Acetylation Disfavors Tau Phase Separation


 , , and
, , and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
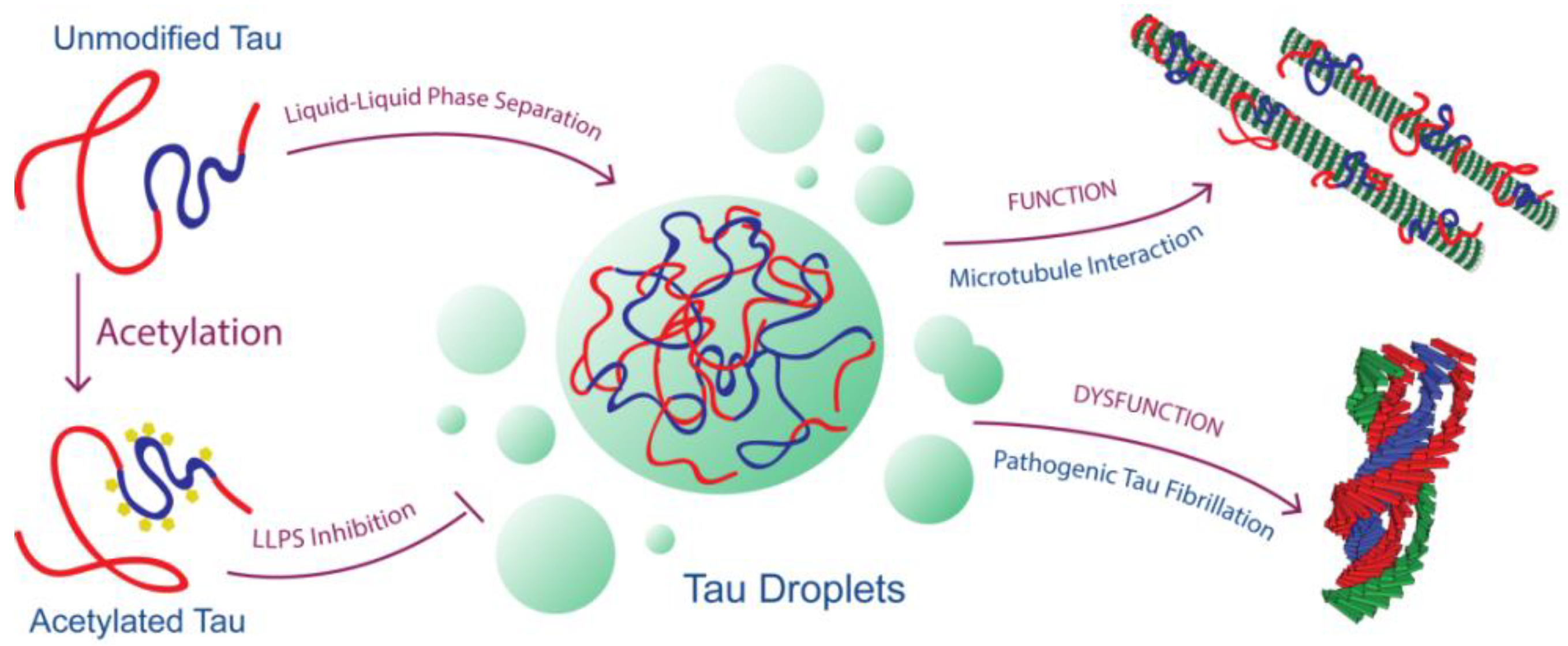{kind=link}
Abstract
1. Introduction
2. Results and Discussion
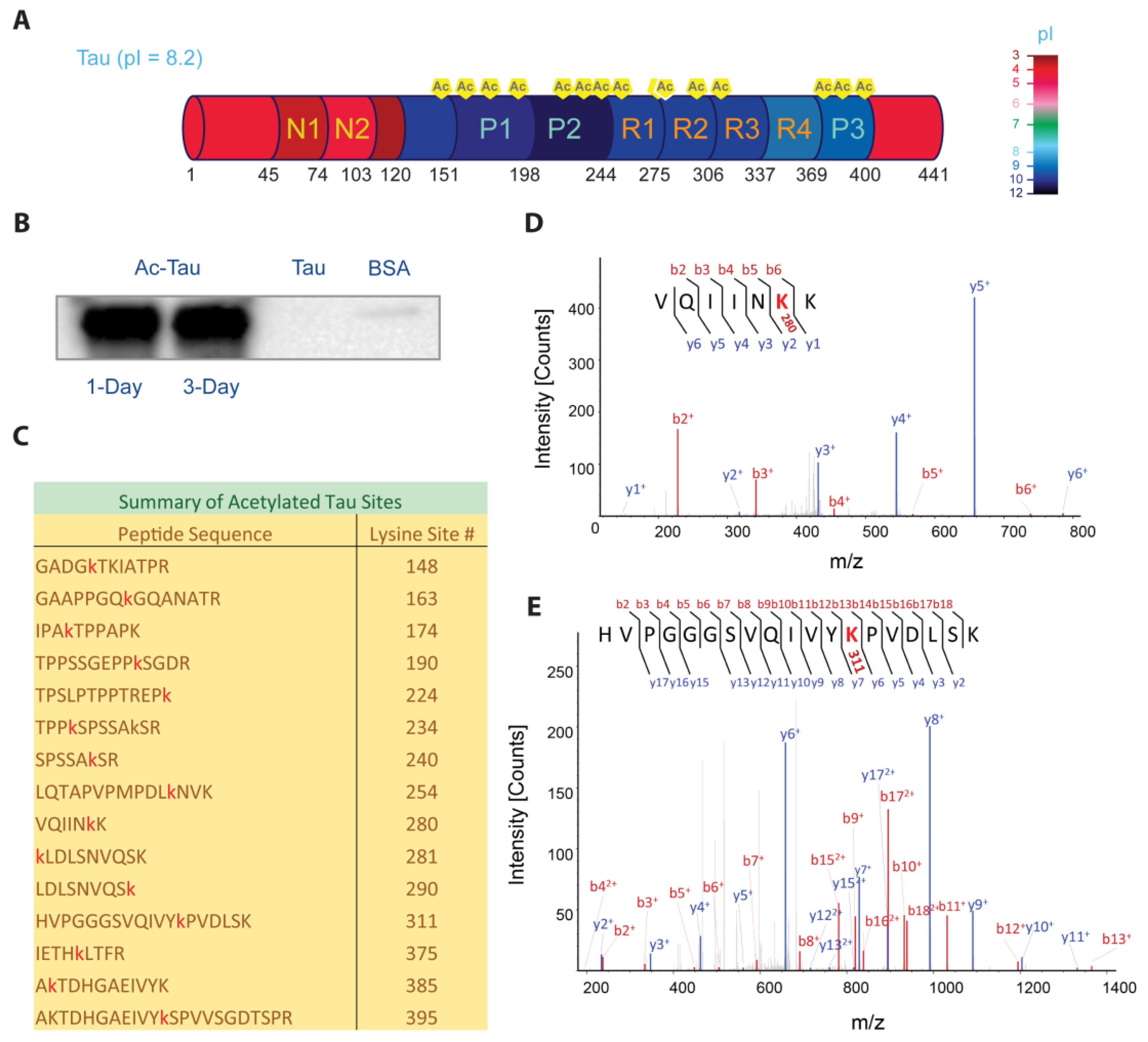
2.1. p300-Mediated Acetylation of Tau
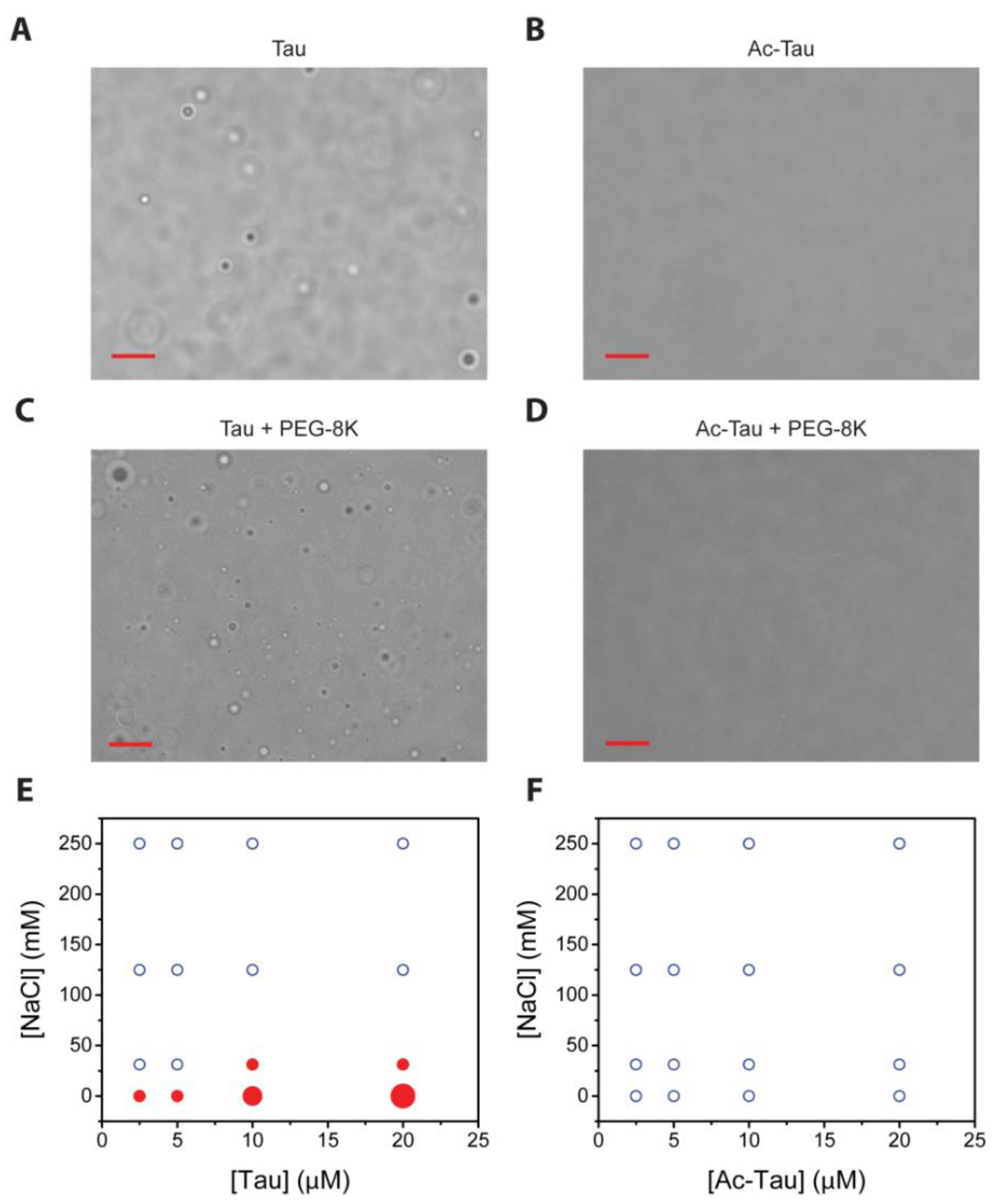
2.2. Acetylation Changes Tau Phase Behavior
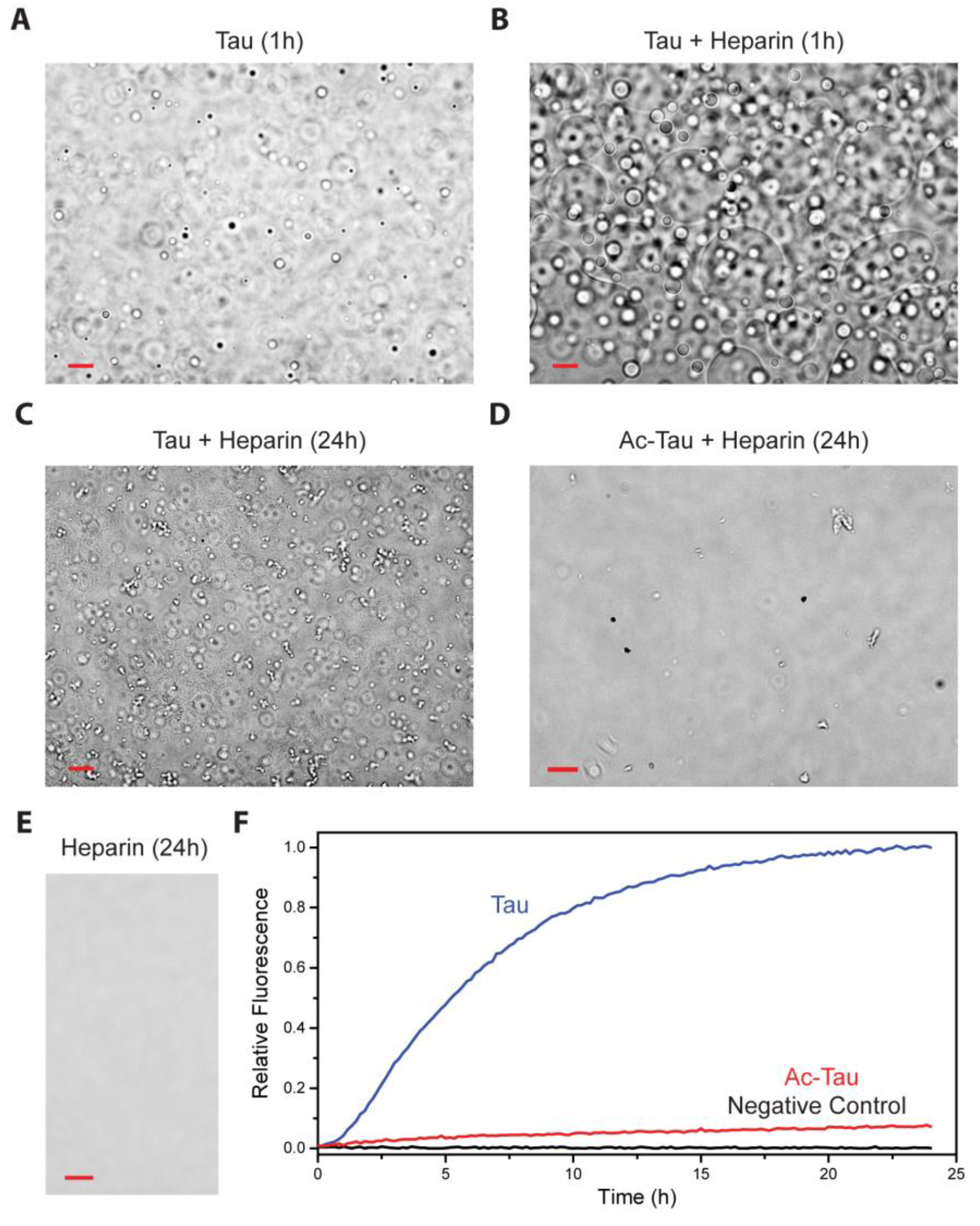
2.3. Acetylation of Tau Inhibits Heparin-Induced Aggregation
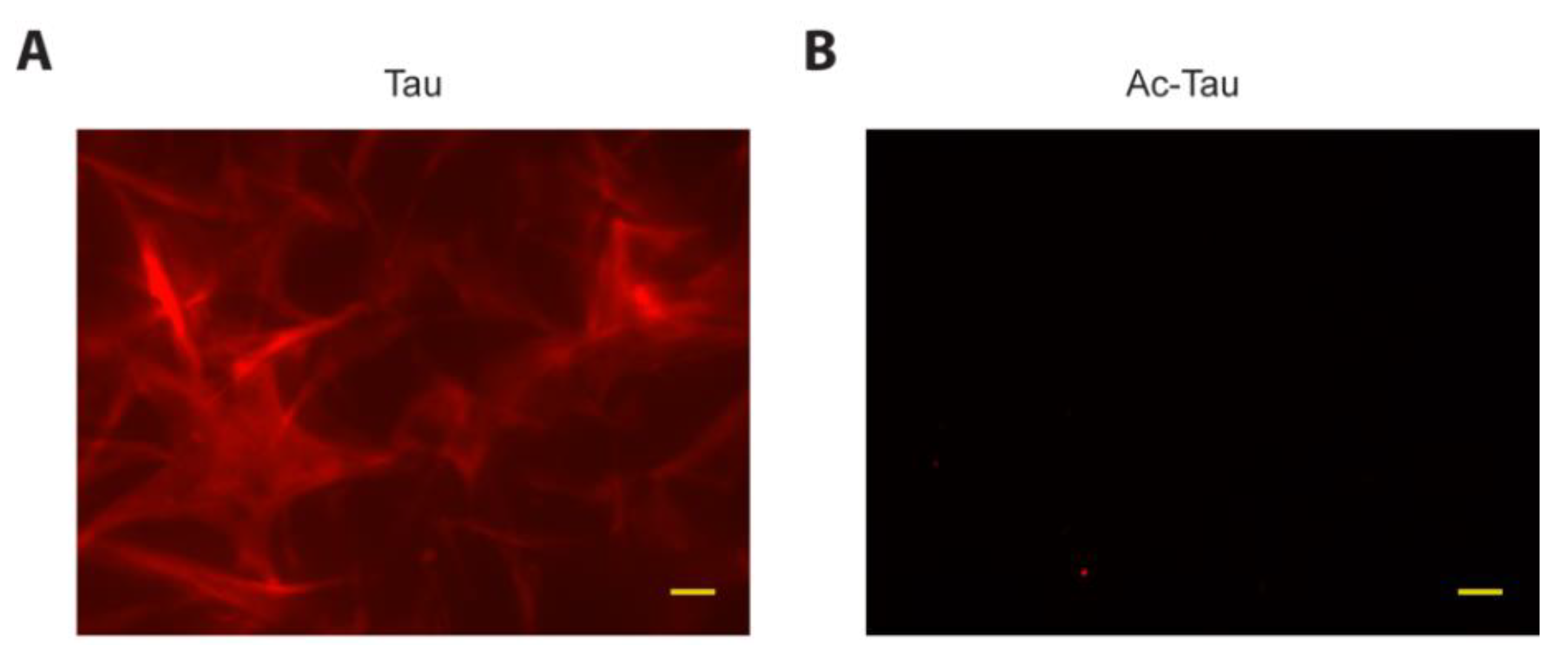
2.4. Acetylation of Tau Prevents Access to LLPS-Mediated Microtubule Assembly
3. Materials and Methods
3.1. Tau Expression and Purification
3.2. p300 Histone Acetyltransferase (HAT) Domain Expression and Purification
3.3. In Vitro p300 HAT-Mediated Acetylation Reactions
3.4. Western Blot of Acetylated Tau
3.5. Mass Spectrometry of Acetylated Tau
3.6. Microscopy Imaging
3.7. Thioflavin T (Th T) Aggregation Assay
3.8. Microtubule Assembly Assay
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| Ac-Tau | Hyperacetylated Tau |
| AD | Alzheimer’s disease |
| IDP | Intrinsically disordered protein |
| HAT | Histone acetyltransferase |
| LLPS | Liquid-liquid phase separation |
| PTMs | Post-translational modifications |
| wt | Wild-type |
References
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Dickson, D.W. Tau and synuclein and their role in neuropathology. Brain Pathol. 1999, 9, 657–661. [Google Scholar] [CrossRef] [PubMed]
- Holtzman, D.M.; Morris, J.C.; Goate, A.M. Alzheimer’s disease: The challenge of the second century. Sci. Transl. Med. 2011, 3, 77sr71. [Google Scholar] [CrossRef] [PubMed]
- Ghetti, B.; Oblak, A.L.; Boeve, B.F.; Johnson, K.A.; Dickerson, B.C.; Goedert, M. Invited review: Frontotemporal dementia caused by microtubule-associated protein tau gene (MAPT) mutations: A chameleon for neuropathology and neuroimaging. Neuropathol. Appl. Neurobiol. 2015, 41, 24–46. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Lee, V.M. Pathological tau: A loss of normal function or a gain in toxicity? Nat. Neurosci. 2005, 8, 1136–1137. [Google Scholar] [CrossRef] [PubMed]
- Feinstein, S.C.; Wilson, L. Inability of tau to properly regulate neuronal microtubule dynamics: A loss-of-function mechanism by which tau might mediate neuronal cell death. Biochim. Biophys. Acta 2005, 1739, 268–279. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, K.; Alonso Adel, C.; Chen, S.; Chohan, M.O.; El-Akkad, E.; Gong, C.X.; Khatoon, S.; Li, B.; Liu, F.; Rahman, A.; et al. Tau pathology in alzheimer disease and other tauopathies. Biochim. Biophys. Acta 2005, 1739, 198–210. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Lee, V.M. Characterization of two VQIXXK motifs for tau fibrillization in vitro. Biochemistry 2006, 45, 15692–15701. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Bibow, S.; Korukottu, J.; Jeganathan, S.; Biernat, J.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Structural polymorphism of 441-residue tau at single residue resolution. PLoS Biol. 2009, 7, e34. [Google Scholar] [CrossRef] [PubMed]
- Von Bergen, M.; Friedhoff, P.; Biernat, J.; Heberle, J.; Mandelkow, E.M.; Mandelkow, E. Assembly of tau protein into alzheimer paired helical filaments depends on a local sequence motif ((306)VQIVYK(311)) forming beta structure. Proc. Natl. Acad. Sci. USA 2000, 97, 5129–5134. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, G.; Udgaonkar, J.B. Mechanistic studies unravel the complexity inherent in tau aggregation leading to alzheimer’s disease and the tauopathies. Biochemistry 2013, 52, 4107–4126. [Google Scholar] [CrossRef] [PubMed]
- Cripps, D.; Thomas, S.N.; Jeng, Y.; Yang, F.; Davies, P.; Yang, A.J. Alzheimer disease-specific conformation of hyperphosphorylated paired helical filament-tau is polyubiquitinated through Lys-48, Lys-11, and Lys-6 ubiquitin conjugation. J. Biol. Chem. 2006, 281, 10825–10838. [Google Scholar] [CrossRef] [PubMed]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hubschmann, S.; Redecke, L.; Mandelkow, E.M.; Muller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Moran, C.R.; Ferreon, J.C.; Deniz, A.A. Alteration of the alpha-synuclein folding landscape by a mutation related to parkinson’s disease. Angew. Chem. Int. Ed. Engl. 2010, 49, 3469–3472. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, A.C.; Ferreon, J.C.; Wright, P.E.; Deniz, A.A. Modulation of allostery by protein intrinsic disorder. Nature 2013, 498, 390–394. [Google Scholar] [CrossRef] [PubMed]
- Ferreon, J.C.; Lee, C.W.; Arai, M.; Martinez-Yamout, M.A.; Dyson, H.J.; Wright, P.E. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl. Acad. Sci. USA 2009, 106, 6591–6596. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Latypova, X.; Terro, F. Post-translational modifications of tau protein: Implications for alzheimer’s disease. Neurochem. Int. 2011, 58, 458–471. [Google Scholar] [CrossRef] [PubMed]
- Kamah, A.; Huvent, I.; Cantrelle, F.X.; Qi, H.; Lippens, G.; Landrieu, I.; Smet-Nocca, C. Nuclear magnetic resonance analysis of the acetylation pattern of the neuronal tau protein. Biochemistry 2014, 53, 3020–3032. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the kxgs motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Min, S.W.; Chen, X.; Tracy, T.E.; Li, Y.; Zhou, Y.; Wang, C.; Shirakawa, K.; Minami, S.S.; Defensor, E.; Mok, S.A.; et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat. Med. 2015, 21, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Brangwynne, C.P. Phase transitions and size scaling of membrane-less organelles. J. Cell Biol. 2013, 203, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Feric, M.; Vaidya, N.; Harmon, T.S.; Mitrea, D.M.; Zhu, L.; Richardson, T.M.; Kriwacki, R.W.; Pappu, R.V.; Brangwynne, C.P. Coexisting liquid phases underlie nucleolar subcompartments. Cell 2016, 165, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Han, T.W.; Xie, S.; Shi, K.; Du, X.; Wu, L.C.; Mirzaei, H.; Goldsmith, E.J.; Longgood, J.; Pei, J.; et al. Cell-free formation of rna granules: Low complexity sequence domains form dynamic fibers within hydrogels. Cell 2012, 149, 753–767. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Banjade, S.; Cheng, H.C.; Kim, S.; Chen, B.; Guo, L.; Llaguno, M.; Hollingsworth, J.V.; King, D.S.; Banani, S.F.; et al. Phase transitions in the assembly of multivalent signalling proteins. Nature 2012, 483, 336–340. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Protter, D.S.; Rosen, M.K.; Parker, R. Formation and maturation of phase-separated liquid droplets by rna-binding proteins. Mol. Cell 2015, 60, 208–219. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.; Lee, H.O.; Jawerth, L.; Maharana, S.; Jahnel, M.; Hein, M.Y.; Stoynov, S.; Mahamid, J.; Saha, S.; Franzmann, T.M.; et al. A liquid-to-solid phase transition of the als protein fus accelerated by disease mutation. Cell 2015, 162, 1066–1077. [Google Scholar] [CrossRef] [PubMed]
- Conicella, A.E.; Zerze, G.H.; Mittal, J.; Fawzi, N.L. Als mutations disrupt phase separation mediated by alpha-helical structure in the TDP-43 low-complexity C-terminal domain. Structure 2016, 24, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.R.; King, O.D.; Shorter, J.; Gitler, A.D. Stress granules as crucibles of als pathogenesis. J. Cell Biol. 2013, 201, 361–372. [Google Scholar] [CrossRef] [PubMed]
- Tsoi, P.S.; Choi, K.J.; Leonard, P.G.; Sizovs, A.; Moosa, M.M.; MacKenzie, K.R.; Ferreon, J.C.; Ferreon, A.C.M. The N-terminal domain of ALS-linked TDP-43 assembles without misfolding. Angew. Chem. Int. Ed. Engl. 2017, 56, 12590–12593. [Google Scholar] [CrossRef] [PubMed]
- Molliex, A.; Temirov, J.; Lee, J.; Coughlin, M.; Kanagaraj, A.P.; Kim, H.J.; Mittag, T.; Taylor, J.P. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell 2015, 163, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Vega, A.; Braun, M.; Scharrel, L.; Jahnel, M.; Wegmann, S.; Hyman, B.T.; Alberti, S.; Diez, S.; Hyman, A.A. Local nucleation of microtubule bundles through tubulin concentration into a condensed tau phase. Cell Rep. 2017, 20, 2304–2312. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Ambadipudi, S.; Biernat, J.; Riedel, D.; Mandelkow, E.; Zweckstetter, M. Liquid-liquid phase separation of the microtubule-binding repeats of the alzheimer-related protein tau. Nat. Commun. 2017, 8, 275. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lin, Y.; Eschmann, N.A.; Zhou, H.; Rauch, J.N.; Hernandez, I.; Guzman, E.; Kosik, K.S.; Han, S. Rna stores tau reversibly in complex coacervates. PLoS Biol. 2017, 15, e2002183. [Google Scholar] [CrossRef] [PubMed]
- Mukrasch, M.D.; Biernat, J.; von Bergen, M.; Griesinger, C.; Mandelkow, E.; Zweckstetter, M. Sites of tau important for aggregation populate {beta}-structure and bind to microtubules and polyanions. J. Biol. Chem. 2005, 280, 24978–24986. [Google Scholar] [CrossRef] [PubMed]
- Fitzpatrick, A.W.P.; Falcon, B.; He, S.; Murzin, A.G.; Murshudov, G.; Garringer, H.J.; Crowther, R.A.; Ghetti, B.; Goedert, M.; Scheres, S.H.W. Cryo-em structures of tau filaments from alzheimer’s disease. Nature 2017, 547, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.J.; Lee, J.H.; Rubinsztein, D.C. Tau degradation: The ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 2013, 105, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. Sirt1 deacetylase protects against neurodegeneration in models for alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Barghorn, S.; Davies, P.; Mandelkow, E. Tau paired helical filaments from alzheimer’s disease brain and assembled in vitro are based on beta-structure in the core domain. Biochemistry 2004, 43, 1694–1703. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.R.; Wang, D.; Wang, L.; Fulco, M.; Pediconi, N.; Zhang, D.; An, W.; Ge, Q.; Roeder, R.G.; Wong, J.; et al. Regulation of the p300 hat domain via a novel activation loop. Nat. Struct. Mol. Biol. 2004, 11, 308–315. [Google Scholar] [CrossRef] [PubMed]





© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreon, J.C.; Jain, A.; Choi, K.-J.; Tsoi, P.S.; MacKenzie, K.R.; Jung, S.Y.; Ferreon, A.C. Acetylation Disfavors Tau Phase Separation. Int. J. Mol. Sci. 2018, 19, 1360. https://doi.org/10.3390/ijms19051360
Ferreon JC, Jain A, Choi K-J, Tsoi PS, MacKenzie KR, Jung SY, Ferreon AC. Acetylation Disfavors Tau Phase Separation. International Journal of Molecular Sciences. 2018; 19(5):1360. https://doi.org/10.3390/ijms19051360
Chicago/Turabian StyleFerreon, Josephine C., Antrix Jain, Kyoung-Jae Choi, Phoebe S. Tsoi, Kevin R. MacKenzie, Sung Yun Jung, and Allan Chris Ferreon. 2018. "Acetylation Disfavors Tau Phase Separation" International Journal of Molecular Sciences 19, no. 5: 1360. https://doi.org/10.3390/ijms19051360
APA StyleFerreon, J. C., Jain, A., Choi, K.-J., Tsoi, P. S., MacKenzie, K. R., Jung, S. Y., & Ferreon, A. C. (2018). Acetylation Disfavors Tau Phase Separation. International Journal of Molecular Sciences, 19(5), 1360. https://doi.org/10.3390/ijms19051360