Oxygen-Induced Retinopathy from Recurrent Intermittent Hypoxia Is Not Dependent on Resolution with Room Air or Oxygen, in Neonatal Rats

 ,
, 
Abstract
1. Introduction
2. Results
2.1. Somatic Growth
2.2. Eye Opening
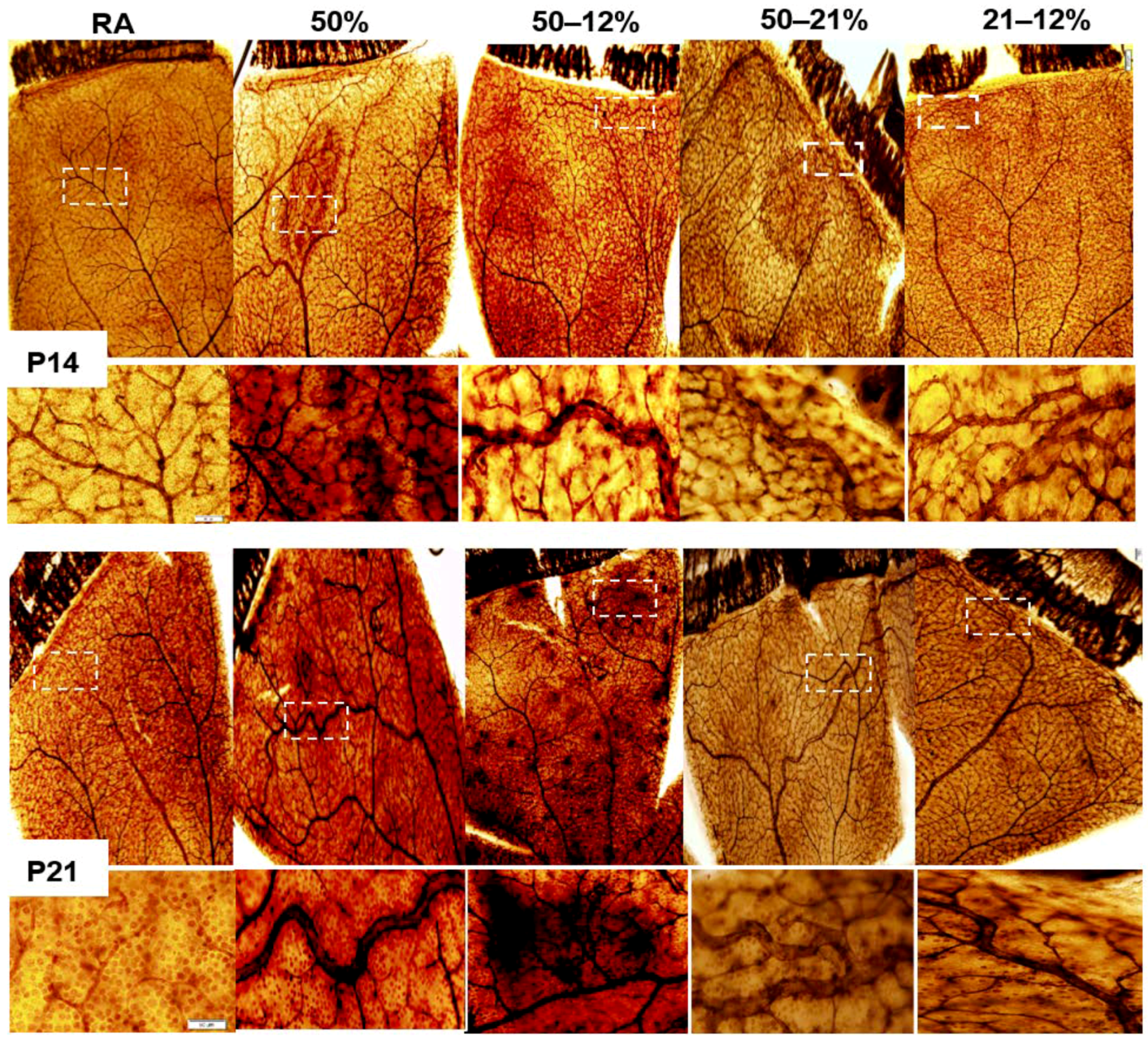
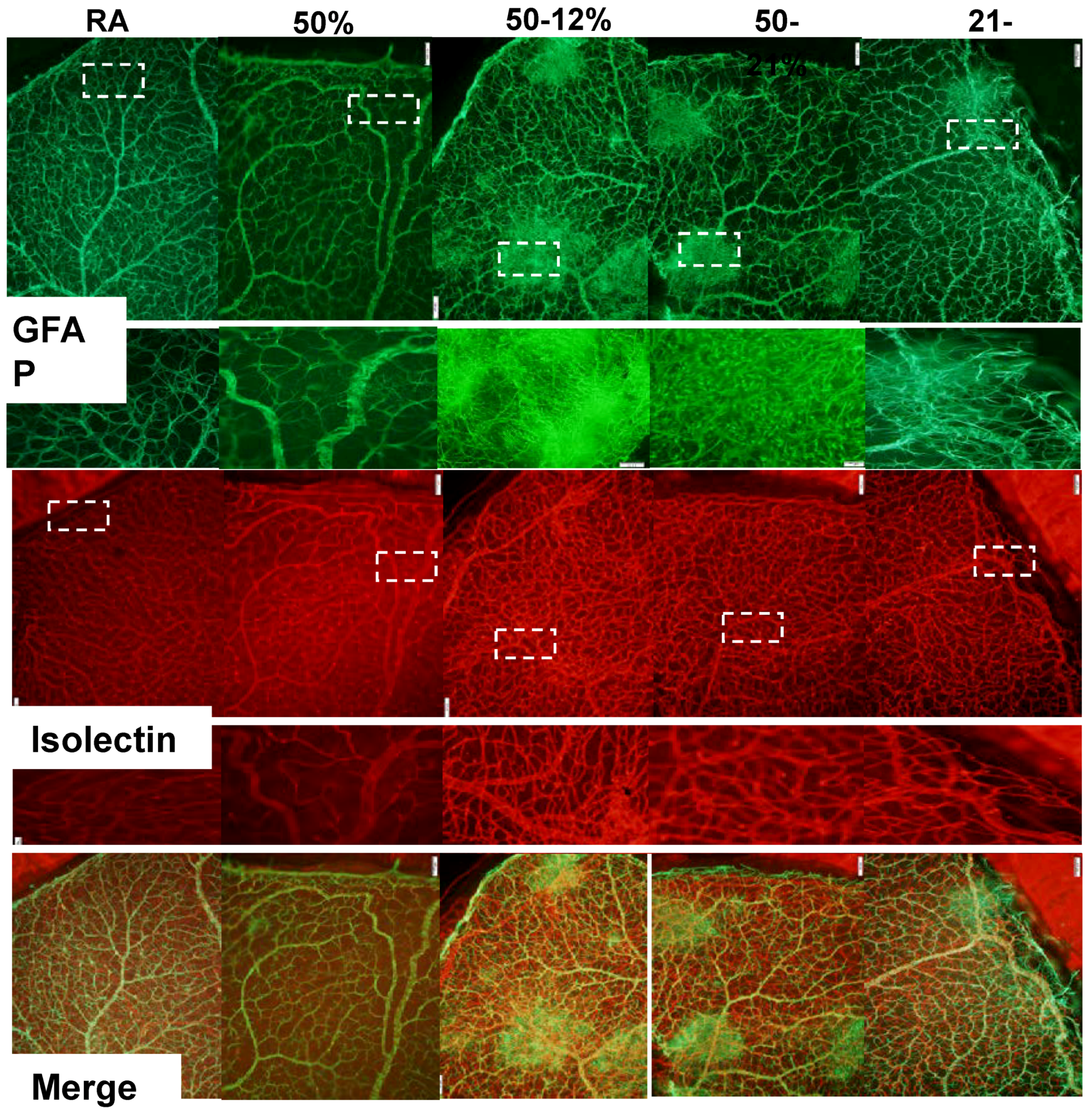
2.3. Retinal Vascular and Astrocyte Integrity
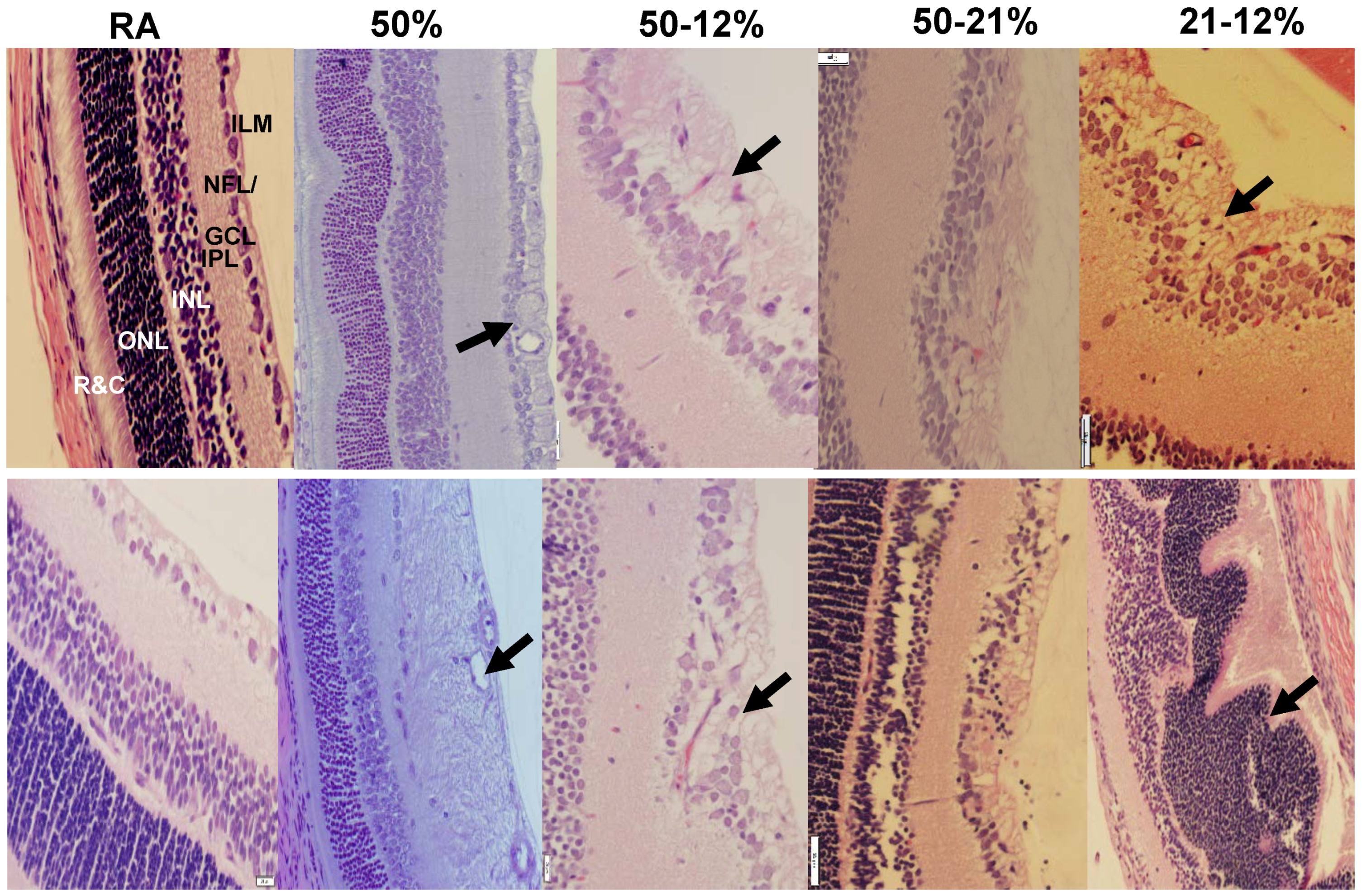
2.4. Retinal Morphometry
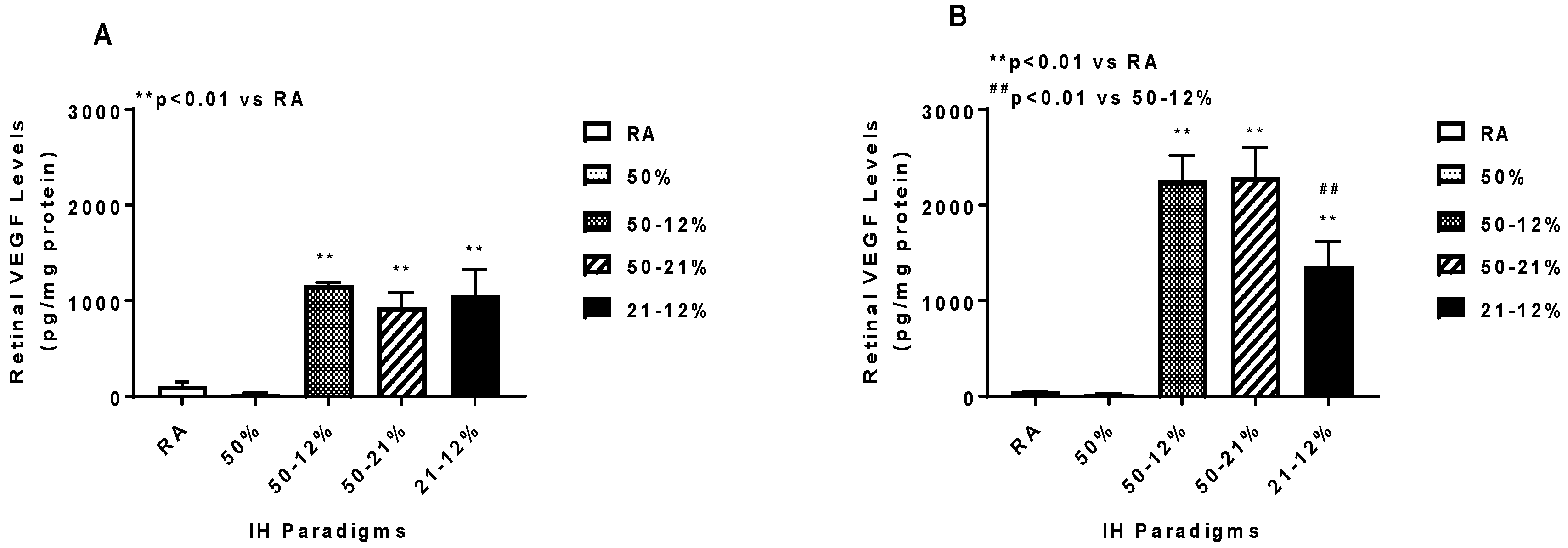
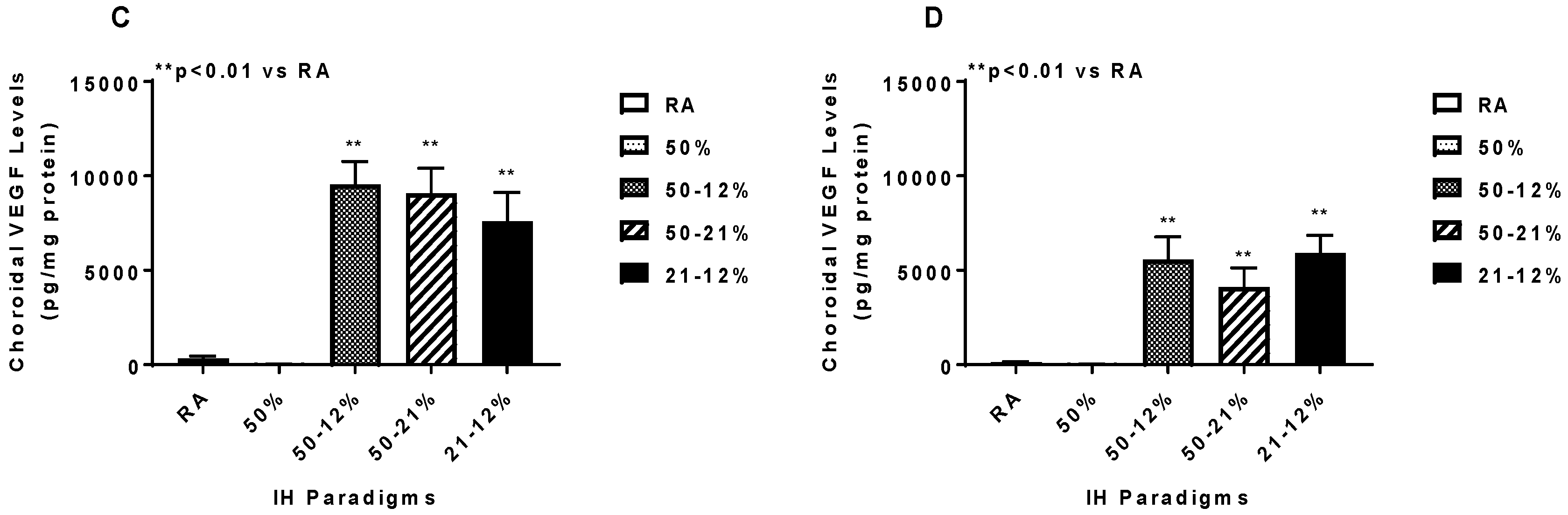
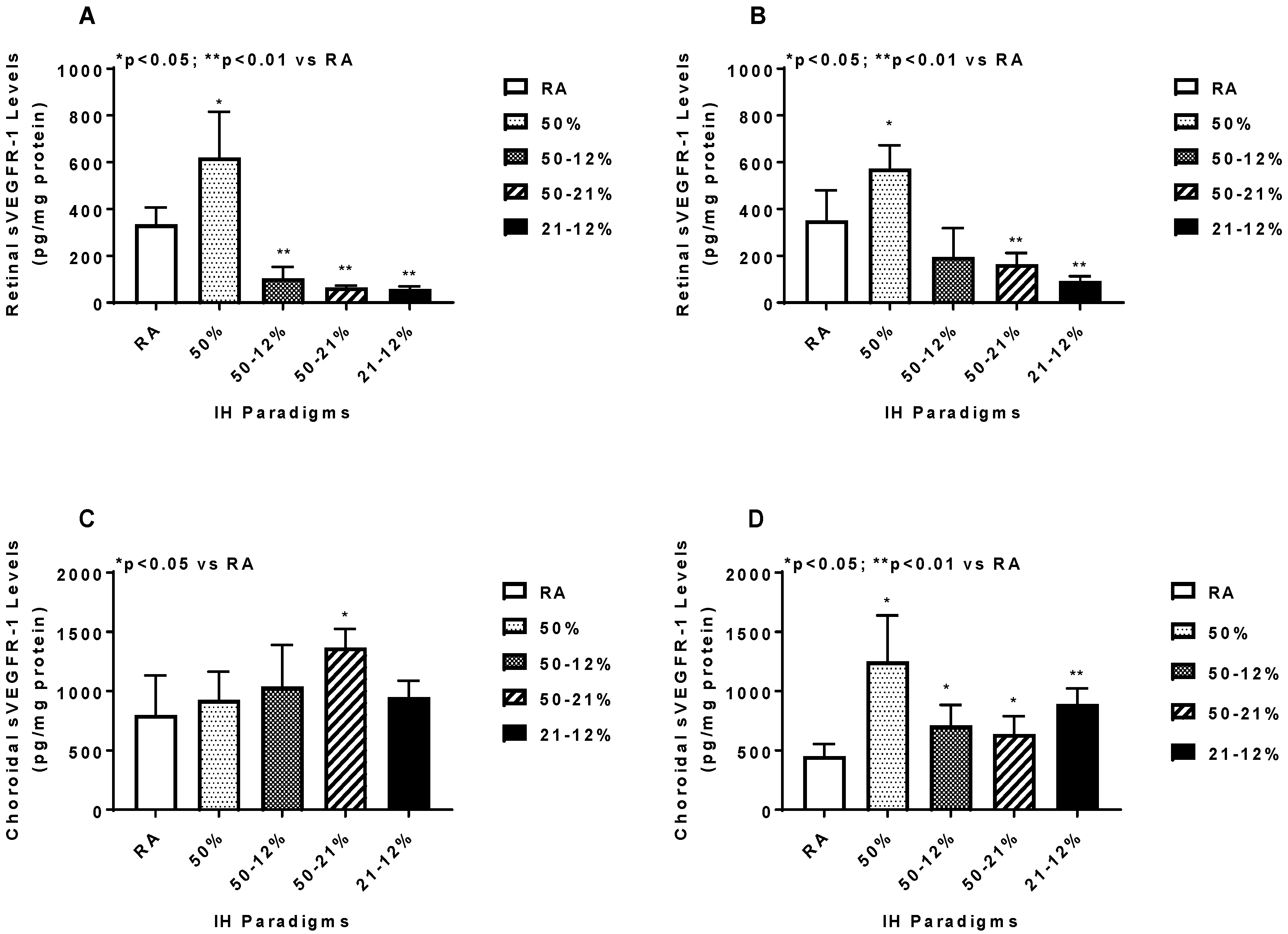
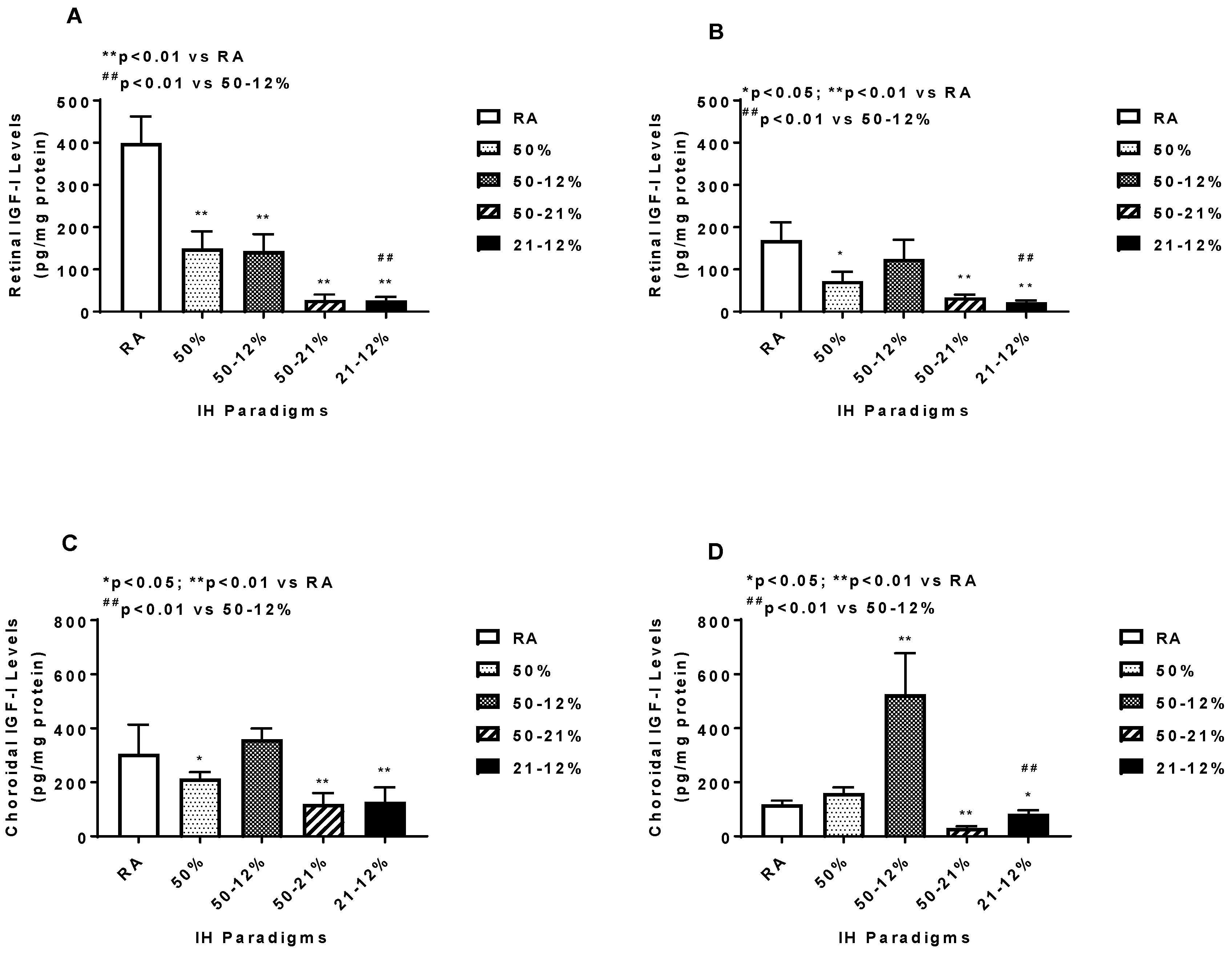
2.5. Angiogenesis Biomarkers in the Ocular and Systemic Compartments
3. Discussion
4. Material and Methods
4.1. Animals
4.2. Experimental Design
4.3. IH Profiles
4.4. Sample Collection and Processing
4.5. Retinal Flatmounts
4.6. Retinal Angiogenesis Quantification
4.7. Assay of Angiogenesis Biomarkers
4.8. Total Cellular Protein Levels
4.9. Statistical Analysis
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| ROP | Retinopathy of prematurity |
| ELGANS | Extremely low gestational age neonates |
| IH | Intermittent hypoxia |
| Hx | Hyperoxia |
| IGF-I | Insulin-like growth factor-1 |
| VEGF | Vascular endothelial growth factor |
| ECs | Endothelial cells |
| INL | Inner nuclear layer |
| NFL | Nerve fiber layer |
| GCL | Ganglion cell layer |
| ONL | Outer nuclear layer |
| ILM | Inner limiting membrane |
| GFAP | Glial fibrillary acidic protein |
| P14 | Postnatal day 14 |
| P21 | Postnatal day 21 |
| sVEGFR-1 | Soluble vascular endothelial growth factor receptor-1 |
| O2 | Oxygen |
| HIF1α | Hypoxia inducible factor-1α |
| ROS | Reactive oxygen species |
| H2O2 | Hydrogen peroxide |
| OIR | Oxygen-induced retinopathy |
References
- Kong, L.; Fry, M.; Al-Samarraie, M.; Gilbert, C.; Steinkuller, P.G. An update on progress and the changing epidemiology of causes of childhood blindness worldwide. J. AAPOS 2012, 16, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Smith, B.T.; Tasman, W.S. Retinopathy of prematurity: Late complications in the baby boomer generation (1946–1964). Trans. Am. Ophthalmol. Soc. 2005, 103, 225–234. [Google Scholar] [PubMed]
- Baker, P.S.; Tasman, W. Myopia in adults with retinopathy of prematurity. Am. J. Ophthalmol. 2008, 145, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, M.I.; Russ, R.; Brennan, K.A.; Williams, C.J.; Berrones, D.; Patel, B.; Martinez-Castellanos, M.A.; Fernandes, A.; Hubbard, G.B., 3rd; Chan, R.V.P.; et al. The Economic Model of Retinopathy of Prematurity (EcROP) Screening and Treatment: Mexico and the United States. Am. J. Ophthalmol. 2016, 168, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Dave, H.B.; Gordillo, L.; Yang, Z.; Zhang, M.S.; Hubbard, G.B., 3rd; Olsen, T.W. The societal burden of blindness secondary to retinopathy of prematurity in Lima, Peru. Am. J. Ophthalmol. 2012, 154, 750–755. [Google Scholar] [CrossRef] [PubMed]
- Eckert, K.A.; Carter, M.J.; Lansingh, V.C.; Wilson, D.A.; Furtado, J.M.; Frick, K.D.; Resnikoff, S. A Simple Method for Estimating the Economic Cost of Productivity Loss Due to Blindness and Moderate to Severe Visual Impairment. Ophthalmic Epidemiol. 2015, 22, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Frick, K.D.; Foster, A. The magnitude and cost of globalblindness: An increasing problem that can be alleviated. Am. J. Ophthalmol. 2003, 135, 471–476. [Google Scholar] [CrossRef]
- Smith, L. Pathogenesis of Retinopathy of Prematurity. Growth Horm. IGF Res. 2004, 14, 140–144. [Google Scholar] [CrossRef] [PubMed]
- Cringle, S.J.; Yu, D.Y. Oxygen supply and consumption in the retina: Implications for studies of retinopathy of prematurity. Doc. Ophthalmol. 2010, 120, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Phelps, D.L. Retinopathy of prematurity: History, classification, and pathophysiology. Neoreviews 2001, 2, e153–e166. [Google Scholar] [CrossRef]
- Phelps, D.L. Retinopathy of prematurity. In Neonatal Perinatal Medicine: Diseases of the Fetus and Infant, 7th ed.; Fanaroff, A.A., Martin, R.J., Eds.; Mosby: St. Louis, MO, USA, 2008; Volume 2, pp. 1595–1599. [Google Scholar]
- Rhein, L.M.; Dobson, N.R.; Darnall, R.A.; Corwin, M.J.; Heeren, T.C.; Poets, C.F.; McEntire, B.L.; Hunt, C.E.; Caffeine Pilot Study Group. Effects of caffeine on intermittent hypoxia in infants born prematurely: A randomized clinical trial. JAMA Pediatr. 2014, 168, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, J.M.; Martin, R.J.; Gauda, E.B. Apnea of prematurity-perfect storm. Respir. Physiol. Neurobiol. 2013, 189, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Gonzalez, F.; Mu, D. Apnea of prematurity: From cause to treatment. Eur. J. Pediatr. 2011, 170, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Stokowski, L.A. A primer on apnea of prematurity. Adv. Neonatal. Care 2005, 5, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Martin, R.J.; Di Fiore, J.M.; Macfarlane, P.M.; Wilson, C.G. Physiologic basis for intermittent hypoxic episodes in preterm infants. Adv. Exp. Med. Biol. 2012, 758, 351–358. [Google Scholar] [PubMed]
- York, J.R.; Landers, S.; Kirby, R.S.; Arbogast, P.G.; Penn, J.S. Arterial oxygen fluctuation and retinopathy of prematurity in very-low-birth-weight infants. J. Perinatol. 2004, 24, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, J.M.; Bloom, J.N.; Orge, F.; Schutt, A.; Schluchter, M.; Cheruvu, V.K.; Walsh, M.; Finer, N.; Martin, R.J. A higher incidence of intermittent hypoxemic episodes is associated with severe retinopathy of prematurity. J. Pediatr. 2010, 157, 69–73. [Google Scholar] [CrossRef] [PubMed]
- Di Fiore, J.M.; Kaffashi, F.; Loparo, K.; Sattar, A.; Schluchter, M.; Foglyano, R.; Martin, R.J.; Wilson, C.G. The relationship between patterns of intermittent hypoxia and retinopathy of prematurity in preterm infants. Pediatr. Res. 2012, 72, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.G.; Benitz, W.E.; Madan, A. Retinopathy of prematurity and pulse oximetry: A national survey of recent practices. J. Perinatol. 2004, 24, 164–168. [Google Scholar] [CrossRef] [PubMed]
- Castillo, A.; Deulofeut, R.; Critz, A.; Sola, A. Prevention of retinopathy of prematurity in preterm infants through changes in clinical practice and SpO2 technology. Acta Paediatr. 2011, 100, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, S.; Fleck, B.W.; Elton, R.A.; McIntosh, N. Transcutaneous oxygen levels in retinopathy of prematurity. Lancet 1995, 346, 1464–1465. [Google Scholar] [CrossRef]
- Saito, Y.; Omoto, T.; Cho, Y.; Hatsukawa, Y.; Fujimura, M.; Takeuchi, T. The progression of retinopathy of prematurity and fluctuation in blood gas tension. Graefe’s Arch. Clin. Exp. Ophthalmol. 1993, 231, 151–156. [Google Scholar] [CrossRef]
- Joshi, R.; van Pul, C.; Atallah, L.; Feijs, L.; Van Huffel, S.; Andriessen, P. Pattern discovery in critical alarms originating from neonates under intensive care. Physiol. Meas. 2016, 37, 564–579. [Google Scholar] [CrossRef] [PubMed]
- Hellström, A.; Engström, E.; Hård, A.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Löfqvist, C.; Svensson, E.; Holm, S.; Ewald, U.; et al. Postnatal serum insulin-like growth factor I deficiency is associated with retinopathy of prematurity and other complications of premature birth. Pediatrics 2003, 112, 1016–1020. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.; Carlsson, B.; Niklasson, A.; Segnestam, K.; Boguszewski, M.; de Lacerda, L.; Savage, M.; Svensson, E.; Smith, L.; Weinberger, D.; et al. IGF-I is critical for normal vascularization of the human retina. J. Clin. Endocrinol. Metab. 2002, 87, 3413–3416. [Google Scholar] [CrossRef] [PubMed]
- Hellstrom, A.; Perruzzi, C.; Ju, M.; Engstrom, E.; Hard, A.L.; Liu, J.L.; Albertsson-Wikland, K.; Carlsson, B.; Niklasson, A.; Sjodell, L.; et al. Low IGF-I suppresses VEGF-survival signaling in retinal endothelial cells: Direct correlation with clinical retinopathy of prematurity. Proc. Natl. Acad. Sci. USA 2001, 98, 5804–5808. [Google Scholar] [CrossRef] [PubMed]
- Gerhardt, H.; Golding, M.; Fruttiger, M.; Ruhrberg, C.; Lundkvist, A.B.; Abramsson, A.; Jeltsch, M.; Mitchell, C.; Alitalo, K.; Shima, D.; et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J. Cell Biol. 2003, 161, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Houck, K.A.; Ferrara, N.; Winer, J.; Cachianes, G.; Li, B.; Leung, D.W. The vascular endothelial growth factor family: Identification of a fourth molecular species and characterization of alternative splicing of RNA. Mol. Endocrinol. 1991, 5, 1806–1814. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Hoareau-Aveilla, C.; Oltean, S.; Harper, S.J.; Bates, D.O. The anti-angiogenic isoforms of VEGF in health and disease. Biochem. Soc. Trans. 2009, 37, 1207–1213. [Google Scholar] [CrossRef] [PubMed]
- Holderfield, M.T.; Hughes, C.C.W. Cross-talk between vascular endothelial growth factor, Notch, and transforming growth factor-2 in vascular morphogenesis. Circ. Res. 2008, 102, 637–652. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N. Binding to the extracellular matrix and proteolytic processing: Two key mechanisms regulating vascular endothelial growth factor action. Mol. Biol. Cell 2010, 21, 687–690. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Collen, D. Role of vascular endothelial growth factor and vascular endothelial growth factor receptors in vascular development. Curr. Top. Microbiol. Immunol. 1999, 237, 133–158. [Google Scholar] [PubMed]
- Ruhrberg, C.; Gerhardt, H.; Golding, M.; Watson, R.; Ioannidou, S.; Fujisawa, H.; Betsholtz, C.; Shima, D.T. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002, 16, 2684–2698. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.; Yang, H.; Chan-Ling, T. Vascularization of the human fetal retina: Roles of vasculogenesis and angiogenesis. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1217–1228. [Google Scholar]
- Smith, L.E.; Hard, A.L.; Hellström, A. The biology of retinopathy of prematurity: How knowledge of pathogenesis guides treatment. Clin. Perinatol. 2013, 40, 201–214. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.; Itin, A.; Alon, T.; Pe’er, J.; Gnessin, H.; Chan-Ling, T.; Keshet, E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J. Neurosci. 1995, 15, 4738–4747. [Google Scholar] [CrossRef] [PubMed]
- Weidemann, A.; Krohne, T.U.; Aguilar, E.; Kurihara, T.; Takeda, N.; Dorrell, M.I.; Simon, M.C.; Haase, V.H.; Friedlander, M.; Johnson, R.S. Astrocyte hypoxic response is essential for pathological but not developmental angiogenesis of the retina. Glia 2010, 58, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Luna, G.; Keeley, P.; Reese, B.E.; Linberg, K.A.; Lewis, G.P.; Fisher, S.K. Astrocyte structural reactivity and plasticity in models of retinal detachment. Exp. Eye Res. 2016, 150, 4–21. [Google Scholar] [CrossRef] [PubMed]
- Huxlin, K.R.; Sefton, A.J.; Furby, J.H. The origin and development of retinal astrocytes in the mouse. J. Neurocytol. 1992, 21, 530–544. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Raff, M.C. Retinal astrocytes are immigrants from the optic nerve. Nature 1988, 332, 834–837. [Google Scholar] [CrossRef] [PubMed]
- Chan-Ling, T.; Stone, J. Degeneration of astrocytes in feline retinopathy of prematurity causes failure of the blood-retinal barrier. Investig. Ophthalmol. Vis. Sci. 1992, 33, 2148–2159. [Google Scholar]
- Shweiki, D.; Itin, A.; Soffer, D.; Keshet, E. Vascular endothelial growth factor induced bvhvpoxia may mediate hypoxia-initiated angiogenesis. Nature 1992, 359, 843–845. [Google Scholar] [CrossRef] [PubMed]
- Ashton, N. Oxygen and the growth and development of retinal vessels. Am. J. Ophthalmol. 1966, 62, 412–435. [Google Scholar] [CrossRef]
- Beharry, K.D.; Cai, C.L.; Ahmad, T.; Guzel, S.; Valencia, G.B.; Aranda, J.V. Impact of chronic neonatal intermittent hypoxia on severity of retinal damage in a rat model of oxygen-induced retinopathy. J. Nat. Sci. (JNSCI) 2018, 4, e488. [Google Scholar] [PubMed]
- Beharry, K.D.; Cai, C.L.; Valencia, G.B.; Lazzaro, D.; Valencia, A.M.; Salomone, F.; Aranda, J.V. Human retinal endothelial cells and astrocytes cultured on 3-D scaffolds for ocular drug discovery and development. Prostaglandins Lipid Mediat. 2018, 134, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Aranda, J.V.; Cai, C.L.; Ahmad, T.; Bronshtein, V.; Sadeh, J.; Valencia, G.B.; Lazzaro, D.R.; Beharry, K.D. Pharmacologic synergism of ocular ketorolac and systemic caffeine citrate in rat oxygen-induced retinopathy. Pediatr. Res. 2016, 80, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Beharry, K.D.; Shen, X.; Li, J.; Wang, L.; Aranda, J.V.; Qu, J. Proteomic profiling of the retinas in a neonatal rat model of oxygen-induced retinopathy with a reproducible ion-current-based MS1 approach. J. Proteome Res. 2015, 14, 2109–2120. [Google Scholar] [CrossRef] [PubMed]
- Jivabhai Patel, S.; Bany-Mohammed, F.; McNally, L.; Valencia, G.B.; Lazzaro, D.R.; Aranda, J.V.; Beharry, K.D. Exogenous superoxide dismutase mimetic without scavenging H2O2 causes photoreceptor damage in a rat model for oxygen-induced retinopathy. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1665–1677. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Beharry, K.D.; Cai, C.L.; Sharma, P.; Bronshtein, V.; Valencia, G.B.; Lazzaro, D.R.; Aranda, J.V. Hydrogen peroxide accumulation in the choroid during intermittent hypoxia increases risk of severe oxygen-induced retinopathy in neonatal rats. Investig. Ophthalmol. Vis. Sci. 2013, 54, 7644–7657. [Google Scholar] [CrossRef] [PubMed]
- Brock, R.S.; Gebrekristos, B.H.; Kuniyoshi, K.M.; Modanlou, H.D.; Falcao, M.C.; Beharry, K.D. Biomolecular effects of JB1 (an IGF-I peptide analog) in a rat model of oxygen-induced retinopathy. Pediatr. Res. 2011, 69, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Coleman, R.J.; Beharry, K.D.; Brock, R.; Abad-Santos, P.; Abad-Santos, M.; Modanlou, H.D. Effects of brief, clustered versus dispersed hypoxic episodes on systemic and ocular growth factors in a rat model of oxygen-induced retinopathy. Pediatr. Res. 2008, 64, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.J.; Cai, C.L.; Shrier, E.M.; McNally, L.; Lazzaro, D.R.; Aranda, J.V.; Beharry, K.D. Ocular adverse effects of intravitreal bevacizumab are potentiated by intermittent hypoxia in a rat model of oxygen-induced retinopathy. J. Ophthalmol. 2017, 2017, 4353129. [Google Scholar] [CrossRef] [PubMed]
- Tenhola, S.; Halonen, P.; Jaaskelainen, J.; Voutilainen, R. Serum markers of GH and insulin action in 12-year-old children born small for gestational age. Eur. J. Endocrinol. 2005, 152, 335–340. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okada, T.; Takahashi, S.; Nagano, N.; Yoshikawa, K.; Usukura, Y.; Hosono, S. Early postnatal alteration of body composition in preterm and small-for-gestational-age infants: Implications of catch-upfat. Pediatr. Res. 2015, 77, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Rocco, M.; D’Itri, L.; De Bels, D.; Corazza, F.; Balestra, C. The “normobaric oxygen paradox”: A new tool for the anesthetist? Minerva Anestesiol. 2014, 80, 366–372. [Google Scholar] [PubMed]
- Semenza, G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999, 15, 551–578. [Google Scholar] [CrossRef] [PubMed]
- Wenger, R. Mammalian oxygen sensing, signalling and gene regulation. J. Exp. Biol. 2000, 203, 1253–1263. [Google Scholar] [PubMed]
- Bell, E.L.; Emerling, B.M.; Chandel, N.S. Mitochondrial regulation of oxygen sensing. Mitochondrion 2005, 5, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S.; Schumacker, P.T. Cellular oxygen sensing by mitochondria: Old questions, new insight. J. Appl. Physiol. 2000, 88, 1880–1889. [Google Scholar] [CrossRef] [PubMed]
- Guzy, R.D.; Schumacker, P.T. Oxygen sensing by mitochondria at complex III: The paradox of increased reactive oxygen species during hypoxia. Exp. Physiol. 2006, 91, 807–819. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.E.; Gu, J.; Schau, M.; Bunn, H.F. Regulation of hypoxia inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc. Natl. Acad. Sci. USA 1998, 95, 7987–7992. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Gorlach, A. Reactive oxygen species in the control of hypoxia-inducible factor-mediated gene expression. Semin. Cell Dev. Biol. 2005, 16, 474–486. [Google Scholar] [CrossRef] [PubMed]
- Donati, A.; Damiani, E.; Zuccari, S.; Domizi, R.; Scorcella, C.; Girardis, M.; Giulietti, A.; Vignini, A.; Adrario, E.; Romano, R.; Mazzanti, L.; Pelaia, P.; Singer, M. Effects of short-term hyperoxia on erythropoietin levels and microcirculation in critically Ill patients: A prospective observational pilot study. BMC Anesthesiol. 2017, 17, 49. [Google Scholar] [CrossRef] [PubMed]
- Cimino, F.; Balestra, C.; Germonpré, P.; De Bels, D.; Tillmans, F.; Saija, A.; Speciale, A.; Virgili, F. Pulsed high oxygen induces a hypoxic-like response in human umbilical endothelial cells and in humans. J. Appl. Physiol. 2012, 113, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Livne-Bar, I.; Lam, S.; Chan, D.; Guo, X.; Askar, I.; Nahirnyj, A.; Flanagan, J.G.; Sivak, J.M. Pharmacologic inhibition of reactive gliosis blocks TNF-α-mediated neuronal apoptosis. Cell Death Dis. 2016, 7, e2386. [Google Scholar] [CrossRef] [PubMed]
- Formichella, C.R.; Abella, S.K.; Sims, S.M.; Cathcart, H.M.; Sappington, R.M. Astrocyte reactivity: A biomarker for retinal ganglion cell health in retinal neurodegeneration. J. Clin. Cell. Immunol. 2014, 5, 188. [Google Scholar] [CrossRef] [PubMed]
- Son, J.L.; Soto, I.; Oglesby, E.; Lopez-Roca, T.; Pease, M.E.; Quigley, H.A.; Marsh-Armstrong, N. Glaucomatous optic nerve injury involves early astrocyte reactivity and late oligodendrocyte loss. Glia 2010, 58, 780–789. [Google Scholar] [CrossRef] [PubMed]
- Pekny, M.; Pekna, M. Astrocyte reactivity and reactive astrogliosis: Costs and benefits. Physiol. Rev. 2014, 94, 1077–1098. [Google Scholar] [CrossRef] [PubMed]
- McLeod, S.D.; D’Anna, S.A.; Lutty, G.A. Clinical and histopathologic features of canine oxygen-induced proliferative retinopathy. Investig. Ophthalmol. Vis. Sci. 1998, 39, 1918–1932. [Google Scholar]
- Secondi, R.; Kong, J.; Blonska, A.M.; Staurenghi, G.; Sparrow, J.R. Fundus autofluorescence findings in a mouse model of retinal detachment. Investig. Ophthalmol. Vis. Sci. 2012, 53, 5190–5197. [Google Scholar] [CrossRef] [PubMed]
- Romano, M.R.; Biagioni, F.; Besozzi, G.; Carrizzo, A.; Vecchione, C.; Fornai, F.; Lograno, M.D. Effects of bevacizumab on neuronal viability of retinal ganglion cells in rats. Brain Res. 2012, 1478, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Penn, J.S.; Tolman, B.L.; Lowery, L.A.; Koutz, C.A. Oxygen-induced retinopathy in the rat: Hemorrhages and dysplasias may lead to retinal detachment. Curr. Eye Res. 1992, 11, 939–953. [Google Scholar] [CrossRef] [PubMed]
- Maslim, J.; Valter, K.; Egensperger, R.; Hollander, H.; Stone, J. Tissue oxygen during a critical developmental period controls the death and survival of photoreceptors. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1667–1677. [Google Scholar]
- Maldonado, R.S.; O’Connell, R.; Ascher, S.B.; Sarin, N.; Freedman, S.F.; Wallace, D.K.; Chiu, S.J.; Farsiu, S.; Cotton, M.; Toth, C.A. Spectral-domain optical coherence tomographic assessment of severity of cystoid macular edema in retinopathy of prematurity. Arch. Ophthalmol. 2012, 130, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Vogler, S.; Pannicke, T.; Hollborn, M.; Grosche, A.; Busch, S.; Hoffmann, S.; Wiedemann, P.; Reichenbach, A.; Hammes, H.P.; Bringmann, A. Müller cell reactivity in response to photoreceptor degeneration in rats with defective polycystin-2. PLoS ONE 2013, 8, e61631. [Google Scholar] [CrossRef] [PubMed]
- Binnes, K.E.; Salt, T.E. Post eye-opening maturation of visual receptive field diameters in superior colliculus of normal- and dark-reared rats. Dev. Brain Res. 1997, 99, 263–266. [Google Scholar] [CrossRef]
- Robinson, S.R.; Dreher, B. The visual pathways of eutherian mammals and marsupials develop according to a common timetable. Brain Behav. Evol. 1990, 36, 177–195. [Google Scholar] [CrossRef] [PubMed]
- Dreher, B.; Robinson, S.R. Development of the retinofugal pathway in birds and mammals: Evidence for a common “timetable”. Brain Behav. Evol. 1988, 31, 369–390. [Google Scholar] [CrossRef] [PubMed]
- Nakano, A.; Nakahara, T.; Mori, A.; Ushikubo, H.; Sakamoto, K.; Ishii, K. Short-term treatment with VEGF receptor inhibitors induces retinopathy of prematurity-like abnormal vascular growth in neonatal rats. Exp. Eye Res. 2016, 143, 120–131. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| RA | 50% | 50–12% | 50–21% | 21–12% | |
|---|---|---|---|---|---|
| P14: | |||||
| %Change Wt. | 216.0 ± 8.6 | 241.1 ± 13.4 ** | 198.9 ± 6.6 ** | 236.1 ± 13.6 ** | 251.2 ± 12.0 **# |
| %Change Lgth. | 72.0 ± 2.2 | 62.0 ± 2.8 ** | 45.8 ± 1.4 ** | 55.3 ± 1.4 ** | 54.3 ± 2.0 **## |
| Brain/Body Wt. Ratios | 0.054 ± 0.0016 | 0.052 ± 0.002 | 0.05 ± 0.0014 * | 0.051 ± 0.0015 | 0.048 ± 0.0015 * |
| Lung/Body Wt. Ratios | 0.018 ± 0.0005 | 0.016 ± 0.0007 * | 0.02 ± 0.0009 | 0.02 ± 0.0007 | 0.018 ± 0.0007 |
| P21: | |||||
| %Change Wt. | 317.4 ± 17.4 | 493.9 ± 17.6 ** | 402.1 ± 10.9 ** | 443.1 ± 15.5 ** | 482.2 ± 13.8 **## |
| %Change Lgth. | 94.8 ± 4.0 | 99.1 ± 1.4 ** | 72.3 ± 2.2 ** | 94.2 ± 2.0 | 94.5 ± 3.7 ## |
| Brain/Body Wt. Ratios | 0.05 ± 0.002 | 0.035 ± 0.0009 ** | 0.042 ± 0.0006 ** | 0.037 ± 0.0012 ** | 0.034 ± 0.001 **## |
| Lung/Body Wt. Ratios | 0.014 ± 0.0004 | 0.011 ± 0.0006 ** | 0.013 ± 0.0004 | 0.011 ± 0.0006 ** | 0.01 ± 0.0006 **## |
| RA | 50% | 50–12% | 50–21% | 21–12% | |
|---|---|---|---|---|---|
| Left Eye | 36/36 (100%) | 25/36 (69%) ** | 5/36 (14%) ** | 25/36 (69%) ** | 29/36 ## (81%) *## |
| Right Eye | 36/36 (100%) | 26/36 (72%) ** | 3/36 (8%) ** | 25/36 (69%) ** | 27/36 ## (75%) **## |
| Both Eyes | 36/36 (100%) | 20/36 (56%) ** | 1/36 (3%) ** | 23/36 (64%) ** | 27/36 ## (75%) **## |
| Vascular Parameters | RA | 50% | 50–12% | 50–21% | 21–12% |
|---|---|---|---|---|---|
| Vascular Density (%) | 45.6 ± 0.9 | 48.4 ± 1.5 | 53.6 ± 1.9 ** | 55.2 ± 1.2 ** | 49.0.2 ± 1.2 # |
| Total Vascular Area | 28,525.3 ± 618.4 | 30,242 ± 1062 | 33,440.4 ± 1325.2 ** | 32,104.6 ± 581.5 * | 31,514.7 ± 561.0 * |
| No. Branching Points | 922.4 ± 38.4 | 956.8 ± 67.8 | 1261.8 ± 92.5 ** | 1145.7 ± 32.7 * | 1174.4 ± 48.0 * |
| No. Segments | 1549.8 ± 59.7 | 1631.0 ± 104.4 | 2091.4 ± 150.9 ** | 1897.3 ± 75.6 | 1837.3 ± 109.0 |
| Mean Segment Length | 16.0 ± 0.4 | 16.5 ± 0.65 | 19.2 ± 0.69 ** | 18.9 ± 0.46 ** | 17.9 ± 0.49 * |
| Measurements | RA | 50% | 50–12% | 50–21% | 21–12% |
|---|---|---|---|---|---|
| Tortuosity Index | 1.08 ± 0.01 | 1.26 ± 0.02 ** | 1.51 ± 0.01 ** | 1.36 ± 0.02 ** | 1.30 ± 0.02 **## |
| Diameter of Arteries | 45.2 ± 1.7 | 37.8 ± 1.3 ** | 58.6 ± 1.0 ** | 48.8 ± 1.3 | 48.9 ± 1.1 ## |
| Diameter of Veins | 28.0 ± 0.6 | 25.0 ± 0.6 ** | 24.9 ± 0.6 ** | 27.4 ± 0.6 | 31.6 ± 0.9 *## |
| Number Cells in NFL/GCL | 17.9 ± 0.8 | 35.9 ± 3.0 ** | 47.6 ± 2.0 ** | 56.8 ± 5.1 ** | 50.9 ± 3.1 ** |
| Total Retinal Thickness (µm) | 283.1 ± 14.7 | 225.7 ± 3.1 | 468.7 ± 60.2 ** | 412.7 ± 3.1 ** | 420.5 ± 7.1 ** |
| NFL/GCL Thickness (µm) | 39.9 ± 3.7 | 56.8 ± 1.6 | 90.1 ± 15.6 ** | 69.3 ± 2.5 * | 66.3 ± 1.4 * |
| IPL Thickness (µm) | 48.4 ± 6.1 | 45.8 ± 1.3 | 103.5 ± 24.5 ** | 59.7 ± 0.6 | 69.4 ± 2.7 # |
| INL Thickness (µm) | 70.8 ± 5.7 | 63.4 ± 0.9 | 99.5 ± 34.4 * | 63.6 ± 1.7 | 99.0 ± 3.2 |
| ONL Thickness (µm) | 90.3 ± 9.2 | 40.9 ± 1.2 ** | 151.3 ± 17.5 ** | 96.4 ± 4.6 | 134.3 ± 6.3 ** |
| RA | 50% | 50–12% | 50–21% | 21–12% | |
|---|---|---|---|---|---|
| P14: | |||||
| VEGF (pg/mL) | 40.7 ± 5.4 | 22.7 ± 1.3 ** | 256.6 ± 15.3 ** | 232.4 ± 6.2 ** | 237.2 ± 6.3 ** |
| IGF-I (pg/mL) | 3619.1 ± 166.9 | 4732.8 ± 158.0 ** | 6567.0 ± 136.0 ** | 848.1 ± 160.8 ** | 779.5 ± 102.6 **## |
| sVEGFR (pg/mL) | 615.8 ± 84.1 | 528.8 ± 82.4 | 486.5 ± 25.2* | 334.9 ± 72.0 | 452.4 ± 102.0 |
| P21: | |||||
| VEGF (pg/mL) | 45.7 ± 7.7 | 11.02 ± 0.67 * | 211.0 ± 3.8 ** | 201.5 ± 9.7 ** | 210.3 ± 13.4 ** |
| IGF-I (pg/mL) | 5512.1 ± 212.1 | 4748.9 ± 123.3 ** | 7679.3 ± 121.5 ** | 1248.7 ± 135.5 ** | 1219.7 ± 121.2 **## |
| sVEGFR (pg/mL) | 704.8 ± 62.6 | 199.6 ± 67.2 ** | 128.9 ± 22.0 ** | 334.9 ± 19.7 ** | 315.9 ± 10.4 **# |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beharry, K.D.; Cai, C.L.; Skelton, J.; Siddiqui, F.; D’Agrosa, C.; Calo, J.; Valencia, G.B.; Aranda, J.V. Oxygen-Induced Retinopathy from Recurrent Intermittent Hypoxia Is Not Dependent on Resolution with Room Air or Oxygen, in Neonatal Rats. Int. J. Mol. Sci. 2018, 19, 1337. https://doi.org/10.3390/ijms19051337
Beharry KD, Cai CL, Skelton J, Siddiqui F, D’Agrosa C, Calo J, Valencia GB, Aranda JV. Oxygen-Induced Retinopathy from Recurrent Intermittent Hypoxia Is Not Dependent on Resolution with Room Air or Oxygen, in Neonatal Rats. International Journal of Molecular Sciences. 2018; 19(5):1337. https://doi.org/10.3390/ijms19051337
Chicago/Turabian StyleBeharry, Kay D., Charles L. Cai, Jacqueline Skelton, Faisal Siddiqui, Christina D’Agrosa, Johanna Calo, Gloria B. Valencia, and Jacob V. Aranda. 2018. "Oxygen-Induced Retinopathy from Recurrent Intermittent Hypoxia Is Not Dependent on Resolution with Room Air or Oxygen, in Neonatal Rats" International Journal of Molecular Sciences 19, no. 5: 1337. https://doi.org/10.3390/ijms19051337
APA StyleBeharry, K. D., Cai, C. L., Skelton, J., Siddiqui, F., D’Agrosa, C., Calo, J., Valencia, G. B., & Aranda, J. V. (2018). Oxygen-Induced Retinopathy from Recurrent Intermittent Hypoxia Is Not Dependent on Resolution with Room Air or Oxygen, in Neonatal Rats. International Journal of Molecular Sciences, 19(5), 1337. https://doi.org/10.3390/ijms19051337