Recent Advances in Prostate Cancer Treatment and Drug Discovery


Abstract
1. Introduction
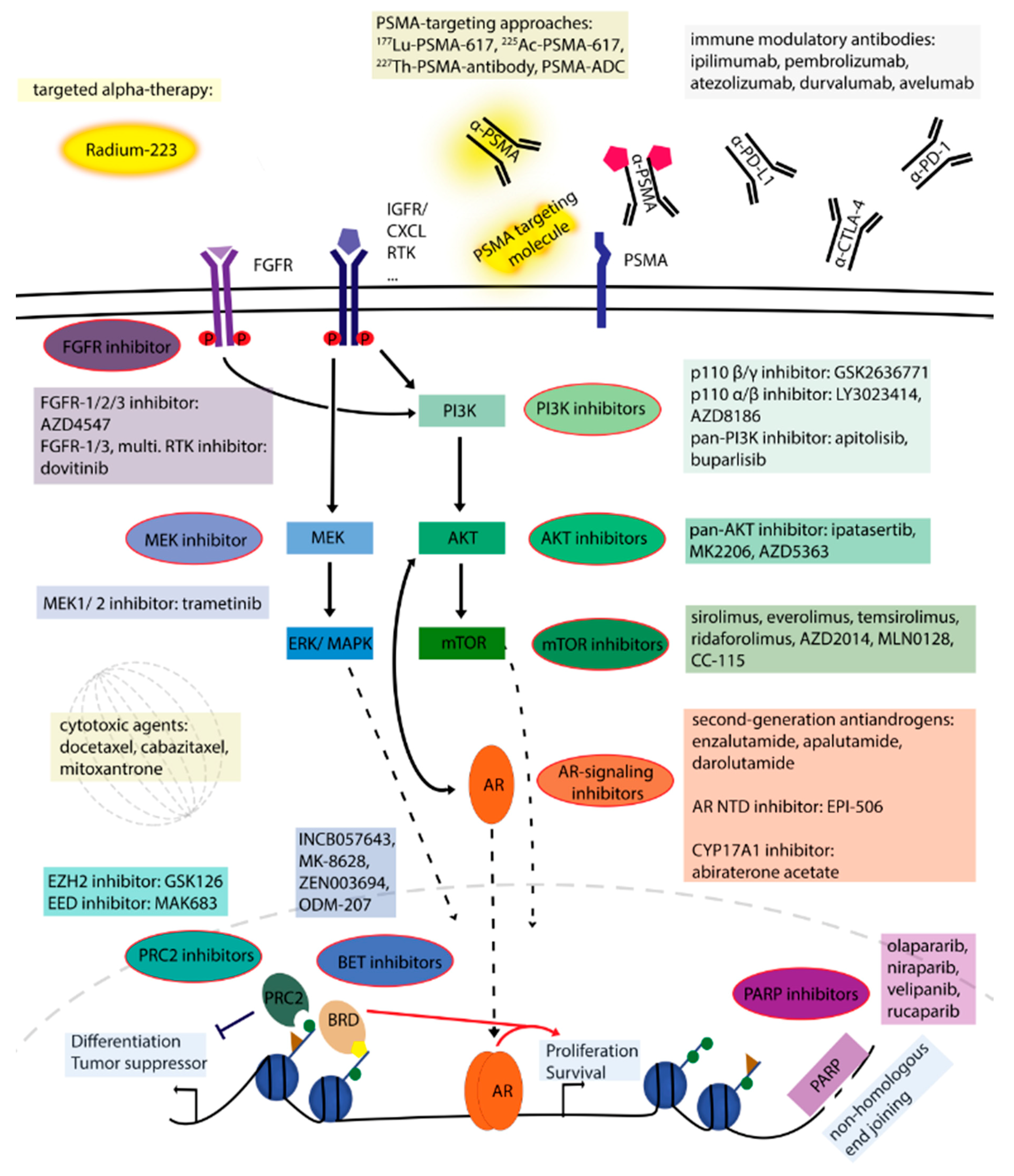
2. Treatment Options and Potential Novel Therapies
2.1. Compounds Targeting Androgen Signaling
2.2. Signaling Pathway Inhibitors
2.3. DNA Damage Repair Pathway
2.4. Epigenetic Mechanisms
2.5. Targeted Alpha Therapy Approach
2.6. Prostate-Specific Membrane Antigen (PSMA) Targeting Approaches
2.7. Chemotherapy
2.8. Immunotherapy
3. Advances in Molecular Characterization of Prostate Cancer through “Omics” Technologies
3.1. Primary Prostate Cancer
3.2. Advanced Prostate Cancer
3.3. Clonal Evolution and Dynamics
3.4. Proteomics and Metabolomics
4. Conclusions and Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Dai, C.; Heemers, H.; Sharifi, N. Androgen signaling in prostate cancer. Cold Spring Harb. Perspect. Med. 2017, 7, a030452. [Google Scholar] [CrossRef] [PubMed]
- Yap, T.A.; Smith, A.D.; Ferraldeschi, R.; Al-Lazikani, B.; Workman, P.; de Bono, J.S. Drug discovery in advanced prostate cancer: Translating biology into therapy. Nat. Rev. Drug Discov. 2016, 15, 699–718. [Google Scholar] [CrossRef] [PubMed]
- Nadal, M.; Prekovic, S.; Gallastegui, N.; Helsen, C.; Abella, M.; Zielinska, K.; Gay, M.; Vilaseca, M.; Taules, M.; Houtsmuller, A.B.; et al. Structure of the homodimeric androgen receptor ligand-binding domain. Nat. Commun. 2017, 8, 14388. [Google Scholar] [CrossRef] [PubMed]
- Van Royen, M.E.; van Cappellen, W.A.; de Vos, C.; Houtsmuller, A.B.; Trapman, J. Stepwise androgen receptor dimerization. J. Cell Sci. 2012, 125, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Grosse, A.; Bartsch, S.; Baniahmad, A. Androgen receptor-mediated gene repression. Mol. Cell. Endocrinol. 2012, 352, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Van der Steen, T.; Tindall, D.J.; Huang, H. Posttranslational modification of the androgen receptor in prostate cancer. Int. J. Mol. Sci. 2013, 14, 14833–14859. [Google Scholar] [CrossRef] [PubMed]
- Faus, H.; Haendler, B. Post-translational modifications of steroid receptors. Biomed. Pharmacother. Biomed. Pharmacother. 2006, 60, 520–528. [Google Scholar] [CrossRef] [PubMed]
- Wadosky, K.M.; Koochekpour, S. Molecular mechanisms underlying resistance to androgen deprivation therapy in prostate cancer. Oncotarget 2016, 7, 64447–64470. [Google Scholar] [CrossRef] [PubMed]
- Keyes, M.; Crook, J.; Morton, G.; Vigneault, E.; Usmani, N.; Morris, W.J. Treatment options for localized prostate cancer. Can. Fam. Phys. Med. Fam. Can. 2013, 59, 1269–1274. [Google Scholar]
- Sumanasuriya, S.; de Bono, J. Treatment of advanced prostate cancer-A review of current therapies and future promise. Cold Spring Harb. Perspect. Med. 2017. [Google Scholar] [CrossRef] [PubMed]
- De Maeseneer, D.J.; van Praet, C.; Lumen, N.; Rottey, S. Battling resistance mechanisms in antihormonal prostate cancer treatment: Novel agents and combinations. Urol. Oncol. 2015, 33, 310–321. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, I.; Day, T.K.; Tilley, W.D.; Selth, L.A. Androgen receptor signaling in castration-resistant prostate cancer: A lesson in persistence. Endocr.-Relat. Cancer 2016, 23, T179–T197. [Google Scholar] [CrossRef] [PubMed]
- Kluetz, P.G.; Pierce, W.; Maher, V.E.; Zhang, H.; Tang, S.; Song, P.; Liu, Q.; Haber, M.T.; Leutzinger, E.E.; Al-Hakim, A.; et al. Radium Ra 223 dichloride injection: U.S. Food and Drug Administration drug approval summary. Clin. Cancer Res. 2014, 20, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017, 9, E34. [Google Scholar] [CrossRef] [PubMed]
- Wise, H.M.; Hermida, M.A.; Leslie, N.R. Prostate cancer, PI3K, PTEN and prognosis. Clin. Sci. 2017, 131, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Schweizer, M.T.; Antonarakis, E.S. Prognostic and therapeutic implications of DNA repair gene mutations in advanced prostate cancer. Clin. Adv. Hematol. Oncol. 2017, 15, 785–795. [Google Scholar] [PubMed]
- Ramakrishnan Geethakumari, P.; Schiewer, M.J.; Knudsen, K.E.; Kelly, W.K. PARP inhibitors in prostate cancer. Curr. Treat. Opt. Oncol. 2017, 18, 37. [Google Scholar] [CrossRef] [PubMed]
- Ngollo, M.; Dagdemir, A.; Karsli-Ceppioglu, S.; Judes, G.; Pajon, A.; Penault-Llorca, F.; Boiteux, J.P.; Bignon, Y.J.; Guy, L.; Bernard-Gallon, D.J. Epigenetic modifications in prostate cancer. Epigenomics 2014, 6, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Nowacka-Zawisza, M.; Wisnik, E. DNA methylation and histone modifications as epigenetic regulation in prostate cancer. Oncol. Rep. 2017, 38, 2587–2596. [Google Scholar] [CrossRef] [PubMed]
- Massie, C.E.; Mills, I.G.; Lynch, A.G. The importance of DNA methylation in prostate cancer development. J. Ster. Biochem. Mol. Biol. 2017, 166, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Gelato, K.A.; Shaikhibrahim, Z.; Ocker, M.; Haendler, B. Targeting epigenetic regulators for cancer therapy: Modulation of bromodomain proteins, methyltransferases, demethylases, and microRNAs. Expert Opin. Ther. Targets 2016, 20, 783–799. [Google Scholar] [CrossRef] [PubMed]
- Davies, M. How checkpoint inhibitors are changing the treatment paradigm in solid tumors: What advanced practitioners in oncology need to know. J. Adv. Pract. Oncol. 2016, 7, 498–509. [Google Scholar] [PubMed]
- Alaia, C.; Boccellino, M.; Zappavigna, S.; Amler, E.; Quagliuolo, L.; Rossetti, S.; Facchini, G.; Caraglia, M. Ipilimumab for the treatment of metastatic prostate cancer. Expert Opin. Biol. Ther. 2018, 18, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, T.; Neal, D.E. The genomic evolution of human prostate cancer. Br. J. Cancer 2015, 113, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Research Network. The molecular taxonomy of primary prostate cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Armenia, J.; Wankowicz, S.A.M.; Liu, D.; Gao, J.; Kundra, R.; Reznik, E.; Chatila, W.K.; Chakravarty, D.; Han, G.C.; Coleman, I.; et al. The long tail of oncogenic drivers in prostate cancer. Nat. Genet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Bangma, C.H.; Bjartell, A.; Catto, J.W.; Culig, Z.; Gronberg, H.; Luo, J.; Visakorpi, T.; Rubin, M.A. The mutational landscape of prostate cancer. Eur. Urol. 2013, 64, 567–576. [Google Scholar] [CrossRef] [PubMed]
- You, S.; Knudsen, B.S.; Erho, N.; Alshalalfa, M.; Takhar, M.; Al-Deen Ashab, H.; Davicioni, E.; Karnes, R.J.; Klein, E.A.; Den, R.B.; et al. Integrated classification of prostate cancer reveals a novel luminal subtype with poor outcome. Cancer Res. 2016, 76, 4948–4958. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.G.; Chang, S.L.; Erho, N.; Yu, M.; Lehrer, J.; Alshalalfa, M.; Speers, C.; Cooperberg, M.R.; Kim, W.; Ryan, C.J.; et al. Associations of luminal and basal subtyping of prostate cancer with prognosis and response to androgen deprivation therapy. JAMA Oncol. 2017, 3, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Bluemn, E.G.; Coleman, I.M.; Lucas, J.M.; Coleman, R.T.; Hernandez-Lopez, S.; Tharakan, R.; Bianchi-Frias, D.; Dumpit, R.F.; Kaipainen, A.; Corella, A.N.; et al. Androgen receptor pathway-independent prostate cancer is sustained through FGF signaling. Cancer Cell 2017, 32, 474–489. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Abiraterone acetate: A review in metastatic castration-resistant prostrate cancer. Drugs 2017, 77, 1565–1576. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Spears, M.R.; Sydes, M.R. Abiraterone in metastatic prostate cancer. N. Engl. J. Med. 2017, 377, 1696–1697. [Google Scholar] [PubMed]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Ozguroglu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone plus prednisone in metastatic, castration-sensitive prostate cancer. N. Engl. J. Med. 2017, 377, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Rydzewska, L.H.M.; Burdett, S.; Vale, C.L.; Clarke, N.W.; Fizazi, K.; Kheoh, T.; Mason, M.D.; Miladinovic, B.; James, N.D.; Parmar, M.K.B.; et al. Adding abiraterone to androgen deprivation therapy in men with metastatic hormone-sensitive prostate cancer: A systematic review and meta-analysis. Eur. J. Cancer 2017, 84, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Alex, A.B.; Pal, S.K.; Agarwal, N. CYP17 inhibitors in prostate cancer: Latest evidence and clinical potential. Ther. Adv. Med. Oncol. 2016, 8, 267–275. [Google Scholar] [CrossRef] [PubMed]
- Ciccarese, C.; Nobili, E.; Grilli, D.; Casolari, L.; Rihawi, K.; Gelsomino, F.; Tortora, G.; Massari, F. The safety and efficacy of enzalutamide in the treatment of advanced prostate cancer. Expert Rev. Anticancer Ther. 2016, 16, 681–696. [Google Scholar] [CrossRef] [PubMed]
- Moreira, R.B.; Debiasi, M.; Francini, E.; Nuzzo, P.V.; Velasco, G.; Maluf, F.C.; Fay, A.P.; Bellmunt, J.; Choueiri, T.K.; Schutz, F.A. Differential side effects profile in patients with mCRPC treated with abiraterone or enzalutamide: A meta-analysis of randomized controlled trials. Oncotarget 2017, 8, 84572–84578. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Liao, R.; Su, C.; Liang, D.; Wu, J.; Qiu, K.; Li, J. Toxicity profile characteristics of novel androgen-deprivation therapy agents in patients with prostate cancer: A meta-analysis. Expert Rev. Anticancer Ther. 2018, 18, 193–198. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Laviec, H.; Castel, H.; Heutte, N.; Leconte, A.; Leger, I.; Giffard, B.; Capel, A.; Dubois, M.; Clarisse, B.; et al. Impact of new generation hormone-therapy on cognitive function in elderly patients treated for a metastatic prostate cancer: Cog-Pro trial protocol. BMC Cancer 2017, 17, 549. [Google Scholar] [CrossRef] [PubMed]
- Schepisi, G.; Farolfi, A.; Conteduca, V.; Martignano, F.; De Lisi, D.; Ravaglia, G.; Rossi, L.; Menna, C.; Bellia, S.R.; Barone, D.; et al. Immunotherapy for prostate cancer: Where we are headed. Int. J. Mol. Sci. 2017, 18, E2627. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.E.; Antonarakis, E.S.; Shore, N.D.; Tutrone, R.F.; Alumkal, J.J.; Ryan, C.J.; Saleh, M.; Hauke, R.J.; Bandekar, R.; Maneval, E.C.; et al. Safety and antitumor activity of apalutamide (ARN-509) in metastatic castration-resistant prostate cancer with and without prior abiraterone acetate and prednisone. Clin. Cancer Res. 2017, 23, 3544–3551. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide treatment and metastasis-free survival in prostate cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Fizazi, K.; Albiges, L.; Loriot, Y.; Massard, C. ODM-201: A new-generation androgen receptor inhibitor in castration-resistant prostate cancer. Expert Rev. Anticancer Ther. 2015, 15, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Shore, N.D. Darolutamide (ODM-201) for the treatment of prostate cancer. Expert Opin. Pharmacother. 2017, 18, 945–952. [Google Scholar] [CrossRef] [PubMed]
- Moilanen, A.M.; Riikonen, R.; Oksala, R.; Ravanti, L.; Aho, E.; Wohlfahrt, G.; Nykanen, P.S.; Tormakangas, O.P.; Palvimo, J.J.; Kallio, P.J. Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies. Sci. Rep. 2015, 5, 12007. [Google Scholar] [CrossRef] [PubMed]
- Borgmann, H.; Lallous, N.; Ozistanbullu, D.; Beraldi, E.; Paul, N.; Dalal, K.; Fazli, L.; Haferkamp, A.; Lejeune, P.; Cherkasov, A.; et al. Moving towards precision urologic oncology: Targeting enzalutamide-resistant prostate cancer and mutated forms of the androgen receptor using the novel inhibitor darolutamide (ODM-201). Eur. Urol. 2018, 73, 4–8. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Banuelos, C.A.; Mawji, N.R.; Wang, J.; Kato, M.; Haile, S.; McEwan, I.J.; Plymate, S.; Sadar, M.D. Targeting androgen receptor activation function-1 with EPI to overcome resistance mechanisms in castration-resistant prostate cancer. Clin. Cancer Res. 2016, 22, 4466–4477. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Chandhasin, C.; Osbourne, E.; Luo, J.; Sadar, M.D.; Perabo, F. Targeting the N-terminal domain of the androgen receptor: A new approach for the treatment of advanced prostate cancer. Oncologist 2016, 21, 1427–1435. [Google Scholar] [CrossRef] [PubMed]
- Dalal, K.; Che, M.; Que, N.S.; Sharma, A.; Yang, R.; Lallous, N.; Borgmann, H.; Ozistanbullu, D.; Tse, R.; Ban, F.; et al. Bypassing drug resistance mechanisms of prostate cancer with small molecules that target androgen receptor-chromatin interactions. Mol. Cancer Ther. 2017, 16, 2281–2291. [Google Scholar] [CrossRef] [PubMed]
- Munuganti, R.S.; Hassona, M.D.; Leblanc, E.; Frewin, K.; Singh, K.; Ma, D.; Ban, F.; Hsing, M.; Adomat, H.; Lallous, N.; et al. Identification of a potent antiandrogen that targets the BF3 site of the androgen receptor and inhibits enzalutamide-resistant prostate cancer. Chem. Biol. 2014, 21, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; de Stanchina, E.; Baselga, J.; et al. Feedback suppression of PI3Kalpha signaling in PTEN-mutated tumors is relieved by selective inhibition of PI3Kbeta. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef] [PubMed]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, M.A.; Kura, Y.; Yoshikawa, K.; Nishio, K.; Davies, B.R.; Uemura, H. Efficacy of targeted AKT inhibition in genetically engineered mouse models of PTEN-deficient prostate cancer. Oncotarget 2016, 7, 15959–15976. [Google Scholar] [CrossRef] [PubMed]
- Hancox, U.; Cosulich, S.; Hanson, L.; Trigwell, C.; Lenaghan, C.; Ellston, R.; Dry, H.; Crafter, C.; Barlaam, B.; Fitzek, M.; et al. Inhibition of PI3Kbeta signaling with AZD8186 inhibits growth of PTEN-deficient breast and prostate tumors alone and in combination with docetaxel. Mol. Cancer Ther. 2015, 14, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wallin, J.J.; Edgar, K.A.; Guan, J.; Berry, M.; Prior, W.W.; Lee, L.; Lesnick, J.D.; Lewis, C.; Nonomiya, J.; Pang, J.; et al. GDC-0980 is a novel class I PI3K/mTOR kinase inhibitor with robust activity in cancer models driven by the PI3K pathway. Mol. Cancer Ther. 2011, 10, 2426–2436. [Google Scholar] [CrossRef] [PubMed]
- Marques, R.B.; Aghai, A.; de Ridder, C.M.A.; Stuurman, D.; Hoeben, S.; Boer, A.; Ellston, R.P.; Barry, S.T.; Davies, B.R.; Trapman, J.; et al. High efficacy of combination therapy using PI3K/AKT inhibitors with androgen deprivation in prostate cancer preclinical models. Eur. Urol. 2015, 67, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Chee, K.G.; Longmate, J.; Quinn, D.I.; Chatta, G.; Pinski, J.; Twardowski, P.; Pan, C.X.; Cambio, A.; Evans, C.P.; Gandara, D.R.; et al. The AKT inhibitor perifosine in biochemically recurrent prostate cancer: A phase II California/Pittsburgh cancer consortium trial. Clin. Genitourin. Cancer 2007, 5, 433–437. [Google Scholar] [CrossRef] [PubMed]
- Massard, C.; Chi, K.N.; Castellano, D.; de Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Statz, C.M.; Patterson, S.E.; Mockus, S.M. mTOR inhibitors in castration-resistant prostate cancer: A Systematic review. Target. Oncol. 2017, 12, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Saura, C.; Roda, D.; Rosello, S.; Oliveira, M.; Macarulla, T.; Perez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-Garcia, J.M.; Musib, L.; Budha, N.; et al. A first-in-human phase I study of the ATP-competitive AKT inhibitor ipatasertib demonstrates robust and safe targeting of AKT in patients with solid tumors. Cancer Discov. 2017, 7, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, D.N.; Boysen, G.; Sumanasuriya, S.; Seed, G.; Marzo, A.M.; de Bono, J. The molecular underpinnings of prostate cancer: Impacts on management and pathology practice. J. Pathol. 2017, 241, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Carracedo, A.; Ma, L.; Teruya-Feldstein, J.; Rojo, F.; Salmena, L.; Alimonti, A.; Egia, A.; Sasaki, A.T.; Thomas, G.; Kozma, S.C.; et al. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J. Clin. Investig. 2008, 118, 3065–3074. [Google Scholar] [CrossRef] [PubMed]
- Butler, D.E.; Marlein, C.; Walker, H.F.; Frame, F.M.; Mann, V.M.; Simms, M.S.; Davies, B.R.; Collins, A.T.; Maitland, N.J. Inhibition of the PI3K/AKT/mTOR pathway activates autophagy and compensatory Ras/Raf/MEK/ERK signalling in prostate cancer. Oncotarget 2017, 8, 56698–56713. [Google Scholar] [CrossRef] [PubMed]
- Corn, P.G.; Wang, F.; McKeehan, W.L.; Navone, N. Targeting fibroblast growth factor pathways in prostate cancer. Clin. Cancer Res. 2013, 19, 5856–5866. [Google Scholar] [CrossRef] [PubMed]
- Gallick, G.E.; Corn, P.G.; Zurita, A.J.; Lin, S.H. Small-molecule protein tyrosine kinase inhibitors for the treatment of metastatic prostate cancer. Fut. Med. Chem. 2012, 4, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Shao, L.; Castro, P.; Coleman, I.; Nelson, P.S.; Smith, P.D.; Davies, B.R.; Ittmann, M. Combination treatment of prostate cancer with FGF receptor and AKT kinase inhibitors. Oncotarget 2017, 8, 6179–6192. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.J.; Kim, H.S.; Park, S.H.; Kim, B.S.; Kim, K.H.; Lee, H.J.; Song, H.S.; Shin, D.Y.; Lee, H.Y.; Kim, H.G.; et al. Phase II study of dovitinib in patients with castration-resistant prostate cancer (KCSG-GU11-05). Cancer Res. Treat. 2018. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.A.; Logan, S.K. Revisiting the role of Wnt/beta-catenin signaling in prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Canesin, G.; Evans-Axelsson, S.; Hellsten, R.; Krzyzanowska, A.; Prasad, C.P.; Bjartell, A.; Andersson, T. Treatment with the WNT5A-mimicking peptide Foxy-5 effectively reduces the metastatic spread of WNT5A-low prostate cancer cells in an orthotopic mouse model. PLoS ONE 2017, 12, e0184418. [Google Scholar] [CrossRef] [PubMed]
- Jefferies, M.T.; Cox, A.C.; Shorning, B.Y.; Meniel, V.; Griffiths, D.; Kynaston, H.G.; Smalley, M.J.; Clarke, A.R. PTEN loss and activation of K-RAS and beta-catenin cooperate to accelerate prostate tumourigenesis. J. Pathol. 2017, 243, 442–456. [Google Scholar] [CrossRef] [PubMed]
- Syed Khaja, A.S.; Helczynski, L.; Edsjo, A.; Ehrnstrom, R.; Lindgren, A.; Ulmert, D.; Andersson, T.; Bjartell, A. Elevated level of Wnt5a protein in localized prostate cancer tissue is associated with better outcome. PLoS ONE 2011, 6, e26539. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Dehm, S.M.; Hillman, D.W.; Sicotte, H.; Tan, W.; Gormley, M.; Bhargava, V.; Jimenez, R.; Xie, F.; Yin, P.; et al. A prospective genome-wide study of prostate cancer metastases reveals association of wnt pathway activation and increased cell cycle proliferation with primary resistance to abiraterone acetate-prednisone. Ann. Oncol. 2018, 29, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-repair defects and olaparib in metastatic prostate cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of CHD1 causes DNA repair defects and enhances prostate cancer therapeutic responsiveness. EMBO Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. CHD1 loss sensitizes prostate cancer to DNA damaging therapy by promoting error-prone double-strand break repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.A.; Chen, A.H.; Parikh, K. A novel use of olaparib for the treatment of metastatic castration-recurrent prostate cancer. Pharmacotherapy 2017, 37, 1406–1414. [Google Scholar] [CrossRef] [PubMed]
- Hussain, M.; Daignault-Newton, S.; Twardowski, P.W.; Albany, C.; Stein, M.N.; Kunju, L.P.; Siddiqui, J.; Wu, Y.M.; Robinson, D.; Lonigro, R.J.; et al. Targeting androgen receptor and DNA repair in metastatic castration-resistant prostate cancer: Results from NCI 9012. J. Clin. Oncol. 2018, 36, 991–999. [Google Scholar] [CrossRef] [PubMed]
- Thompson, T.C.; Li, L. Connecting androgen receptor signaling and the DNA damage response: Development of new therapies for advanced prostate cancer. Mol. Cell. Oncol. 2017, 4, e1321167. [Google Scholar] [CrossRef] [PubMed]
- Wengner, A.M.; Siemeister, G.; Luecking, U.; Lefranc, J.; Lienau, P.; Deeg, G.; Lagkadinou, E.; Liu, L.; Golfier, S.; Schatz, C.; et al. ATR inhibitor BAY 1895344 shows potent anti-tumor efficacy in monotherapy and strong combination potential with the targeted alpha therapy radium-223 dichloride in preclinical tumor models. Cancer Res. 2017, 77, 836. [Google Scholar] [CrossRef]
- Karanika, S.; Karantanos, T.; Li, L.; Wang, J.; Park, S.; Yang, G.; Zuo, X.; Song, J.H.; Maity, S.N.; Manyam, G.C.; et al. Targeting DNA damage response in prostate cancer by inhibiting androgen receptor-CDC6-ATR-Chk1 signaling. Cell Rep. 2017, 18, 1970–1981. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wu, Q.; Li, L. Functional and therapeutic significance of EZH2 in urological cancers. Oncotarget 2017, 8, 38044–38055. [Google Scholar] [CrossRef] [PubMed]
- Meyer, H.A.; Ahrens-Fath, I.; Sommer, A.; Haendler, B. Novel molecular aspects of prostate carcinogenesis. Biomed. Pharmacother. Biomed. Pharmacother. 2004, 58, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.A.; Yu, J. EZH2, an epigenetic driver of prostate cancer. Protein Cell 2013, 4, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.; Wu, Z.J.; Groner, A.C.; He, H.H.; Cai, C.; Lis, R.T.; Wu, X.; Stack, E.C.; Loda, M.; Liu, T.; et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science 2012, 338, 1465–1469. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.; Otte, A.P.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.S.; Schaarschuch, K.; Dalimov, Z.; Lasorsa, E.; Ku, S.; Ramakrishnan, S.; Hu, Q.; Azabdaftari, G.; Wang, J.; Pili, R.; et al. Top2a identifies and provides epigenetic rationale for novel combination therapeutic strategies for aggressive prostate cancer. Oncotarget 2015, 6, 3136–3146. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Jin, X.; Yang, J.; Yang, Y.; He, Y.; Ding, L.; Pan, Y.; Chen, S.; Jiang, J.; Huang, H. Inhibition of EZH2 by chemo- and radiotherapy agents and small molecule inhibitors induces cell death in castration-resistant prostate cancer. Oncotarget 2016, 7, 3440–3452. [Google Scholar] [CrossRef] [PubMed]
- Garapaty-Rao, S.; Nasveschuk, C.; Gagnon, A.; Chan, E.Y.; Sandy, P.; Busby, J.; Balasubramanian, S.; Campbell, R.; Zhao, F.; Bergeron, L.; et al. Identification of EZH2 and EZH1 small molecule inhibitors with selective impact on diffuse large B cell lymphoma cell growth. Chem. Biol. 2013, 20, 1329–1339. [Google Scholar] [CrossRef] [PubMed]
- Qi, W.; Zhao, K.; Gu, J.; Huang, Y.; Wang, Y.; Zhang, H.; Zhang, M.; Zhang, J.; Yu, Z.; Li, L.; et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat. Chem. Biol. 2017, 13, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, S.J.; Haendler, B. Exploiting epigenetic alterations in prostate cancer. Int. J. Mol. Sci. 2017, 18, E1017. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Malik, R.; Khan, A.P.; Asangani, I.A.; Cieslik, M.; Prensner, J.R.; Wang, X.; Iyer, M.K.; Jiang, X.; Borkin, D.; Escara-Wilke, J.; et al. Targeting the MLL complex in castration-resistant prostate cancer. Nat. Med. 2015, 21, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Ahmed, M.; Guo, H.; Soares, F.; Hua, J.T.; Gao, S.; Lu, C.; Poon, C.; Han, W.; Langstein, J.; et al. LSD1-mediated epigenetic reprogramming drives CENPE expression and prostate cancer progression. Cancer Res. 2017, 77, 5479–5490. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.C.; Ma, J.; Wang, Z.; Li, J.; Jiang, B.; Zhou, W.; Shi, X.; Wang, X.; Zhao, W.; Liu, H.M. A systematic review of histone lysine-dpecific demethylase 1 and its inhibitors. Med. Res. Rev. 2015, 35, 1032–1071. [Google Scholar] [CrossRef] [PubMed]
- Sehrawat, A.; Gao, L.; Wang, Y.; Bankhead, A., 3rd; McWeeney, S.K.; King, C.J.; Schwartzman, J.; Urrutia, J.; Bisson, W.H.; Coleman, D.J.; et al. LSD1 activates a lethal prostate cancer gene network independently of its demethylase function. Proc. Natl. Acad. Sci. USA 2018. [Google Scholar] [CrossRef] [PubMed]
- Fu, X.; Zhang, P.; Yu, B. Advances toward LSD1 inhibitors for cancer therapy. Fut. Med. Chem. 2017, 9, 1227–1242. [Google Scholar] [CrossRef] [PubMed]
- Di Micco, R.; Fontanals-Cirera, B.; Low, V.; Ntziachristos, P.; Yuen, S.K.; Lovell, C.D.; Dolgalev, I.; Yonekubo, Y.; Zhang, G.; Rusinova, E.; et al. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014, 9, 234–247. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Gelato, K.A.; Fernandez-Montalvan, A.; Siegel, S.; Haendler, B. Targeting BET bromodomains for cancer treatment. Epigenomics 2015, 7, 487–501. [Google Scholar] [CrossRef] [PubMed]
- Zuber, V.; Bettella, F.; Witoelar, A.; Consortium, P.; Cruk, G.; Consortium, B.; Consortium, T.; Andreassen, O.A.; Mills, I.G.; Urbanucci, A. Bromodomain protein 4 discriminates tissue-specific super-enhancers containing disease-specific susceptibility loci in prostate and breast cancer. BMC Genom. 2017, 18, 270. [Google Scholar] [CrossRef] [PubMed]
- Wyce, A.; Degenhardt, Y.; Bai, Y.; Le, B.; Korenchuk, S.; Crouthame, M.C.; McHugh, C.F.; Vessella, R.; Creasy, C.L.; Tummino, P.J.; et al. Inhibition of BET bromodomain proteins as a therapeutic approach in prostate cancer. Oncotarget 2013, 4, 2419–2429. [Google Scholar] [CrossRef] [PubMed]
- Asangani, I.A.; Dommeti, V.L.; Wang, X.; Malik, R.; Cieslik, M.; Yang, R.; Escara-Wilke, J.; Wilder-Romans, K.; Dhanireddy, S.; Engelke, C.; et al. Therapeutic targeting of BET bromodomain proteins in castration-resistant prostate cancer. Nature 2014, 510, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.C.; Selth, L.A.; Li, Y.; Nyquist, M.D.; Miao, L.; Bradner, J.E.; Raj, G.V.; Tilley, W.D.; Dehm, S.M. Targeting chromatin binding regulation of constitutively active AR variants to overcome prostate cancer resistance to endocrine-based therapies. Nucleic Acids Res. 2015, 43, 5880–5897. [Google Scholar] [CrossRef] [PubMed]
- Faivre, E.J.; Wilcox, D.; Lin, X.; Hessler, P.; Torrent, M.; He, W.; Uziel, T.; Albert, D.H.; McDaniel, K.; Kati, W.; et al. Exploitation of castration-resistant prostate cancer transcription factor dependencies by the novel BET inhibitor ABBV-075. Mol. Cancer Res. 2017, 15, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Raina, K.; Lu, J.; Qian, Y.; Altieri, M.; Gordon, D.; Rossi, A.M.; Wang, J.; Chen, X.; Dong, H.; Siu, K.; et al. PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 7124–7129. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, D.; Zhao, Y.; Ren, S.; Gao, K.; Ye, Z.; Wang, S.; Pan, C.W.; Zhu, Y.; Yan, Y.; et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. Nat. Med. 2017, 23, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.; Wang, Z.; Wei, W. SPOP-mediated degradation of BRD4 dictates cellular sensitivity to BET inhibitors. Cell Cycle 2017, 16, 2326–2329. [Google Scholar] [CrossRef] [PubMed]
- Janouskova, H.; El Tekle, G.; Bellini, E.; Udeshi, N.D.; Rinaldi, A.; Ulbricht, A.; Bernasocchi, T.; Civenni, G.; Losa, M.; Svinkina, T.; et al. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. Nat. Med. 2017, 23, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Markowski, M.C.; de Marzo, A.M.; Antonarakis, E.S. BET inhibitors in metastatic prostate cancer: Therapeutic implications and rational drug combinations. Expert Opin. Investig. Drugs 2017, 26, 1391–1397. [Google Scholar] [CrossRef] [PubMed]
- Culig, Z. Androgen receptor coactivators in regulation of growth and differentiation in prostate cancer. J. Cell. Physiol. 2016, 231, 270–274. [Google Scholar] [CrossRef] [PubMed]
- Lasko, L.M.; Jakob, C.G.; Edalji, R.P.; Qiu, W.; Montgomery, D.; Digiammarino, E.L.; Hansen, T.M.; Risi, R.M.; Frey, R.; Manaves, V.; et al. Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Garcia, J.; Chan, E.; de la Cruz, C.; Segal, E.; Merchant, M.; Kharbanda, S.; Raisner, R.; Haverty, P.M.; Modrusan, Z.; et al. Therapeutic targeting of the CBP/p300 bromodomain blocks the growth of castration-resistant prostate cancer. Cancer Res. 2017, 77, 5564–5575. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.M.; Parker, C. The safety and efficacy of radium-223 dichloride for the treatment of advanced prostate cancer. Expert Rev. Anticancer Ther. 2016, 16, 911–918. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Suominen, M.I.; Rissanen, J.P.; Kakonen, R.; Fagerlund, K.M.; Alhoniemi, E.; Mumberg, D.; Ziegelbauer, K.; Halleen, J.M.; Kakonen, S.M.; Scholz, A. Survival benefit with radium-223 dichloride in a mouse model of breast cancer bone metastasis. J. Natl. Cancer Instit. 2013, 105, 908–916. [Google Scholar] [CrossRef] [PubMed]
- Suominen, M.I.; Fagerlund, K.M.; Rissanen, J.P.; Konkol, Y.M.; Morko, J.P.; Peng, Z.; Alhoniemi, E.J.; Laine, S.K.; Corey, E.; Mumberg, D.; et al. Radium-223 inhibits osseous prostate cancer growth by dual targeting of cancer cells and bone microenvironment in mouse models. Clin. Cancer Res. 2017, 23, 4335–4346. [Google Scholar] [CrossRef] [PubMed]
- Vogelzang, N.J. Radium-223 dichloride for the treatment of castration-resistant prostate cancer with symptomatic bone metastases. Expert Rev. Clin. Pharmacol. 2017, 10, 809–819. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.; Heidenreich, A.; Nilsson, S.; Shore, N. Current approaches to incorporation of radium-223 in clinical practice. Prost. Cancer Prost. Dis. 2018, 21, 37–47. [Google Scholar] [CrossRef] [PubMed]
- Zustovich, F.; Barsanti, R. Targeted alpha therapies for the treatment of bone metastases. Int. J. Mol. Sci. 2017, 19, E74. [Google Scholar] [CrossRef] [PubMed]
- Parker, C.C.; Pascoe, S.; Chodacki, A.; O’Sullivan, J.M.; Germa, J.R.; O’Bryan-Tear, C.G.; Haider, T.; Hoskin, P. A randomized, double-blind, dose-finding, multicenter, phase 2 study of radium chloride (Ra 223) in patients with bone metastases and castration-resistant prostate cancer. Eur. Urol. 2013, 63, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Malamas, A.S.; Gameiro, S.R.; Knudson, K.M.; Hodge, J.W. Sublethal exposure to alpha radiation (223Ra dichloride) enhances various carcinomas’ sensitivity to lysis by antigen-specific cytotoxic T lymphocytes through calreticulin-mediated immunogenic modulation. Oncotarget 2016, 7, 86937–86947. [Google Scholar] [CrossRef] [PubMed]
- Bakht, M.K.; Oh, S.W.; Youn, H.; Cheon, G.J.; Kwak, C.; Kang, K.W. Influence of androgen deprivation therapy on the uptake of PSMA-targeted agents: Emerging opportunities and challenges. Nucl. Med. Mol. Imaging 2017, 51, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Olson, W.C.; Israel, R.J. Antibody-drug conjugates targeting prostate-specific membrane antigen. Front. Biosci. 2014, 19, 12–33. [Google Scholar] [CrossRef]
- DiPippo, V.A.; Nguyen, H.M.; Brown, L.G.; Olson, W.C.; Vessella, R.L.; Corey, E. Addition of PSMA ADC to enzalutamide therapy significantly improves survival in in vivo model of castration resistant prostate cancer. Prostate 2016, 76, 325–334. [Google Scholar] [CrossRef] [PubMed]
- Murga, J.D.; Moorji, S.M.; Han, A.Q.; Magargal, W.W.; DiPippo, V.A.; Olson, W.C. Synergistic co-targeting of prostate-specific membrane antigen and androgen receptor in prostate cancer. Prostate 2015, 75, 242–254. [Google Scholar] [CrossRef] [PubMed]
- Ferdinandus, J.; Violet, J.; Sandhu, S.; Hofman, M.S. Prostate-specific membrane antigen theranostics: Therapy with lutetium-177. Curr. Opin. Urol. 2018, 28, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Kratochwil, C.; Bruchertseifer, F.; Rathke, H.; Hohenfellner, M.; Giesel, F.L.; Haberkorn, U.; Morgenstern, A. Targeted Alpha Therapy of mCRPC with (225)Actinium-PSMA-617: Swimmer-Plot analysis suggests efficacy regarding duration of tumor-control. J. Nucl. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Hammer, S.; Larssen, A.; Ellingsen, C.; Geraudie, S.; Grant, D.; Indrevoll, B.; von Ahsen, O.; Kristian, K.; Hagemann, U.B.; Karlsson, J.; et al. Preclinical pharmacology of the PSMA-targeted thorium-227 conjugate PSMA-TTC: A novel targeted alpha therapeutic for the treatment of prostate cancer. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2017, 77. [Google Scholar] [CrossRef]
- Martin, S.K.; Kyprianou, N. Exploitation of the androgen receptor to overcome taxane resistance in advanced prostate cancer. Adv. Cancer Res. 2015, 127, 123–158. [Google Scholar] [PubMed]
- Thadani-Mulero, M.; Nanus, D.M.; Giannakakou, P. Androgen receptor on the move: Boarding the microtubule expressway to the nucleus. Cancer Res. 2012, 72, 4611–4615. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, O.; Afonso, J.; Vazquez, S.; Campos, B.; Lazaro, M.; Leon, L.; Anton Aparicio, L.M. Metastatic castration-resistant prostate cancer: Changing landscape with cabazitaxel. Anti-Cancer Drugs 2014, 25, 237–243. [Google Scholar] [CrossRef] [PubMed]
- Francini, E.; Sweeney, C.J. Docetaxel activity in the era of life-prolonging hormonal therapies for metastatic castration-resistant prostate cancer. Eur. Urol. 2016, 70, 410–412. [Google Scholar] [CrossRef] [PubMed]
- Van Soest, R.J.; de Wit, R. Irrefutable evidence for the use of docetaxel in newly diagnosed metastatic prostate cancer: Results from the STAMPEDE and CHAARTED trials. BMC Med. 2015, 13, 304. [Google Scholar] [CrossRef] [PubMed]
- Summers, N.; Vanderpuye-Orgle, J.; Reinhart, M.; Gallagher, M.; Sartor, O. Efficacy and safety of post-docetaxel therapies in metastatic castration-resistant prostate cancer: A systematic review of the literature. Curr. Med. Res. Opin. 2017, 33, 1995–2008. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, R.; Bryce, A.; Ryan, C.J.; Harzstark, A.; Derleth, C.; Kim, W.; Friedlander, T.; Lin, A.M.; Rodvelt-Bagchi, T.; Dhawan, M.; et al. A multicenter phase I study of cabazitaxel, mitoxantrone, and prednisone for chemotherapy-naive patients with metastatic castration-resistant prostate cancer: A department of defense prostate cancer clinical trials consortium study. Urol. Oncol. 2017, 35, e7–e13. [Google Scholar] [CrossRef] [PubMed]
- Green, A.K.; Corty, R.W.; Wood, W.A.; Meeneghan, M.; Reeder-Hayes, K.E.; Basch, E.; Milowsky, M.I.; Dusetzina, S.B. Comparative effectiveness of mitoxantrone plus prednisone versus prednisone alone in metastatic castrate-resistant prostate cancer after docetaxel failure. Oncologist 2015, 20, 516–522. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Mulders, P.F.; De Santis, M.; Powles, T.; Fizazi, K. Targeted treatment of metastatic castration-resistant prostate cancer with sipuleucel-T immunotherapy. Cancer Immunol. Immunother. 2015, 64, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Ledford, H. Therapeutic cancer vaccine survives biotech bust. Nature 2015, 519, 17–18. [Google Scholar] [CrossRef] [PubMed]
- Pollard, M.E.; Moskowitz, A.J.; Diefenbach, M.A.; Hall, S.J. Cost-effectiveness analysis of treatments for metastatic castration resistant prostate cancer. Asian J. Urol. 2017, 4, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, T.N.; Schreiber, R.D. Neoantigens in cancer immunotherapy. Science 2015, 348, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Turajlic, S.; Litchfield, K.; Xu, H.; Rosenthal, R.; McGranahan, N.; Reading, J.L.; Wong, Y.N.S.; Rowan, A.; Kanu, N.; Al Bakir, M.; et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: A pan-cancer analysis. Lancet Oncol. 2017, 18, 1009–1021. [Google Scholar] [CrossRef]
- Pasero, C.; Gravis, G.; Guerin, M.; Granjeaud, S.; Thomassin-Piana, J.; Rocchi, P.; Paciencia-Gros, M.; Poizat, F.; Bentobji, M.; Azario-Cheillan, F.; et al. Inherent and tumor-driven immune tolerance in the prostate microenvironment impairs natural killer cell antitumor activity. Cancer Res. 2016, 76, 2153–2165. [Google Scholar] [CrossRef] [PubMed]
- Boibessot, C.; Toren, P. Sex steroids in the tumor microenvironment and prostate cancer progression. Endocr.-Relat. Cancer 2018, 25, R179–R196. [Google Scholar] [CrossRef] [PubMed]
- Maia, M.C.; Hansen, A.R. A comprehensive review of immunotherapies in prostate cancer. Crit. Rev. Oncol./Hematol. 2017, 113, 292–303. [Google Scholar] [CrossRef] [PubMed]
- Cabel, L.; Loir, E.; Gravis, G.; Lavaud, P.; Massard, C.; Albiges, L.; Baciarello, G.; Loriot, Y.; Fizazi, K. Long-term complete remission with Ipilimumab in metastatic castrate-resistant prostate cancer: Case report of two patients. J. Immunother. Cancer 2017, 5, 31. [Google Scholar] [CrossRef] [PubMed]
- Lemery, S.; Keegan, P.; Pazdur, R. First FDA approval agnostic of cancer site—When a biomarker defines the indication. N. Engl. J. Med. 2017, 377, 1409–1412. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The genomic complexity of primary human prostate cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Gasi Tandefelt, D.; Boormans, J.; Hermans, K.; Trapman, J. ETS fusion genes in prostate cancer. Endocr.-Relat. Cancer 2014, 21, R143–R152. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, S.H.; Joung, J.Y.; Lee, G.K.; Hong, E.K.; Kang, K.M.; Yu, A.; Nam, B.H.; Chung, J.; Seo, H.K.; et al. Overexpression of ERG and wild-type PTEN are associated with favorable clinical prognosis and low biochemical recurrence in prostate cancer. PLoS ONE 2015, 10, e0122498. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Qiao, Y.; Asangani, I.A.; Ateeq, B.; Poliakov, A.; Cieslik, M.; Pitchiaya, S.; Chakravarthi, B.; Cao, X.; Jing, X.; et al. Development of peptidomimetic inhibitors of the ERG gene fusion product in prostate cancer. Cancer Cell 2017, 31, 532–548. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.S.; Roshan-Moniri, M.; Hsing, M.; Lau, D.; Kim, A.; Yen, P.; Mroczek, M.; Nouri, M.; Lien, S.; Axerio-Cilies, P.; et al. Discovery and characterization of small molecules targeting the DNA-binding ETS domain of ERG in prostate cancer. Oncotarget 2017, 8, 42438–42454. [Google Scholar] [CrossRef] [PubMed]
- Mikhaylenko, D.S.; Efremov, G.D.; Strelnikov, V.V.; Zaletaev, D.V.; Alekseev, B.Y. Somatic mutation analyses in studies of the clonal evolution and diagnostic targets of prostate cancer. Curr. Genom. 2017, 18, 236–243. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Borno, S.T.; Fischer, A.; Kerick, M.; Falth, M.; Laible, M.; Brase, J.C.; Kuner, R.; Dahl, A.; Grimm, C.; Sayanjali, B.; et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov. 2012, 2, 1024–1035. [Google Scholar] [CrossRef] [PubMed]
- Kron, K.J.; Murison, A.; Zhou, S.; Huang, V.; Yamaguchi, T.N.; Shiah, Y.J.; Fraser, M.; van der Kwast, T.; Boutros, P.C.; Bristow, R.G.; et al. TMPRSS2-ERG fusion co-opts master transcription factors and activates NOTCH signaling in primary prostate cancer. Nat. Genet. 2017, 49, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Aieta, M.; Amadori, D.; De Giorgi, U. Neuroendocrine differentiation in prostate cancer: Current and emerging therapy strategies. Crit. Rev. Oncol./Hematol. 2014, 92, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Priemer, D.S.; Montironi, R.; Wang, L.; Williamson, S.R.; Lopez-Beltran, A.; Cheng, L. Neuroendocrine tumors of the prostate: Emerging insights from molecular data and updates to the 2016 World Health Organization classification. Endocr. Pathol. 2016, 27, 123–135. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; MacDonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted next-generation sequencing of advanced prostate cancer identifies potential therapeutic targets and disease heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef] [PubMed]
- Ylitalo, E.B.; Thysell, E.; Jernberg, E.; Lundholm, M.; Crnalic, S.; Egevad, L.; Stattin, P.; Widmark, A.; Bergh, A.; Wikstrom, P. Subgroups of castration-resistant prostate cancer bone metastases defined through an inverse relationship between androgen receptor activity and immune response. Eur. Urol. 2017, 71, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, M.A.; Duncavage, E.J.; Walter, M.J. Implications of tumor clonal heterogeneity in the era of next-generation sequencing. Trends Cancer 2015, 1, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated evolution of prostate cancer genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.W.; Loeb, L.A.; Salk, J.J. The influence of subclonal resistance mutations on targeted cancer therapy. Nat. Rev. Clin. Oncol. 2016, 13, 335–347. [Google Scholar] [CrossRef] [PubMed]
- Haffner, M.C.; Mosbruger, T.; Esopi, D.M.; Fedor, H.; Heaphy, C.M.; Walker, D.A.; Adejola, N.; Gurel, M.; Hicks, J.; Meeker, A.K.; et al. Tracking the clonal origin of lethal prostate cancer. J. Clin. Investig. 2013, 123, 4918–4922. [Google Scholar] [CrossRef] [PubMed]
- Gundem, G.; Van Loo, P.; Kremeyer, B.; Alexandrov, L.B.; Tubio, J.M.C.; Papaemmanuil, E.; Brewer, D.S.; Kallio, H.M.L.; Hognas, G.; Annala, M.; et al. The evolutionary history of lethal metastatic prostate cancer. Nature 2015, 520, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254ra125. [Google Scholar] [CrossRef] [PubMed]
- Prekovic, S.; Van den Broeck, T.; Moris, L.; Smeets, E.; Claessens, F.; Joniau, S.; Helsen, C.; Attard, G. Treatment-induced changes in the androgen receptor axis: Liquid biopsies as diagnostic/prognostic tools for prostate cancer. Mol. Cell. Endocrinol. 2018, 462, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed]
- Mitsiades, N.; Sung, C.C.; Schultz, N.; Danila, D.C.; He, B.; Eedunuri, V.K.; Fleisher, M.; Sander, C.; Sawyers, C.L.; Scher, H.I. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012, 72, 6142–6152. [Google Scholar] [CrossRef] [PubMed]
- Iglesias-Gato, D.; Wikstrom, P.; Tyanova, S.; Lavallee, C.; Thysell, E.; Carlsson, J.; Hagglof, C.; Cox, J.; Andren, O.; Stattin, P.; et al. The proteome of primary prostate cancer. Eur. Urol. 2016, 69, 942–952. [Google Scholar] [CrossRef] [PubMed]
- Cutruzzola, F.; Giardina, G.; Marani, M.; Macone, A.; Paiardini, A.; Rinaldo, S.; Paone, A. Glucose metabolism in the progression of prostate cancer. Front. Physiol. 2017, 8, 97. [Google Scholar] [CrossRef] [PubMed]
- Halliday, K.R.; Fenoglio-Preiser, C.; Sillerud, L.O. Differentiation of human tumors from nonmalignant tissue by natural-abundance 13C NMR spectroscopy. Magn. Reson. Med. 1988, 7, 384–411. [Google Scholar] [CrossRef] [PubMed]
- Hahn, P.; Smith, I.C.; Leboldus, L.; Littman, C.; Somorjai, R.L.; Bezabeh, T. The classification of benign and malignant human prostate tissue by multivariate analysis of 1H magnetic resonance spectra. Cancer Res. 1997, 57, 3398–3401. [Google Scholar] [PubMed]
- Lima, A.R.; Bastos Mde, L.; Carvalho, M.; Guedes de Pinho, P. Biomarker discovery in human prostate cancer: An update in metabolomics studies. Transl. Oncol. 2016, 9, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Komura, K.; Sweeney, C.J.; Inamoto, T.; Ibuki, N.; Azuma, H.; Kantoff, P.W. Current treatment strategies for advanced prostate cancer. Int. J. Urol. 2018, 25, 220–231. [Google Scholar] [CrossRef] [PubMed]
- De Velasco, M.A.; Uemura, H. Prostate cancer immunotherapy: Where are we and where are we going? Curr. Opin. Urol. 2018, 28, 15–24. [Google Scholar] [CrossRef] [PubMed]
- Bryant, G.; Wang, L.; Mulholland, D.J. Overcoming oncogenic mediated tumor immunity in prostate cancer. Int. J. Mol. Sci. 2017, 18, E1542. [Google Scholar] [CrossRef] [PubMed]
- Annala, M.; Vandekerkhove, G.; Khalaf, D.; Taavitsainen, S.; Beja, K.; Warner, E.W.; Sunderland, K.; Kollmannsberger, C.; Eigl, B.J.; Finch, D.; et al. Circulating tumor DNA genomics correlate with resistance to abiraterone and enzalutamide in prostate cancer. Cancer Discov. 2018, 8, 444–457. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Agents | Population | Status | Identifier |
|---|---|---|---|
| ADT +/− abiraterone acetate + prednisolone ADT +/− abiraterone acetate + prednisolone + enzalutamide | Advancing or metastatic castration-resistant prostate cancer (mCRPC) | Completed | NCT00268476 |
| Abiraterone acetate + prednisone/prednisolone | mCRPC post-chemotherapy | Completed | NCT00638690 |
| Abiraterone acetate + prednisone | Asymptomatic or mildly symptomatic patients with mCRPC | Completed | NCT00887198 |
| ADT +/− abiraterone acetate + prednisone | Metastatic hormone-naive prostate cancer | Active, not recruiting | NCT01715285 |
| ADT + docetaxel +/− local radiation therapy +/− abiraterone acetate + prednisone | Metastatic hormone-naive prostate cancer | Recruiting | NCT01957436 |
| Enzalutamide +/− abiraterone acetate + prednisone | mCRPC | Active, not recruiting | NCT01949337 |
| Enzalutamide | nmCRPC | Active, not recruiting | NCT02003924 |
| Enzalutamide +/− radium-223 | Castration-resistant prostate cancer (CRPC) | Recruiting | NCT02194842 |
| Enzalutamide + ADT | Metastatic prostate cancer | Active, not recruiting | NCT02446405 |
| Enzalutamide + ADT + radiation therapy | High-risk localized prostate cancer | Recruiting | NCT02446444 |
| Apalutamide | nmCRPC | Active, not recruiting | NCT01946204 |
| Abiraterone + prednisone +/− apalutamide | Chemotherapy-naive mCRPC | Active, not recruiting | NCT02257736 |
| ADT +/− apalutamide | mHSPC | Active, not recruiting | NCT02489318 |
| Darolutamide | nmCRPC | Recruiting | NCT02200614 |
| ADT + docetaxel +/− darolutamide | mHSPC | Recruiting | NCT02799602 |
| Agents | Target | Population | Phase | Status | Identifier |
|---|---|---|---|---|---|
| Buparlisib | PI3K | High-risk prostate cancer | 2 | Active, not recruiting | NCT01695473 |
| AZD8186 +/− abiraterone acetate + prednisone | PI3Kβ/δ CYP17A1 | Advanced CRPC | 1 | Recruiting | NCT01884285 |
| GSK2636771 + enzalutamide | PI3Kβ AR | mCRPC | 1 | Recruiting | NCT02215096 |
| Enzalutamide +/− LY3023414 | AR PI3K/mTOR | mCRPC | 2 | Recruiting | NCT02407054 |
| Apitolisib or ipatasertib +/− abiraterone acetate + prednisone/prednisolone | PI3K AKT CYP17A1 | CRPC post-docetaxel | 1/2 | Active, not recruiting | NCT01485861 |
| Ipatasertib +/− abiraterone acetate + prednisone/prednisolone | AKT CYP17A1 | mCRPC | 3 | Recruiting | NCT03072238 |
| MK2206 | AKT | Recurrent prostate cancer | 1 | Active, not recruiting | NCT01480154 |
| Bicalutamide +/− MK2206 | AR AKT | Recurrent prostate cancer | 2 | Active, not recruiting | NCT01251861 |
| Docetaxel +/− AZD5363 | tubulin AKT | mCRPC | 2 | Recruiting | NCT02121639 |
| Enzalutamide +/− AZD5363 | AR AKT | mCRPC | 2 | Recruiting | NCT02525068 |
| Sirolimus + docetaxel + carboplatin | mTOR tubulin DNA | mCRPC | 1/2 | Recruiting | NCT02565901 |
| Ridaforolimus | mTOR | Taxane-resistant androgen-independent prostate cancer | 2 | Completed | NCT00110188 |
| Ridaforolimus + bicalutamide | mTOR AR | Prostate cancer | 2 | Completed | NCT00777959 |
| Temsirolimus | mTOR | High-risk prostate cancer | 2 | Completed | NCT00071968 |
| AZD2014 | mTOR | High-risk prostate cancer | 1 | Active, not recruiting | NCT02064608 |
| MLN0128 | mTOR | Advanced CRPC | 2 | Active, not recruiting | NCT02091531 |
| Temsirolimus + cixutumumab | mTOR IGF-1R | mCRPC | 1/2 | Completed | NCT01026623 |
| Everolimus + docetaxel | mTOR tubulin | mCRPC | 1/2 | Completed | NCT00459186 |
| Everolimus + bevacizumab | mTOR VEGF | Advanced prostate cancer | 1/2 | Completed | NCT00574769 |
| Everolimus + carboplatin | mTOR DNA replication | mCRPC post-docetaxel | 2 | Completed | NCT01051570 |
| Everolimus + radiation therapy | mTOR | Prostate cancer | 1 | Recruiting | NCT01548807 |
| CC-115 + enzalutamide | DNA-PK/ mTOR AR | CRPC | 1 | Recruiting | NCT02833883 |
| Agents | Target | Population | Phase | Status | Identifier |
|---|---|---|---|---|---|
| Olaparib | PARP | High-risk prostate cancer | 2 | Recruiting | NCT03047135 |
| Olaparib + abiraterone acetate + prednisone/prednisolone | PARP CYP17A | mCRPC | 2 | Active, not recruiting | NCT01972217 |
| Olaparib +/− degarelix | PARP GnRH antagonist | Intermediate/high-risk prostate cancer | 1 | Recruiting | NCT02324998 |
| Olaparib +/− cediranib | PARP VEGFR | mCRPC | 2 | Recruiting | NCT02893917 |
| Olaparib +/− abiraterone acetate + prednisone vs. abiraterone acetate + prednisone | PARP CYP17A | mCRPC | 2 | Recruiting | NCT03012321 |
| Olaparib + radium-223 | PARP Hydroxyapatite | mCRPC | 1/2 | Not yet recruiting | NCT03317392 |
| Veliparib +/− temozolomide | PARP Rapidly dividing cells | mCRPC | 1 | Completed | NCT01085422 |
| Abiraterone acetate + prednisone +/− veliparib | CYP17A PARP | mCRPC | 2 | Active, not recruiting | NCT01576172 |
| Rucaparib | PARP | mCRPC | 2 | Recruiting | NCT02952534 |
| Rucaparib vs. abiraterone acetate + prednisone or enzalutamide or docetaxel | PARP CYP17A AR Tubulin | mCRPC with homologous recombination gene deficiency | 3 | Recruiting | NCT02975934 |
| Niraparib | PARP | mCRPC and DNA repair anomalies | 2 | Recruiting | NCT02854436 |
| Niraparib + radium-223 | PARP Hydroxyapatite | CRPC | 1 | Recruiting | NCT03076203 |
| Agents | Target | Population | Phase | Status | Identifier |
|---|---|---|---|---|---|
| Radium-223 + abiraterone acetate + prednisone | Hydroxyapatite CYP17A | mCRPC | 2 | Completed, has results | NCT02097303 |
| Radium-223 + enzalutamide | Hydroxyapatite AR | mCRPC | 2 | Recruiting | NCT02199197 |
| Radium-223 + docetaxel | Hydroxyapatite Tubulin | mCRPC | 2 | Recruiting | NCT03230734 |
| Radium-223 + olaparib | Hydroxyapatite PARP | mCRPC | 1/2 | Active, not recruiting | NCT03317392 |
| Radium-223 + niraparib | Hydroxyapatite PARP | mCRPC | 1 | Active, not recruiting | NCT03076203 |
| Radium-223 + pembrolizumab | Hydroxyapatite PD-1 | mCRPC | 2 | Recruiting | NCT03093428 |
| Radium-223 + atezolizumab | Hydroxyapatite PD-L1 | mCRPC | 1 | Recruiting | NCT02814669 |
| Radium-223 + sipuleucel-T | Hydroxyapatite Immunotherapy | mCRPC | 2 | Recruiting | NCT02463799 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nevedomskaya, E.; Baumgart, S.J.; Haendler, B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. Int. J. Mol. Sci. 2018, 19, 1359. https://doi.org/10.3390/ijms19051359
Nevedomskaya E, Baumgart SJ, Haendler B. Recent Advances in Prostate Cancer Treatment and Drug Discovery. International Journal of Molecular Sciences. 2018; 19(5):1359. https://doi.org/10.3390/ijms19051359
Chicago/Turabian StyleNevedomskaya, Ekaterina, Simon J. Baumgart, and Bernard Haendler. 2018. "Recent Advances in Prostate Cancer Treatment and Drug Discovery" International Journal of Molecular Sciences 19, no. 5: 1359. https://doi.org/10.3390/ijms19051359
APA StyleNevedomskaya, E., Baumgart, S. J., & Haendler, B. (2018). Recent Advances in Prostate Cancer Treatment and Drug Discovery. International Journal of Molecular Sciences, 19(5), 1359. https://doi.org/10.3390/ijms19051359