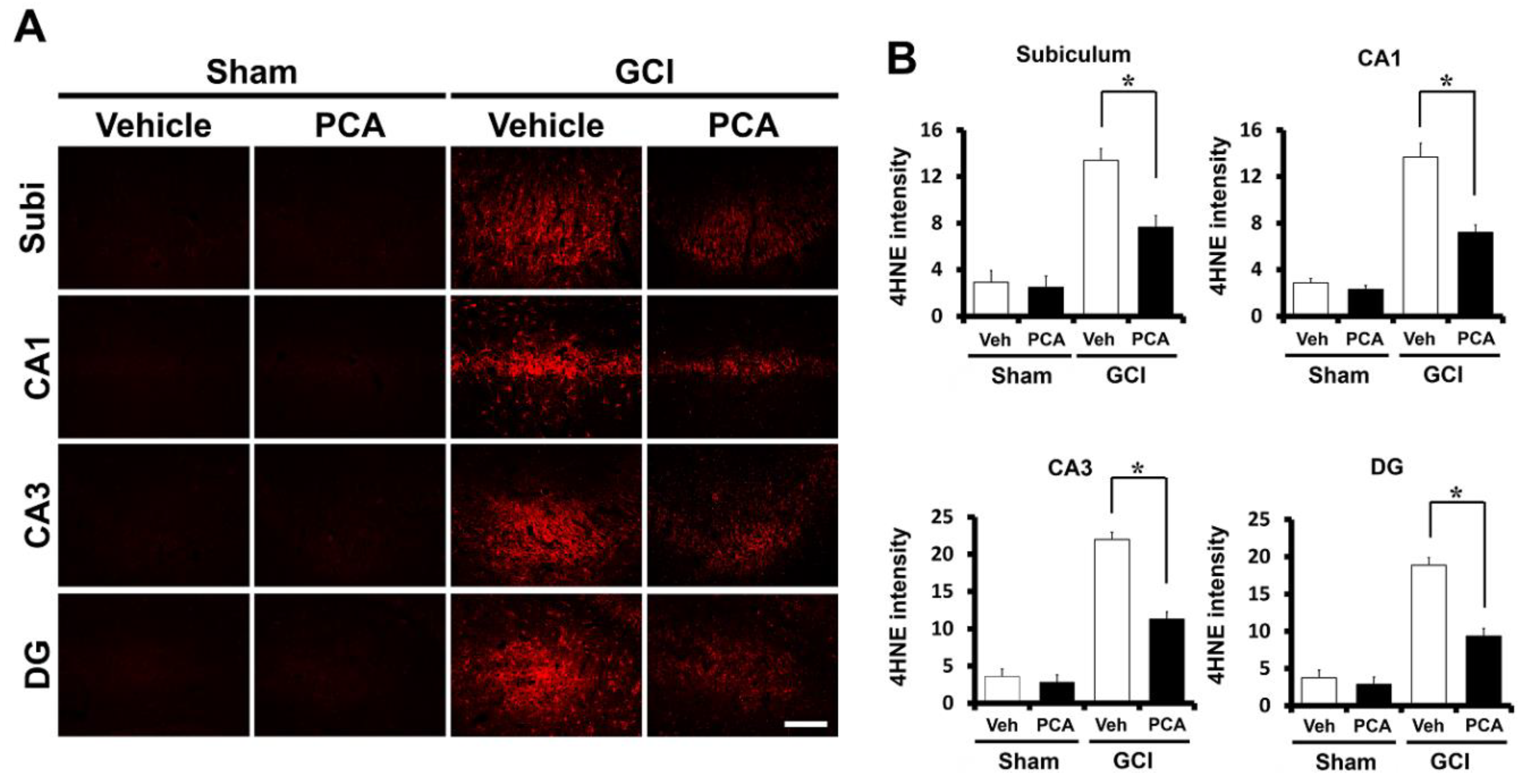
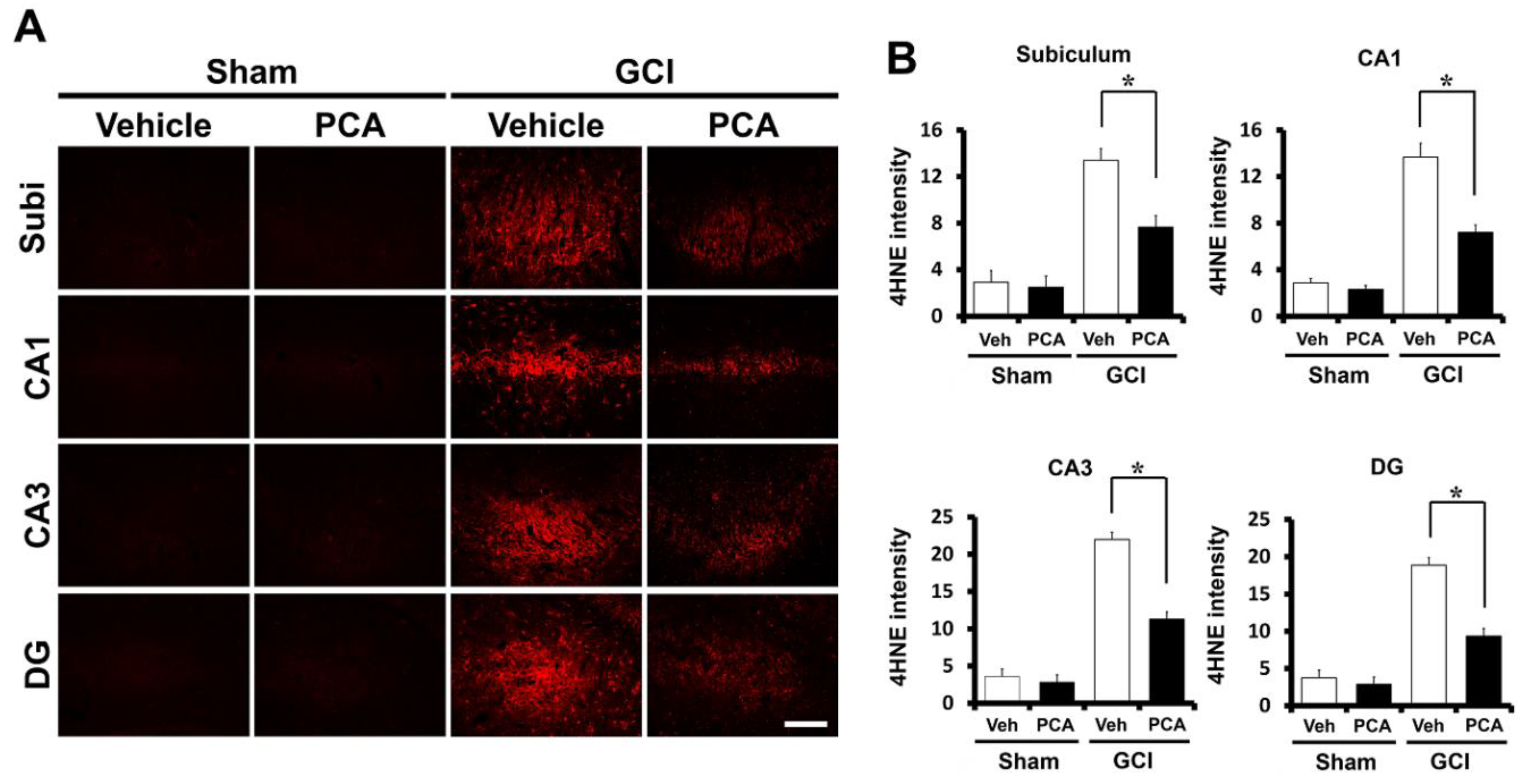
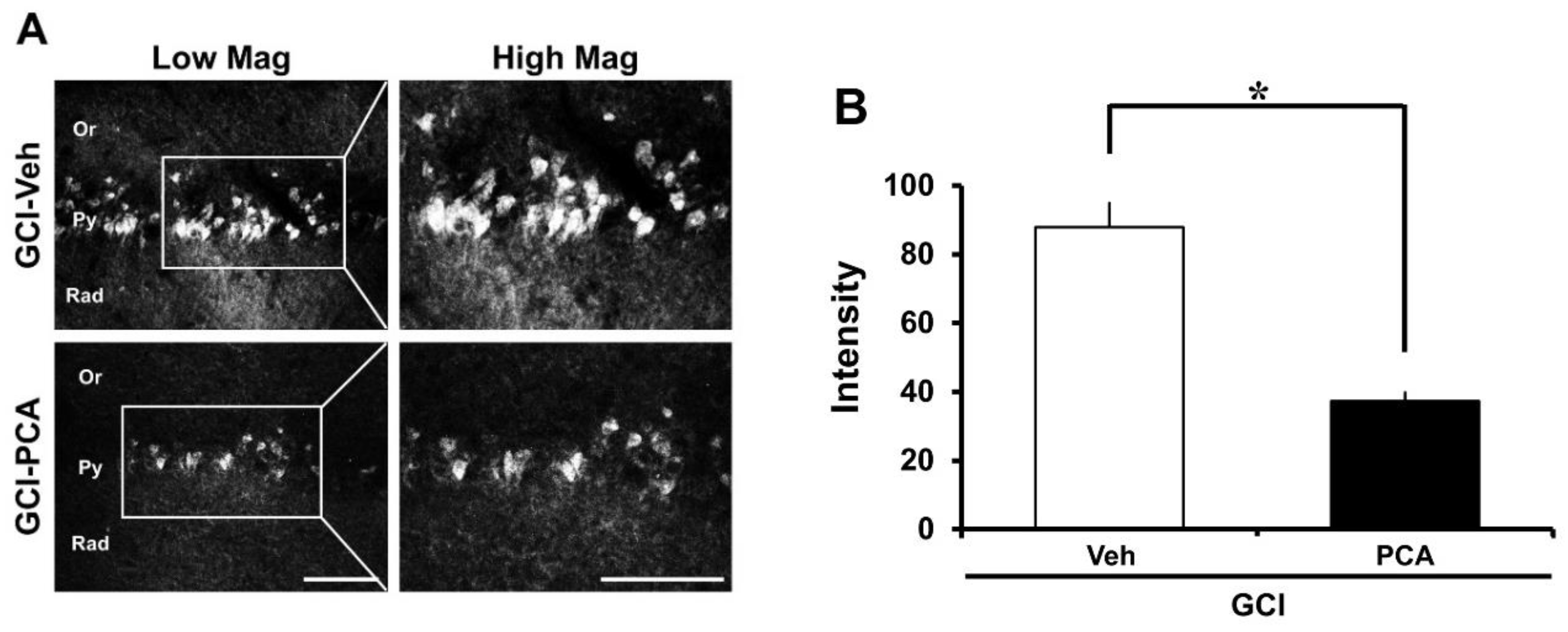
2.2. PCA Reduces Ischemia-Induced Oxidative Injury
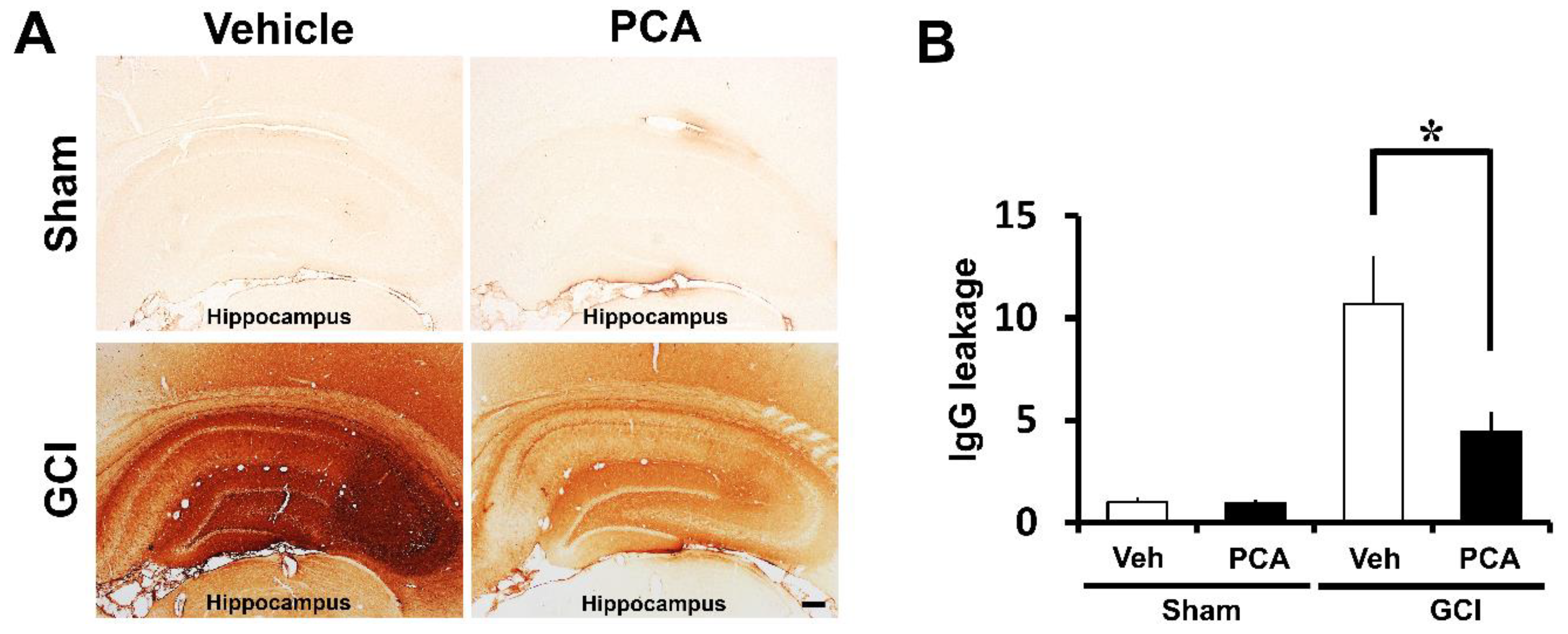
2.3. PCA Prevents Ischemia-Induced Blood–Brain Barrier (BBB) Disruption
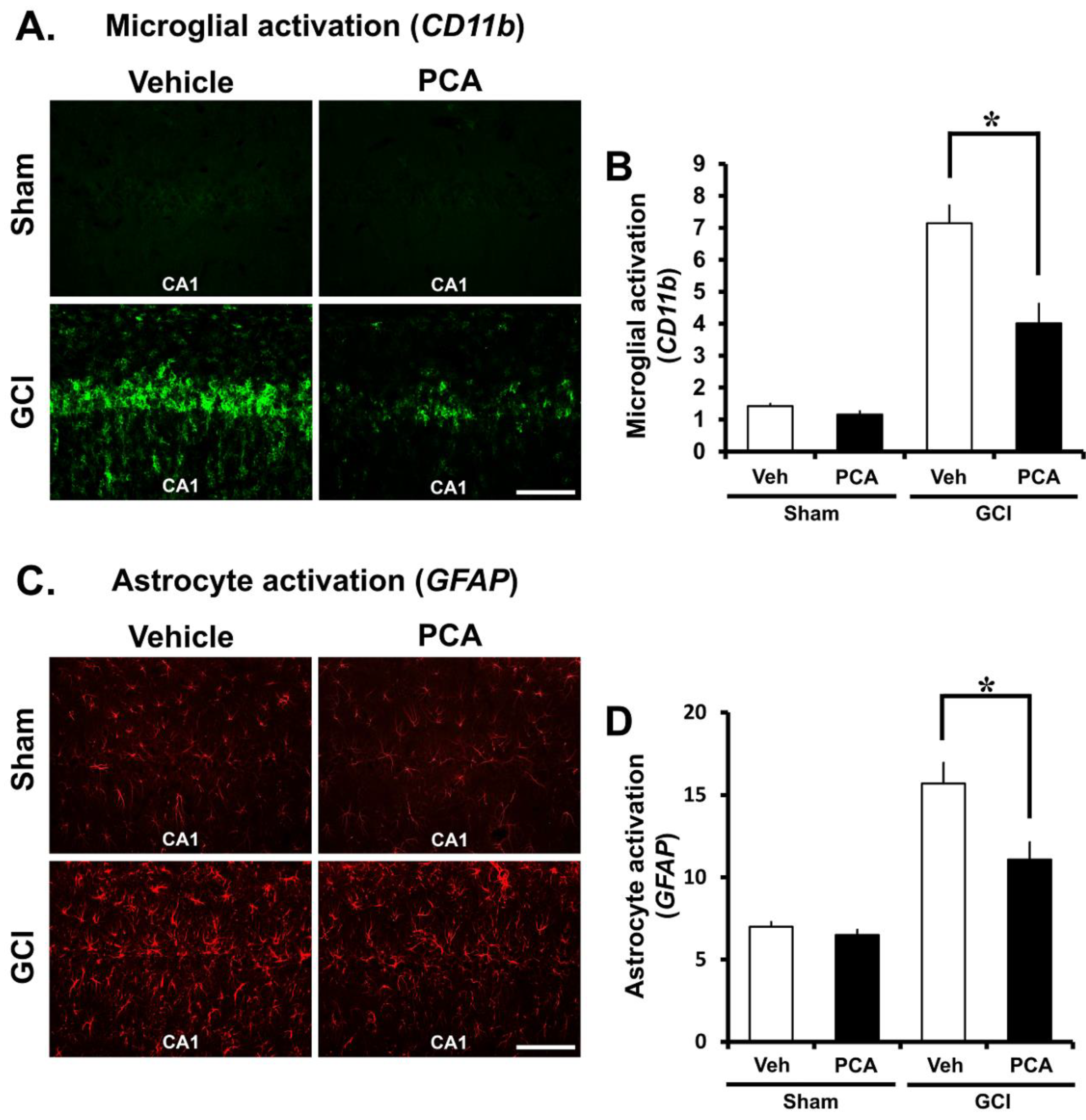
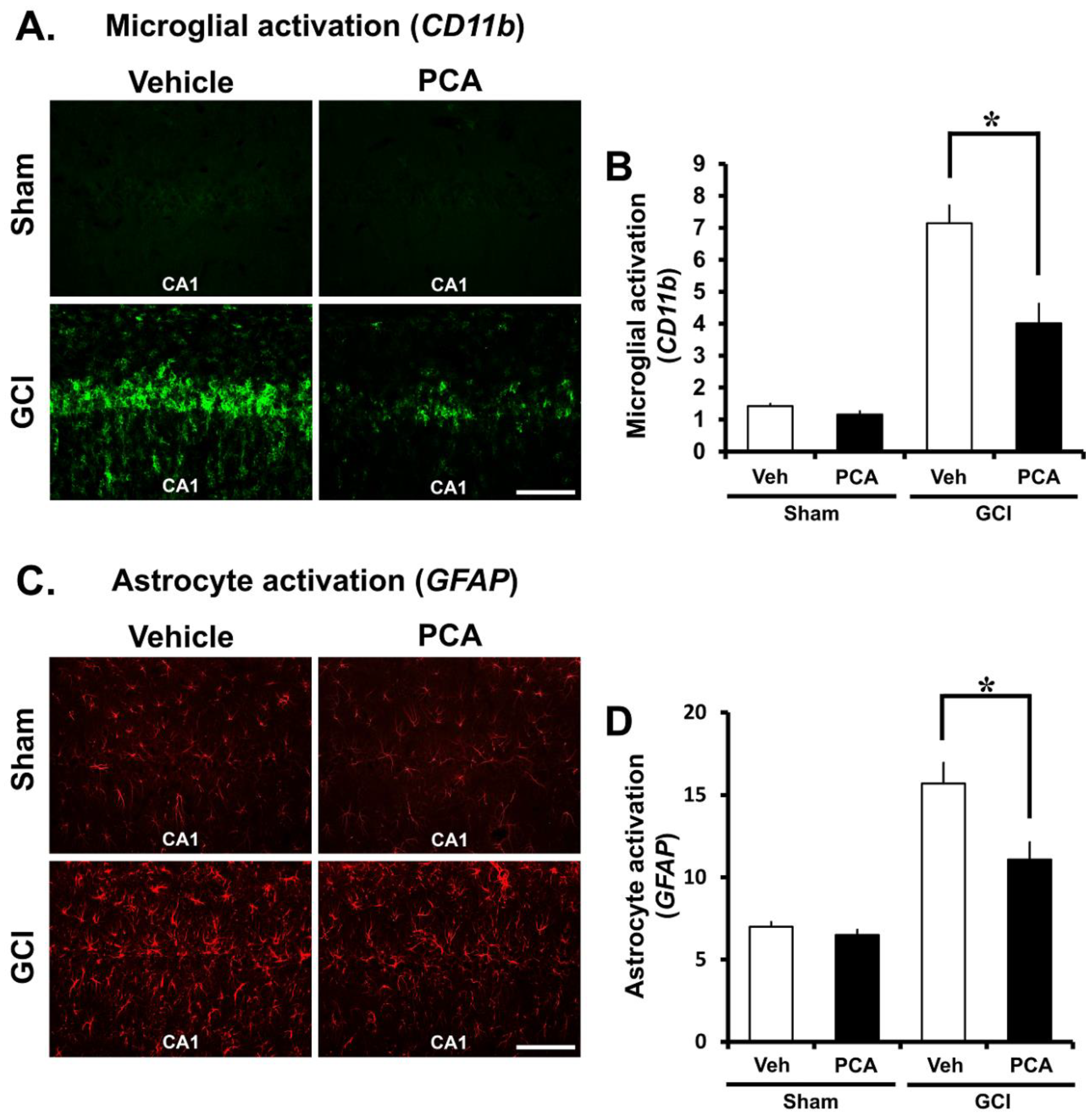
2.4. PCA Decreases Ischemia-Induced Inflammatory Responses Mediated by Microglia and Astrocytes
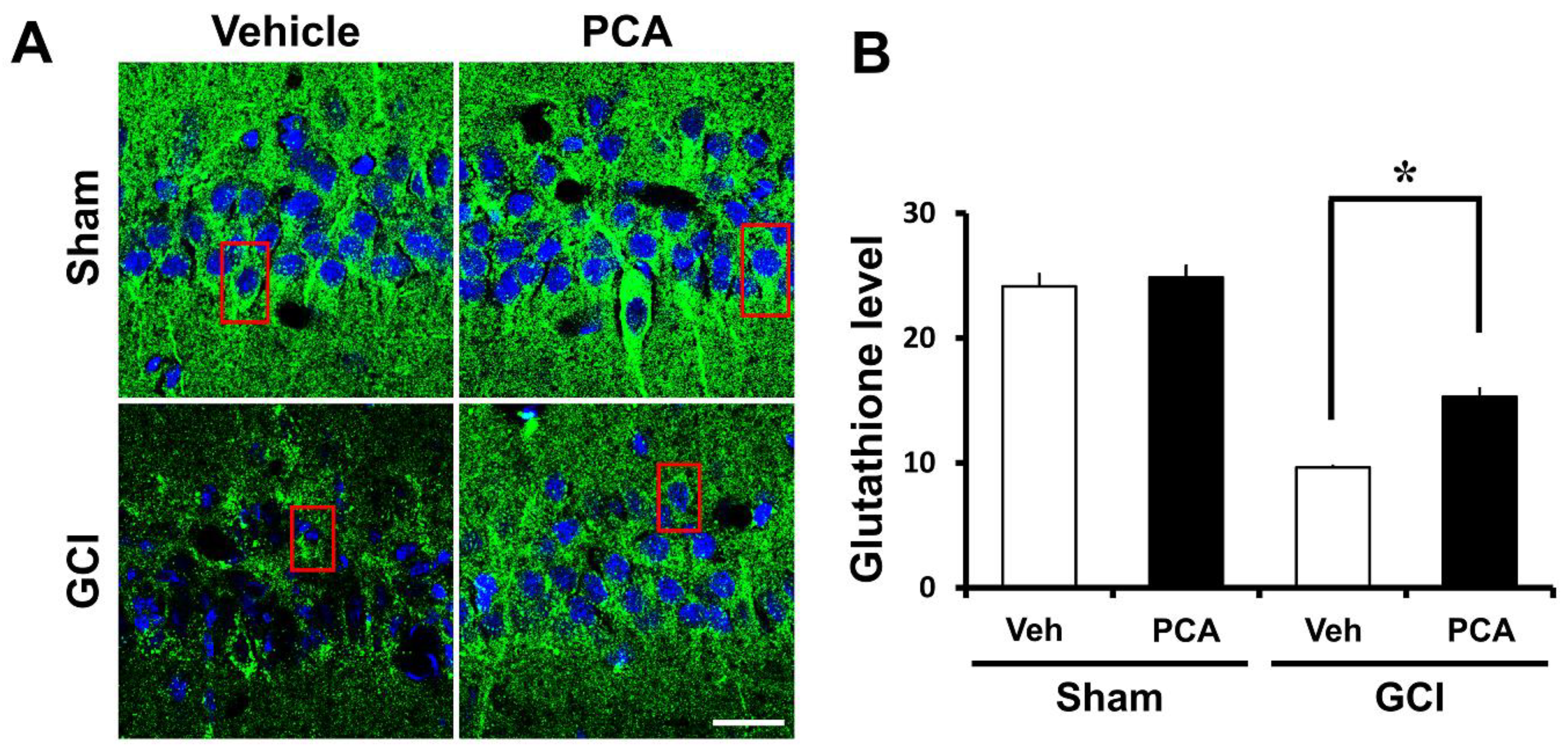
2.5. PCA Reverses Ischemia-Induced Glutathione (GSH) Deprivation
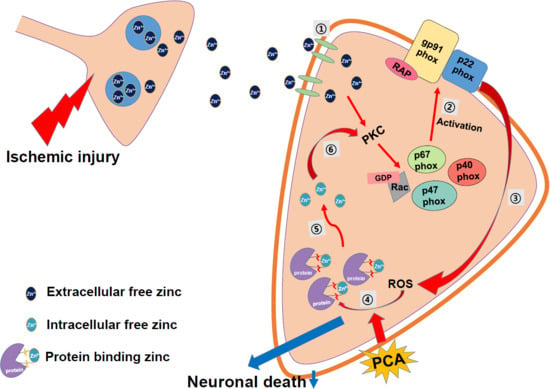
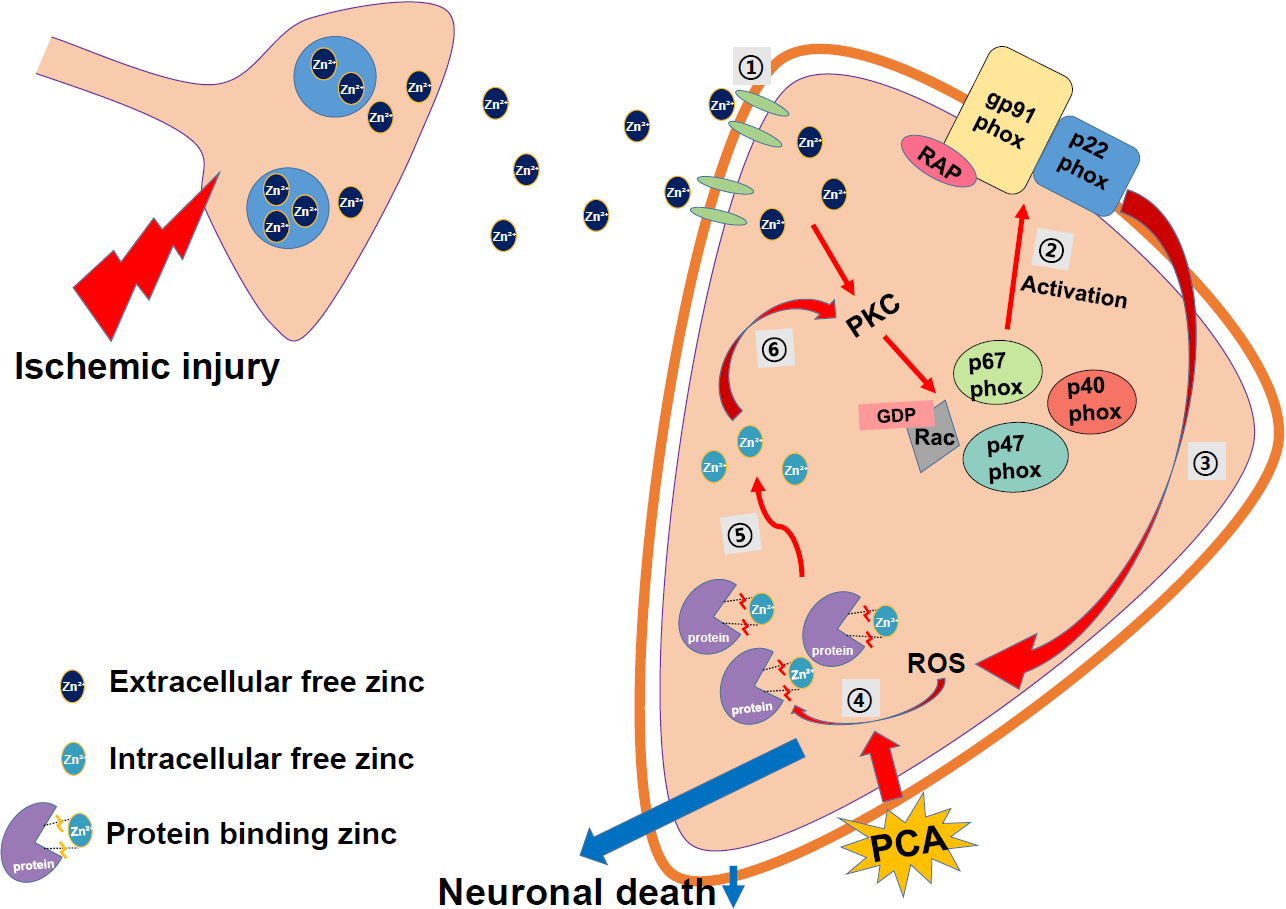
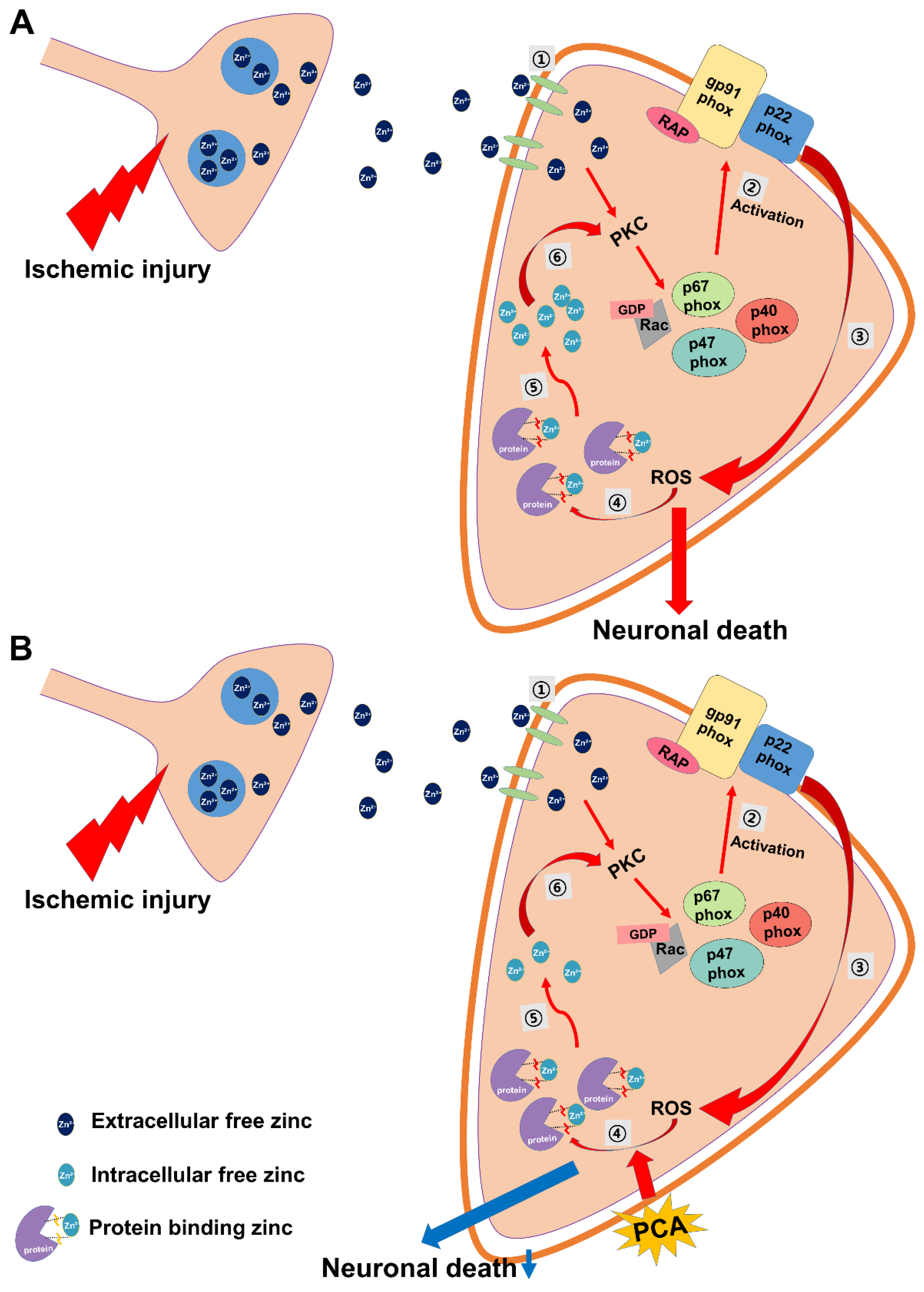
2.6. PCA Reduces Intracellular Free Zinc Level
2.7. PCA Improves Cognitive Function after Ischemia
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Experimental Animals
4.3. Global Cerebral Ischemia Surgery
4.4. PCA (Protocatechuic Acid) Administration
4.5. Brain Sample Procedure
4.6. Analysis of Hippocampal Neuronal Death
4.7. Analysis of Live Neurons
4.8. Analysis of Oxidative Stress
4.9. Analysis of Blood–Brain Barrier Disruption
4.10. Analysis of Microglia and Astrocytes Activation
4.11. Analysis of GSH Levels
4.12. Analysis of Intracellular Free Zinc Level
4.13. Behavioral Test
4.14. Data Analysis
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Ste-Marie, L.; Vachon, P.; Vachon, L.; Bemeur, C.; Guertin, M.C.; Montgomery, J. Hydroxyl radical production in the cortex and striatum in a rat model of focal cerebral ischemia. Can. J. Neurol. Sci. 2000, 27, 152–159. [Google Scholar] [PubMed]
- Wan, L.; Cheng, Y.; Luo, Z.; Guo, H.; Zhao, W.; Gu, Q.; Yang, X.; Xu, J.; Bei, W.; Guo, J. Neuroprotection, learning and memory improvement of a standardized extract from Renshen Shouwu against neuronal injury and vascular dementia in rats with brain ischemia. J. Ethnopharmacol. 2015, 165, 118–126. [Google Scholar] [CrossRef] [PubMed]
- Nagel, S.; Papadakis, M.; Hoyte, L.; Buchan, A.M. Therapeutic hypothermia in experimental models of focal and global cerebral ischemia and intracerebral hemorrhage. Expert Rev. Neurother. 2008, 8, 1255–1268. [Google Scholar] [CrossRef] [PubMed]
- Dirnagl, U.; Lindauer, U.; Them, A.; Schreiber, S.; Pfister, H.W.; Koedel, U.; Reszka, R.; Freyer, D.; Villringer, A. Global cerebral ischemia in the rat: Online monitoring of oxygen free radical production using chemiluminescence in vivo. J. Cereb. Blood Flow Metab. 1995, 15, 929–940. [Google Scholar] [CrossRef] [PubMed]
- Kakkar, S.; Bais, S. A review on protocatechuic Acid and its pharmacological potential. ISRN Pharmacol. 2014, 2014, 952943. [Google Scholar] [CrossRef] [PubMed]
- Adefegha, S.A.; Oboh, G.; Omojokun, O.S.; Adefegha, O.M. Alterations of Na+/K+-ATPase, cholinergic and antioxidant enzymes activity by protocatechuic acid in cadmium-induced neurotoxicity and oxidative stress in Wistar rats. Biomed. Pharmacother. 2016, 83, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Yuksel, M.; Yildar, M.; Basbug, M.; Cavdar, F.; Cikman, O.; Aksit, H.; Aslan, F.; Aksit, D. Does protocatechuic acid, a natural antioxidant, reduce renal ischemia reperfusion injury in rats? Ulus. Travma Acil. Cerrahi Derg. 2017, 23, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Li, G.; Szeto, S.S.W.; Chong, C.M.; Quan, Q.; Huang, C.; Cui, W.; Guo, B.; Wang, Y.; Han, Y.; et al. Examining the neuroprotective effects of protocatechuic acid and chrysin on in vitro and in vivo models of Parkinson disease. Free Radic. Biol. Med. 2015, 84, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Wang, J.M.; Chu, C.Y.; Cheng, M.T.; Tseng, T.H. In vivo protective effect of protocatechuic acid on tert-butyl hydroperoxide-induced rat hepatotoxicity. Food Chem. Toxicol. 2002, 40, 635–641. [Google Scholar] [CrossRef]
- Liu, W.H.; Lin, C.C.; Wang, Z.H.; Mong, M.C.; Yin, M.C. Effects of protocatechuic acid on trans fat induced hepatic steatosis in mice. J. Agric. Food Chem. 2010, 58, 10247–10252. [Google Scholar] [CrossRef] [PubMed]
- Cueva, C.; Mingo, S.; Munoz-Gonzalez, I.; Bustos, I.; Requena, T.; del Campo, R.; Martin-Alvarez, P.J.; Bartolome, B.; Moreno-Arribas, M.V. Antibacterial activity of wine phenolic compounds and oenological extracts against potential respiratory pathogens. Lett. Appl. Microbiol. 2012, 54, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kazlowska, K.; Hsu, T.; Hou, C.C.; Yang, W.C.; Tsai, G.J. Anti-inflammatory properties of phenolic compounds and crude extract from Porphyra dentata. J. Ethnopharmacol. 2010, 128, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Adefegha, S.A.; Omojokun, O.S.; Oboh, G. Modulatory effect of protocatechuic acid on cadmium induced nephrotoxicity and hepatoxicity in rats in vivo. Springerplus 2015, 4, 619. [Google Scholar] [CrossRef] [PubMed]
- Hoane, M.R.; Kaplan, S.A.; Ellis, A.L. The effects of nicotinamide on apoptosis and blood-brain barrier breakdown following traumatic brain injury. Brain Res. 2006, 1125, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Tang, X.N.; Berman, A.E.; Swanson, R.A.; Yenari, M.A. Digitally quantifying cerebral hemorrhage using Photoshop and Image J. J. Neurosci. Methods 2010, 190, 240–243. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Stoll, G.; Jander, S.; Schroeter, M. Inflammation and glial responses in ischemic brain lesions. Prog. Neurobiol. 1998, 56, 149–171. [Google Scholar] [CrossRef]
- Ali, S.M.; Dunn, E.; Oostveen, J.A.; Hall, E.D.; Carter, D.B. Induction of apolipoprotein E mRNA in the hippocampus of the gerbil after transient global ischemia. Brain Res. Mol. Brain Res. 1996, 38, 37–44. [Google Scholar] [CrossRef]
- Safonova, O.A.; Popova, T.N.; Kryl’skii, D.V. Glutathione System Activity in Rat Tissues under Phenylethyl Biguanide Action on the Background of Experimental Brain Ischemia/Reperfusion Development. Eksp. Klin. Farmakol. 2016, 79, 23–27. [Google Scholar] [PubMed]
- Foster, M.; Samman, S. Zinc and redox signaling: Perturbations associated with cardiovascular disease and diabetes mellitus. Antioxid. Redox Signal. 2010, 13, 1549–1573. [Google Scholar] [CrossRef] [PubMed]
- Freret, T.; Bouet, V.; Leconte, C.; Roussel, S.; Chazalviel, L.; Divoux, D.; Schumann-Bard, P.; Boulouard, M. Behavioral deficits after distal focal cerebral ischemia in mice: Usefulness of adhesive removal test. Behav. Neurosci. 2009, 123, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Aronowski, J.; Samways, E.; Strong, R.; Rhoades, H.M.; Grotta, J.C. An alternative method for the quantitation of neuronal damage after experimental middle cerebral artery occlusion in rats: Analysis of behavioral deficit. J. Cereb. Blood Flow Metab. 1996, 16, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Bouet, V.; Boulouard, M.; Toutain, J.; Divoux, D.; Bernaudin, M.; Schumann-Bard, P.; Freret, T. The adhesive removal test: A sensitive method to assess sensorimotor deficits in mice. Nat. Protoc. 2009, 4, 1560–1564. [Google Scholar] [CrossRef] [PubMed]
- Bouet, V.; Freret, T.; Toutain, J.; Divoux, D.; Boulouard, M.; Schumann-Bard, P. Sensorimotor and cognitive deficits after transient middle cerebral artery occlusion in the mouse. Exp. Neurol. 2007, 203, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Freret, T.; Chazalviel, L.; Roussel, S.; Bernaudin, M.; Schumann-Bard, P.; Boulouard, M. Long-term functional outcome following transient middle cerebral artery occlusion in the rat: Correlation between brain damage and behavioral impairment. Behav. Neurosci. 2006, 120, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Chen, J.; Li, Y.; Zhang, Z.G.; Chopp, M. Quantitative measurement of motor and somatosensory impairments after mild (30 min) and severe (2 h) transient middle cerebral artery occlusion in rats. J. Neurol. Sci. 2000, 174, 141–146. [Google Scholar] [CrossRef]
- Suh, S.W.; Shin, B.S.; Ma, H.; Van Hoecke, M.; Brennan, A.M.; Yenari, M.A.; Swanson, R.A. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann. Neurol. 2008, 64, 654–663. [Google Scholar] [CrossRef] [PubMed]
- Neumann, J.T.; Cohan, C.H.; Dave, K.R.; Wright, C.B.; Perez-Pinzon, M.A. Global cerebral ischemia: Synaptic and cognitive dysfunction. Curr. Drug Targets 2013, 14, 20–35. [Google Scholar] [CrossRef] [PubMed]
- Muley, M.M.; Thakare, V.N.; Patil, R.R.; Bafna, P.A.; Naik, S.R. Amelioration of cognitive, motor and endogenous defense functions with silymarin, piracetam and protocatechuic acid in the cerebral global ischemic rat model. Life Sci. 2013, 93, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.Y.; Lee, S.H.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Suh, S.W. Administration of Protocatechuic Acid Reduces Traumatic Brain Injury-Induced Neuronal Death. Int. J. Mol. Sci. 2017, 18, 2510. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Choi, B.Y.; Kho, A.R.; Jeong, J.H.; Hong, D.K.; Lee, S.H.; Lee, S.Y.; Lee, M.W.; Song, H.K.; Choi, H.C.; et al. Protective Effects of Protocatechuic Acid on Seizure-Induced Neuronal Death. Int. J. Mol. Sci. 2018, 19, 187. [Google Scholar] [CrossRef] [PubMed]
- Guan, S.; Ge, D.; Liu, T.Q.; Ma, X.H.; Cui, Z.F. Protocatechuic acid promotes cell proliferation and reduces basal apoptosis in cultured neural stem cells. Toxicol. In Vitro 2009, 23, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.N.; An, C.N.; Zhang, H.N.; Pu, X.P. Protocatechuic acid inhibits neurotoxicity induced by MPTP in vivo. Neurosci. Lett. 2010, 474, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.J.; Wu, L.; Zhang, Q.L.; Li, J.; Yin, F.X.; Yuan, Y. Pharmacokinetics of phenolic compounds of Danshen extract in rat blood and brain by microdialysis sampling. J. Ethnopharmacol. 2011, 136, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Hua, J.S.; Li, L.P.; Zhu, X.M. Effects of moxibustion pretreating on SOD and MDA in the rat of global brain ischemia. J. Tradit. Chin. Med. 2008, 28, 289–292. [Google Scholar] [CrossRef]
- An, L.J.; Guan, S.; Shi, G.F.; Bao, Y.M.; Duan, Y.L.; Jiang, B. Protocatechuic acid from Alpinia oxyphylla against MPP+-induced neurotoxicity in PC12 cells. Food Chem. Toxicol. 2006, 44, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Shi, G.F.; An, L.J.; Jiang, B.; Guan, S.; Bao, Y.M. Alpinia protocatechuic acid protects against oxidative damage in vitro and reduces oxidative stress in vivo. Neurosci. Lett. 2006, 403, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Crews, B.C.; Rowlinson, S.W.; Marnett, A.B.; Kozak, K.R.; Remmel, R.P.; Marnett, L.J. Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: Facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors. Proc. Natl. Acad. Sci. USA 2000, 97, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Das, K.K.; Das, S.N.; DasGupta, S. The influence of ascorbic acid on nickel-induced hepatic lipid peroxidation in rats. J. Basic Clin. Physiol. Pharmacol. 2001, 12, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.K.; Rashid, R.; Fatima, N.; Mahmood, S.; Mir, S.; Khan, S.; Jabeen, N.; Murtaza, G. Pharmacological Activities of Protocatechuic Acid. Acta Pol. Pharm. 2015, 72, 643–650. [Google Scholar] [PubMed]
- Abbott, N.J.; Patabendige, A.A.; Dolman, D.E.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Dalkara, T.; Gursoy-Ozdemir, Y.; Yemisci, M. Brain microvascular pericytes in health and disease. Acta Neuropathol. 2011, 122, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rosenberg, G.A. Blood-brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 2011, 42, 3323–3328. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Loane, D.J. Neuroinflammation after traumatic brain injury: Opportunities for therapeutic intervention. Brain Behav. Immun. 2012, 26, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.B.; Hochfelder, C.G.; Lamberty, B.G.; Meays, B.M.; Morsey, B.M.; Kelso, M.L.; Fox, H.S.; Yelamanchili, S.V. Traumatic brain injury increases levels of miR-21 in extracellular vesicles: Implications for neuroinflammation. FEBS Open Bio 2016, 6, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Britschgi, M.; Wyss-Coray, T. Systemic and acquired immune responses in Alzheimer’s disease. Int. Rev. Neurobiol. 2007, 82, 205–233. [Google Scholar] [PubMed]
- Woodruff, T.M.; Thundyil, J.; Tang, S.C.; Sobey, C.G.; Taylor, S.M.; Arumugam, T.V. Pathophysiology, treatment, and animal and cellular models of human ischemic stroke. Mol. Neurodegener. 2011, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Pham, L.D.; Maki, T.; Liang, A.C.; Arai, K. A radical scavenger edaravone inhibits matrix metalloproteinase-9 upregulation and blood-brain barrier breakdown in a mouse model of prolonged cerebral hypoperfusion. Neurosci. Lett. 2014, 573, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Sifat, A.E.; Vaidya, B.; Abbruscato, T.J. Blood-Brain Barrier Protection as a Therapeutic Strategy for Acute Ischemic Stroke. AAPS J. 2017, 19, 957–972. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Wang, X.; Yang, S.; Huang, J.; Xue, X.; Zheng, Y.; Shang, G.; Tao, J.; Chen, L. Electroacupunctre improves motor impairment via inhibition of microglia-mediated neuroinflammation in the sensorimotor cortex after ischemic stroke. Life Sci. 2016, 151, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Dvoriantchikova, G.; Barakat, D.; Brambilla, R.; Agudelo, C.; Hernandez, E.; Bethea, J.R.; Shestopalov, V.I.; Ivanov, D. Inactivation of astroglial NF-kappa B promotes survival of retinal neurons following ischemic injury. Eur. J. Neurosci. 2009, 30, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Sun, N.; Hao, J.R.; Li, X.Y.; Yin, X.H.; Zong, Y.Y.; Zhang, G.Y.; Gao, C. GluR6-FasL-Trx2 mediates denitrosylation and activation of procaspase-3 in cerebral ischemia/reperfusion in rats. Cell Death Dis. 2013, 4, e771. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Inhibition of GTRAP3-18 may increase neuroprotective glutathione (GSH) synthesis. Int. J. Mol. Sci. 2012, 13, 12017–12035. [Google Scholar] [CrossRef] [PubMed]
- Ohana, E.; Hoch, E.; Keasar, C.; Kambe, T.; Yifrach, O.; Hershfinkel, M.; Sekler, I. Identification of the Zn2+ binding site and mode of operation of a mammalian Zn2+ transporter. J. Biol. Chem. 2009, 284, 17677–17686. [Google Scholar] [CrossRef] [PubMed]
- Beyersmann, D.; Haase, H. Functions of zinc in signaling, proliferation and differentiation of mammalian cells. Biometals 2001, 14, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Miyagi, C.; Fukada, T.; Kagara, N.; Che, Y.S.; Hirano, T. Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 2004, 429, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, S.; Sakata-Sogawa, K.; Hasegawa, A.; Suzuki, T.; Kabu, K.; Sato, E.; Kurosaki, T.; Yamashita, S.; Tokunaga, M.; Nishida, K.; et al. Zinc is a novel intracellular second messenger. J. Cell Biol. 2007, 177, 637–645. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Jung, J.W.; Suh, S.W. The Emerging Role of Zinc in the Pathogenesis of Multiple Sclerosis. Int. J. Mol. Sci. 2017, 18, 2070. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.L.; Auer, R.N.; Siesjo, B.K. The density and distribution of ischemic brain injury in the rat following 2–10 min of forebrain ischemia. Acta Neuropathol. 1984, 64, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Auer, R.N.; Olsson, Y.; Siesjo, B.K. Hypoglycemic brain injury in the rat. Correlation of density of brain damage with the EEG isoelectric time: A quantitative study. Diabetes 1984, 33, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Schmued, L.C.; Hopkins, K.J. Fluoro-Jade B: A high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res. 2000, 874, 123–130. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Kim, J.H.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Sohn, M.; Suh, S.W. Prevention of hypoglycemia-induced hippocampal neuronal death by N-acetyl-l-cysteine (NAC). Amino Acids 2017, 49, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Suh, S.W.; Gum, E.T.; Hamby, A.M.; Chan, P.H.; Swanson, R.A. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J. Clin. Investig. 2007, 117, 910–918. [Google Scholar] [CrossRef] [PubMed]
- Ruth, R.E.; Feinerman, G.S. Foreign and endogenous serum protein extravasation during harmaline tremors or kainic acid seizures in the rat: A comparison. Acta Neuropathol. 1988, 76, 380–387. [Google Scholar] [CrossRef] [PubMed]
- Hsu, S.M.; Raine, L.; Fanger, H. Use of avidin-biotin-peroxidase complex (ABC) in immunoperoxidase techniques: A comparison between ABC and unlabeled antibody (PAP) procedures. J. Histochem. Cytochem. 1981, 29, 577–580. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Swanson, R.A. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J. Immunol. 2005, 174, 2288–2296. [Google Scholar] [CrossRef] [PubMed]
- Kauppinen, T.M.; Higashi, Y.; Suh, S.W.; Escartin, C.; Nagasawa, K.; Swanson, R.A. Zinc triggers microglial activation. J. Neurosci. 2008, 28, 5827–5835. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Kasarskis, E.J.; Ringo, D.; Frederickson, R.E. A quinoline fluorescence method for visualizing and assaying the histochemically reactive zinc (bouton zinc) in the brain. J. Neurosci. Methods 1987, 20, 91–103. [Google Scholar] [CrossRef]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.-H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. Int. J. Mol. Sci. 2018, 19, 1420. https://doi.org/10.3390/ijms19051420
Kho AR, Choi BY, Lee SH, Hong DK, Lee SH, Jeong JH, Park K-H, Song HK, Choi HC, Suh SW. Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. International Journal of Molecular Sciences. 2018; 19(5):1420. https://doi.org/10.3390/ijms19051420
Chicago/Turabian StyleKho, A Ra, Bo Young Choi, Song Hee Lee, Dae Ki Hong, Sang Hwon Lee, Jeong Hyun Jeong, Kyoung-Ha Park, Hong Ki Song, Hui Chul Choi, and Sang Won Suh. 2018. "Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death" International Journal of Molecular Sciences 19, no. 5: 1420. https://doi.org/10.3390/ijms19051420
APA StyleKho, A. R., Choi, B. Y., Lee, S. H., Hong, D. K., Lee, S. H., Jeong, J. H., Park, K.-H., Song, H. K., Choi, H. C., & Suh, S. W. (2018). Effects of Protocatechuic Acid (PCA) on Global Cerebral Ischemia-Induced Hippocampal Neuronal Death. International Journal of Molecular Sciences, 19(5), 1420. https://doi.org/10.3390/ijms19051420