The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function



Abstract
1. Introduction
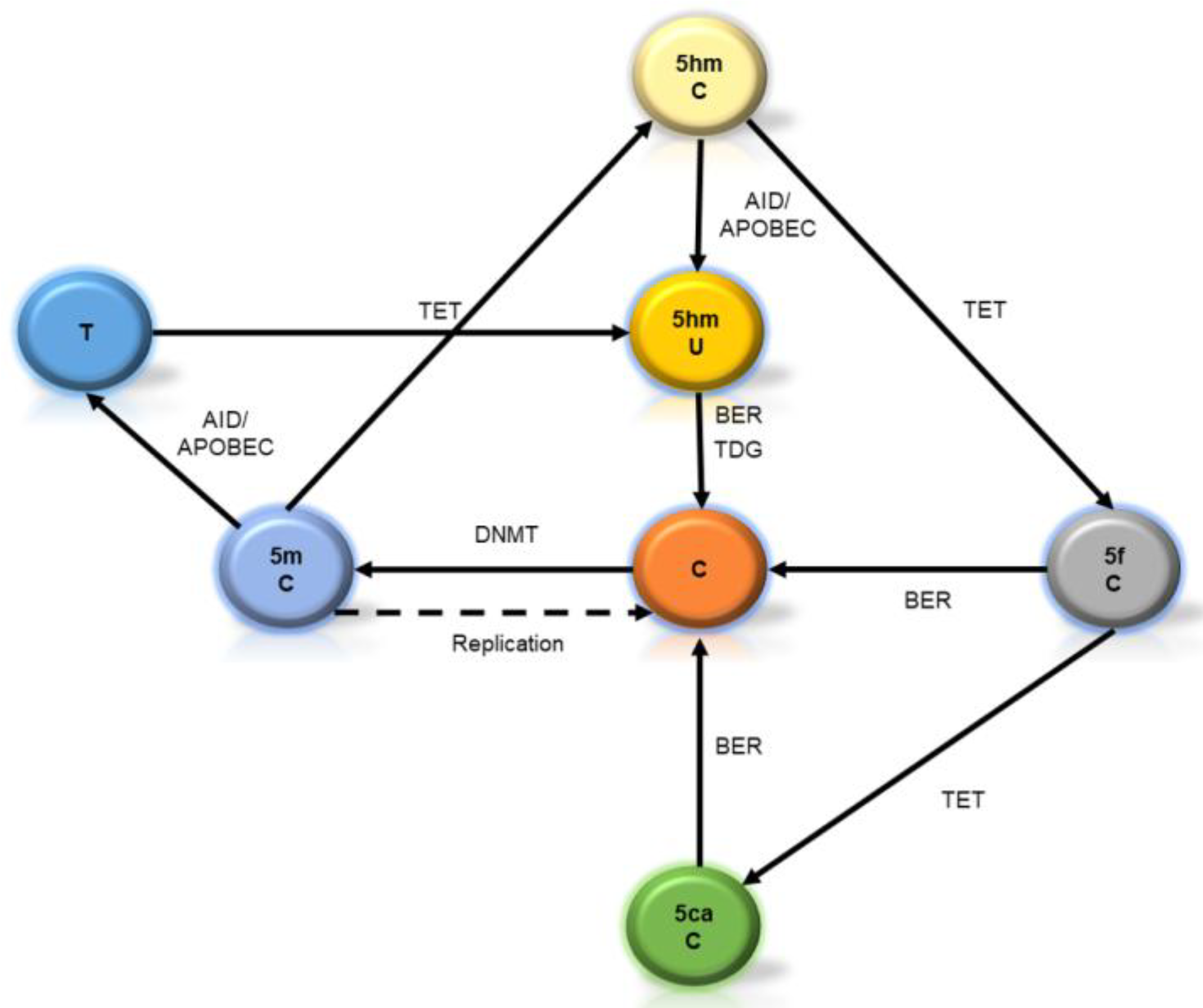
2. Starting from Scratch: Is DNA Methylation a Stable Mark?
Non-CG Methylation
3. Histone Post-Translational Modifications: An Unstable Mark?
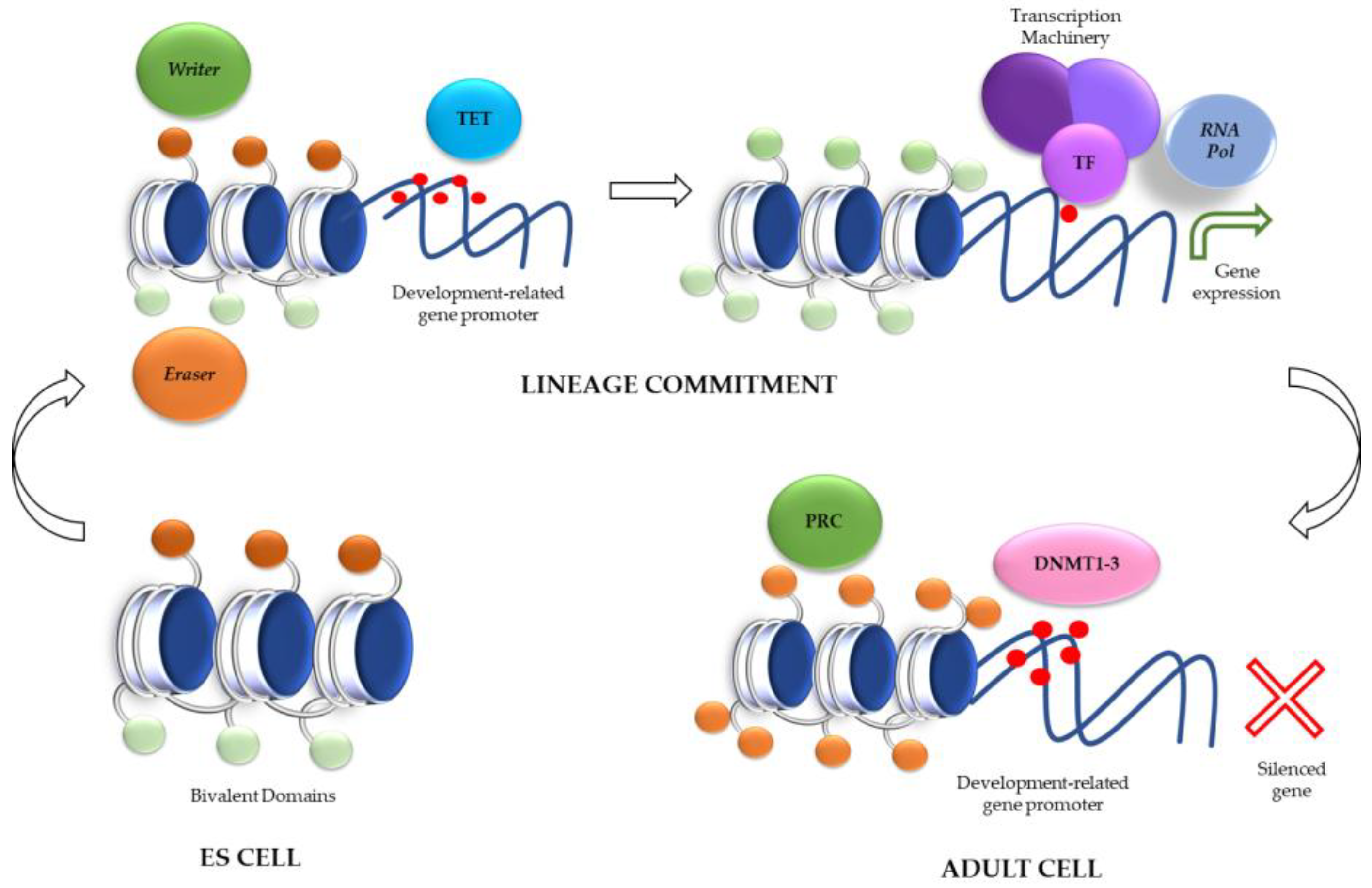
Role of Histone Modifications in ES Cells: Bivalent Domains
4. A Single-Strand Contribution: Epigenetic Mechanisms Mediated by Non-Coding RNA
5. Epigenetics of Early Development
5.1. Epigenetic Remodelling on the Zygote
5.2. Epigenetic Remodelling on Gametes and Somatic Tissues
6. Early Stress and Postnatal Environmental Influences on the Brain: An Epigenetic Answer to Long-Term Effects?
7. Future Perspectives
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviations
| MeSH | Medical Subject Headings |
| 5mC | 5-Methylcytosine |
| EWAS | Epigenome-Wide Association Studies |
| DNMT | DNA methyl-transferases |
| 5hmC | 5-Hydroximethylcitosine |
| BER | Base-excision Repair System |
| ES | Embryonic Stem Cells |
| PTM | Post translational Modifications |
| ChIP | Chromatin Immunoprecipitation |
| PRC | Polycomb-Repressive Complexes |
| EZH2 | Enhancer of zeste 2 polycomb repressive complex 2 subunit |
| lnRNA | Long non-coding RNA |
| miRNA | MicroRNA |
| PGC | Primordial germ cells |
References
- Noble, D. Conrad Waddington and the origin of epigenetics. J. Exp. Biol. 2015, 218, 816–818. [Google Scholar] [CrossRef] [PubMed]
- Leenen, F.A.; Muller, C.P.; Turner, J.D. DNA methylation: Conducting the orchestra from exposure to phenotype? Clin. Epigenetics 2016, 8, 92. [Google Scholar] [CrossRef] [PubMed]
- Roadmap Epigenomics, C.; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef]
- Fullard, J.F.; Halene, T.B.; Giambartolomei, C.; Haroutunian, V.; Akbarian, S.; Roussos, P. Understanding the genetic liability to schizophrenia through the neuroepigenome. Schizophr. Res. 2016, 177, 115–124. [Google Scholar] [CrossRef] [PubMed]
- Portela, A.; Esteller, M. Epigenetic modifications and human disease. Nat. Biotechnol. 2010, 28, 1057–1068. [Google Scholar] [CrossRef] [PubMed]
- Puri, B.; Hall, A.; Ho, R. Revision Notes in Psychiatry, 3rd ed.; CRC Press: New York, NY, USA, 2014. [Google Scholar]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Ooi, S.K.; Bestor, T.H. The colorful history of active DNA demethylation. Cell 2008, 133, 1145–1148. [Google Scholar] [CrossRef] [PubMed]
- Koivunen, P.; Laukka, T. The TET enzymes. Cell. Mol. Life Sci. 2018, 75, 1339–1348. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Zhang, Y.; Hart, R.P.; Chen, J.; Herrup, K.; Li, J. Alteration in 5-hydroxymethylcytosine-mediated epigenetic regulation leads to Purkinje cell vulnerability in ATM deficiency. Brain 2015, 138, 3520–3536. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Inoue, K.; Ono, R.; Ogonuki, N.; Kohda, T.; Kaneko-Ishino, T.; Ogura, A.; Ishino, F. Erasing genomic imprinting memory in mouse clone embryos produced from day 11.5 primordial germ cells. Development 2002, 129, 1807–1817. [Google Scholar] [PubMed]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef] [PubMed]
- Ficz, G.; Branco, M.R.; Seisenberger, S.; Santos, F.; Krueger, F.; Hore, T.A.; Marques, C.J.; Andrews, S.; Reik, W. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature 2011, 473, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Szulwach, K.E.; Li, X.; Li, Y.; Song, C.X.; Wu, H.; Dai, Q.; Irier, H.; Upadhyay, A.K.; Gearing, M.; Levey, A.I.; et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci. 2011, 14, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; Mukamel, E.A.; Nery, J.R.; Urich, M.; Puddifoot, C.A.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global epigenomic reconfiguration during mammalian brain development. Science 2013, 341, 1237905. [Google Scholar] [CrossRef] [PubMed]
- Mellen, M.; Ayata, P.; Dewell, S.; Kriaucionis, S.; Heintz, N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell 2012, 151, 1417–1430. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Li, X.; Yan, L.; Tan, Y.; Li, R.; Zhao, Y.; Wang, Y.; Xie, J.; Zhang, Y.; Song, C.; et al. Whole-genome analysis of 5-hydroxymethylcytosine and 5-methylcytosine at base resolution in the human brain. Genome Biol. 2014, 15, R49. [Google Scholar] [CrossRef] [PubMed]
- Arand, J.; Spieler, D.; Karius, T.; Branco, M.R.; Meilinger, D.; Meissner, A.; Jenuwein, T.; Xu, G.; Leonhardt, H.; Wolf, V.; et al. In vivo control of CpG and non-CpG DNA methylation by DNA methyltransferases. PLoS Genet. 2012, 8, e1002750. [Google Scholar] [CrossRef] [PubMed]
- Ramsahoye, B.H.; Biniszkiewicz, D.; Lyko, F.; Clark, V.; Bird, A.P.; Jaenisch, R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc. Natl. Acad. Sci. USA 2000, 97, 5237–5242. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Muller, F.; Liao, J.; Zhang, Y.; Gu, H.; Bock, C.; Boyle, P.; Epstein, C.B.; Bernstein, B.E.; Lengauer, T.; et al. Genomic distribution and inter-sample variation of non-CpG methylation across human cell types. PLoS Genet. 2011, 7, e1002389. [Google Scholar] [CrossRef] [PubMed]
- Butcher, L.M.; Ito, M.; Brimpari, M.; Morris, T.J.; Soares, F.A.; Ahrlund-Richter, L.; Carey, N.; Vallier, L.; Ferguson-Smith, A.C.; Beck, S. Non-CG DNA methylation is a biomarker for assessing endodermal differentiation capacity in pluripotent stem cells. Nat. Commun. 2016, 7, 10458. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Su, Y.; Shin, J.H.; Shin, J.; Li, H.; Xie, B.; Zhong, C.; Hu, S.; Le, T.; Fan, G.; et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat. Neurosci. 2014, 17, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Park, S.J.; Nakai, K. Differential landscape of non-CpG methylation in embryonic stem cells and neurons caused by DNMT3s. Sci. Rep. 2017, 7, 11295. [Google Scholar] [CrossRef] [PubMed]
- Koyama, M.; Kurumizaka, H. Structural diversity of the nucleosome. J. Biochem. 2018, 163, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sabari, B.R.; Garcia, B.A.; Allis, C.D.; Zhao, Y. SnapShot: Histone modifications. Cell 2014, 159, 458–458.e1. [Google Scholar] [CrossRef] [PubMed]
- Latham, J.A.; Dent, S.Y. Cross-regulation of histone modifications. Nat. Struct. Mol. Biol. 2007, 14, 1017–1024. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.; Schneider, R. Chatting histone modifications in mammals. Brief. Funct. Genom. 2010, 9, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Karch, K.R.; Denizio, J.E.; Black, B.E.; Garcia, B.A. Identification and interrogation of combinatorial histone modifications. Front. Genet. 2013, 4, 264. [Google Scholar] [CrossRef] [PubMed]
- Abshiru, N.; Rajan, R.E.; Verreault, A.; Thibault, P. Unraveling Site-Specific and Combinatorial Histone Modifications Using High-Resolution Mass Spectrometry in Histone Deacetylase Mutants of Fission Yeast. J. Proteome Res. 2016, 15, 2132–2142. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Denu, J.M. Reading the Combinatorial Histone Language. ACS Chem. Biol. 2016, 11, 564–574. [Google Scholar] [CrossRef] [PubMed]
- Fenley, A.T.; Anandakrishnan, R.; Kidane, Y.H.; Onufriev, A.V. Modulation of nucleosomal DNA accessibility via charge-altering post-translational modifications in histone core. Epigenetics Chromatin 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Vogelauer, M.; Wu, J.; Suka, N.; Grunstein, M. Global histone acetylation and deacetylation in yeast. Nature 2000, 408, 495–498. [Google Scholar] [CrossRef] [PubMed]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Mikkelsen, T.S.; Xie, X.; Kamal, M.; Huebert, D.J.; Cuff, J.; Fry, B.; Meissner, A.; Wernig, M.; Plath, K.; et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 2006, 125, 315–326. [Google Scholar] [CrossRef] [PubMed]
- Cui, K.; Zang, C.; Roh, T.Y.; Schones, D.E.; Childs, R.W.; Peng, W.; Zhao, K. Chromatin signatures in multipotent human hematopoietic stem cells indicate the fate of bivalent genes during differentiation. Cell Stem Cell 2009, 4, 80–93. [Google Scholar] [CrossRef] [PubMed]
- Lien, W.H.; Guo, X.; Polak, L.; Lawton, L.N.; Young, R.A.; Zheng, D.; Fuchs, E. Genome-wide maps of histone modifications unwind in vivo chromatin states of the hair follicle lineage. Cell Stem Cell 2011, 9, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Rugg-Gunn, P.J.; Cox, B.J.; Ralston, A.; Rossant, J. Distinct histone modifications in stem cell lines and tissue lineages from the early mouse embryo. Proc. Natl. Acad. Sci. USA 2010, 107, 10783–10790. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, C.; Liu, W.; Li, J.; Li, C.; Kou, X.; Chen, J.; Zhao, Y.; Gao, H.; Wang, H.; et al. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature 2016, 537, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Hannon, G.J.; Brennecke, J. The Piwi-piRNA pathway provides an adaptive defense in the transposon arms race. Science 2007, 318, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Stuwe, E.; Toth, K.F.; Aravin, A.A. Small but sturdy: Small RNAs in cellular memory and epigenetics. Genes Dev. 2014, 28, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Li, L.C. Chromatin remodeling by the small RNA machinery in mammalian cells. Epigenetics 2014, 9, 45–52. [Google Scholar] [CrossRef] [PubMed]
- De Rie, D.; Abugessaisa, I.; Alam, T.; Arner, E.; Arner, P.; Ashoor, H.; Astrom, G.; Babina, M.; Bertin, N.; Burroughs, A.M.; et al. An integrated expression atlas of miRNAs and their promoters in human and mouse. Nat. Biotechnol. 2017, 35, 872–878. [Google Scholar] [CrossRef] [PubMed]
- Itou, D.; Shiromoto, Y.; Yukiho, S.Y.; Ishii, C.; Nishimura, T.; Ogonuki, N.; Ogura, A.; Hasuwa, H.; Fujihara, Y.; Kuramochi-Miyagawa, S.; et al. Induction of DNA methylation by artificial piRNA production in male germ cells. Curr. Biol. 2015, 25, 901–906. [Google Scholar] [CrossRef] [PubMed]
- Saito, K. The epigenetic regulation of transposable elements by PIWI-interacting RNAs in Drosophila. Genes Genet. Syst. 2013, 88, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Aravin, A.A.; Bourc’his, D. Small RNA guides for de novo DNA methylation in mammalian germ cells. Genes Dev. 2008, 22, 970–975. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; O’Carroll, D.; Tan, C.L.; Hillman, D.; Sugimori, M.; Llinas, R.; Greengard, P. Cerebellar neurodegeneration in the absence of microRNAs. J. Exp. Med. 2007, 204, 1553–1558. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Inoue, K.; Ishii, J.; Vanti, W.B.; Voronov, S.V.; Murchison, E.; Hannon, G.; Abeliovich, A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science 2007, 317, 1220–1224. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.M.; Zhang, C.L. TLX: A master regulator for neural stem cell maintenance and neurogenesis. Biochim. Biophys. Acta 2015, 1849, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Goodfellow, M.; Phillips, N.E.; Manning, C.; Galla, T.; Papalopulu, N. MicroRNA input into a neural ultradian oscillator controls emergence and timing of alternative cell states. Nat. Commun. 2014, 5, 3399. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.L.; Ohtsuka, T.; Gonzalez, A.; Kageyama, R. MicroRNA9 regulates neural stem cell differentiation by controlling Hes1 expression dynamics in the developing brain. Genes Cells 2012, 17, 952–961. [Google Scholar] [CrossRef] [PubMed]
- Marson, A.; Levine, S.S.; Cole, M.F.; Frampton, G.M.; Brambrink, T.; Johnstone, S.; Guenther, M.G.; Johnston, W.K.; Wernig, M.; Newman, J.; et al. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 2008, 134, 521–533. [Google Scholar] [CrossRef] [PubMed]
- Jauhari, A.; Singh, T.; Yadav, S. Expression of miR-145 and Its Target Proteins Are Regulated by miR-29b in Differentiated Neurons. Mol. Neurobiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, J.A.; Lau, P.; Maric, D.; Barker, J.L.; Hudson, L.D. Integrating microRNA and mRNA expression profiles of neuronal progenitors to identify regulatory networks underlying the onset of cortical neurogenesis. BMC Neurosci. 2009, 10, 98. [Google Scholar] [CrossRef] [PubMed]
- Clovis, Y.M.; Enard, W.; Marinaro, F.; Huttner, W.B.; de Pietri Tonelli, D. Convergent repression of Foxp2 3′UTR by miR-9 and miR-132 in embryonic mouse neocortex: Implications for radial migration of neurons. Development 2012, 139, 3332–3342. [Google Scholar] [CrossRef] [PubMed]
- Winter, J. MicroRNAs of the miR379-410 cluster: New players in embryonic neurogenesis and regulators of neuronal function. Neurogenesis 2015, 2, e1004970. [Google Scholar] [CrossRef] [PubMed]
- Jossin, Y.; Cooper, J.A. Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nat. Neurosci. 2011, 14, 697–703. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Kim, H.J.; Schafer, S.T.; Paquola, A.; Clemenson, G.D.; Toda, T.; Oh, J.; Pankonin, A.R.; Lee, B.S.; Johnston, S.T.; et al. Functional Implications of miR-19 in the Migration of Newborn Neurons in the Adult Brain. Neuron 2016, 91, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Kocerha, J.; Faghihi, M.A.; Lopez-Toledano, M.A.; Huang, J.; Ramsey, A.J.; Caron, M.G.; Sales, N.; Willoughby, D.; Elmen, J.; Hansen, H.F.; et al. MicroRNA-219 modulates NMDA receptor-mediated neurobehavioral dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 3507–3512. [Google Scholar] [CrossRef] [PubMed]
- Persengiev, S.P.; Kondova, I.I.; Bontrop, R.E. The Impact of MicroRNAs on Brain Aging and Neurodegeneration. Curr. Gerontol. Geriatr. Res. 2012, 2012, 359369. [Google Scholar] [CrossRef] [PubMed]
- Inukai, S.; de Lencastre, A.; Turner, M.; Slack, F. Novel microRNAs differentially expressed during aging in the mouse brain. PLoS ONE 2012, 7, e40028. [Google Scholar] [CrossRef] [PubMed]
- Danka Mohammed, C.P.; Park, J.S.; Nam, H.G.; Kim, K. MicroRNAs in brain aging. Mech. Ageing Dev. 2017, 168, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.K.; Finniss, S.; Cazacu, S.; Xiang, C.; Brodie, C. Mesenchymal stem cells deliver exogenous miRNAs to neural cells and induce their differentiation and glutamate transporter expression. Stem Cells Dev. 2014, 23, 2851–2861. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Junaid, M.A. Lifestyle, pregnancy and epigenetic effects. Epigenomics 2015, 7, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Mondanizadeh, M.; Arefian, E.; Mosayebi, G.; Saidijam, M.; Khansarinejad, B.; Hashemi, S.M. MicroRNA-124 regulates neuronal differentiation of mesenchymal stem cells by targeting Sp1 mRNA. J. Cell. Biochem. 2015, 116, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Makeyev, E.V.; Zhang, J.; Carrasco, M.A.; Maniatis, T. The MicroRNA miR-124 promotes neuronal differentiation by triggering brain-specific alternative pre-mRNA splicing. Mol. Cell 2007, 27, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Adlakha, Y.K.; Saini, N. Brain microRNAs and insights into biological functions and therapeutic potential of brain enriched miRNA-128. Mol. Cancer 2014, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Ye, P.; Murai, K.; Lang, M.F.; Li, S.; Zhang, H.; Li, W.; Fu, C.; Yin, J.; Wang, A.; et al. miR-137 forms a regulatory loop with nuclear receptor TLX and LSD1 in neural stem cells. Nat. Commun. 2011, 2, 529. [Google Scholar] [CrossRef] [PubMed]
- Silber, J.; Lim, D.A.; Petritsch, C.; Persson, A.I.; Maunakea, A.K.; Yu, M.; Vandenberg, S.R.; Ginzinger, D.G.; James, C.D.; Costello, J.F.; et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008, 6, 14. [Google Scholar] [CrossRef] [PubMed]
- Qian, Y.; Song, J.; Ouyang, Y.; Han, Q.; Chen, W.; Zhao, X.; Xie, Y.; Chen, Y.; Yuan, W.; Fan, C. Advances in Roles of miR-132 in the Nervous System. Front. Pharmacol. 2017, 8, 770. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Papagiannakopoulos, T.; Pan, G.; Thomson, J.A.; Kosik, K.S. MicroRNA-145 regulates OCT4, SOX2, and KLF4 and represses pluripotency in human embryonic stem cells. Cell 2009, 137, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Du, Z.W.; Ma, L.X.; Phillips, C.; Zhang, S.C. miR-200 and miR-96 families repress neural induction from human embryonic stem cells. Development 2013, 140, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Dugas, J.C.; Cuellar, T.L.; Scholze, A.; Ason, B.; Ibrahim, A.; Emery, B.; Zamanian, J.L.; Foo, L.C.; McManus, M.T.; Barres, B.A. Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron 2010, 65, 597–611. [Google Scholar] [CrossRef] [PubMed]
- Lichner, Z.; Pall, E.; Kerekes, A.; Pallinger, E.; Maraghechi, P.; Bosze, Z.; Gocza, E. The miR-290-295 cluster promotes pluripotency maintenance by regulating cell cycle phase distribution in mouse embryonic stem cells. Differentiation 2011, 81, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Wakayama, T.; Perry, A.C.; Zuccotti, M.; Johnson, K.R.; Yanagimachi, R. Full-term development of mice from enucleated oocytes injected with cumulus cell nuclei. Nature 1998, 394, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Horsthemke, B. In brief: Genomic imprinting and imprinting diseases. J. Pathol. 2014, 232, 485–487. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Solter, D. Completion of mouse embryogenesis requires both the maternal and paternal genomes. Cell 1984, 37, 179–183. [Google Scholar] [CrossRef]
- Beaujean, N.; Taylor, J.; Gardner, J.; Wilmut, I.; Meehan, R.; Young, L. Effect of limited DNA methylation reprogramming in the normal sheep embryo on somatic cell nuclear transfer. Biol. Reprod. 2004, 71, 185–193. [Google Scholar] [CrossRef] [PubMed]
- Beaujean, N.; Hartshorne, G.; Cavilla, J.; Taylor, J.; Gardner, J.; Wilmut, I.; Meehan, R.; Young, L. Non-conservation of mammalian preimplantation methylation dynamics. Curr. Biol. 2004, 14, R266–R267. [Google Scholar] [CrossRef] [PubMed]
- Fulka, H.; Mrazek, M.; Tepla, O.; Fulka, J., Jr. DNA methylation pattern in human zygotes and developing embryos. Reproduction 2004, 128, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Wu, F.R.; Zhang, Y.; Ding, B.; Lei, X.H.; Huang, J.C.; Wang, C.H.; Liu, Y.; Wang, R.; Li, W.Y. H3K27me3 may be associated with Oct4 and Sox2 in mouse preimplantation embryos. Genet. Mol. Res. 2014, 13, 10121–10129. [Google Scholar] [CrossRef] [PubMed]
- Doherty, A.S.; Bartolomei, M.S.; Schultz, R.M. Regulation of stage-specific nuclear translocation of Dnmt1o during preimplantation mouse development. Dev. Biol. 2002, 242, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Ratnam, S.; Mertineit, C.; Ding, F.; Howell, C.Y.; Clarke, H.J.; Bestor, T.H.; Chaillet, J.R.; Trasler, J.M. Dynamics of Dnmt1 methyltransferase expression and intracellular localization during oogenesis and preimplantation development. Dev. Biol. 2002, 245, 304–314. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Howell, C.Y.; Bestor, T.H.; Ding, F.; Latham, K.E.; Mertineit, C.; Trasler, J.M.; Chaillet, J.R. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell 2001, 104, 829–838. [Google Scholar] [CrossRef]
- Cirio, M.C.; Ratnam, S.; Ding, F.; Reinhart, B.; Navara, C.; Chaillet, J.R. Preimplantation expression of the somatic form of Dnmt1 suggests a role in the inheritance of genomic imprints. BMC Dev. Biol. 2008, 8, 9. [Google Scholar] [CrossRef] [PubMed]
- Hirasawa, R.; Chiba, H.; Kaneda, M.; Tajima, S.; Li, E.; Jaenisch, R.; Sasaki, H. Maternal and zygotic Dnmt1 are necessary and sufficient for the maintenance of DNA methylation imprints during preimplantation development. Genes Dev. 2008, 22, 1607–1616. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Liu, Y.J.; Nakashima, H.; Umehara, H.; Inoue, K.; Matoba, S.; Tachibana, M.; Ogura, A.; Shinkai, Y.; Nakano, T. PGC7 binds histone H3K9me2 to protect against conversion of 5mC to 5hmC in early embryos. Nature 2012, 486, 415–419. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Kalantry, S.; Rao, A. PGC7, H3K9me2 and Tet3: Regulators of DNA methylation in zygotes. Cell Res. 2013, 23, 6–9. [Google Scholar] [CrossRef] [PubMed]
- Maupetit-Mehouas, S.; Montibus, B.; Nury, D.; Tayama, C.; Wassef, M.; Kota, S.K.; Fogli, A.; Cerqueira Campos, F.; Hata, K.; Feil, R.; et al. Imprinting control regions (ICRs) are marked by mono-allelic bivalent chromatin when transcriptionally inactive. Nucleic Acids Res. 2016, 44, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Messerschmidt, D.M.; de Vries, W.; Ito, M.; Solter, D.; Ferguson-Smith, A.; Knowles, B.B. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science 2012, 335, 1499–1502. [Google Scholar] [CrossRef] [PubMed]
- Alexander, K.A.; Wang, X.; Shibata, M.; Clark, A.G.; Garcia-Garcia, M.J. TRIM28 Controls Genomic Imprinting through Distinct Mechanisms during and after Early Genome-wide Reprogramming. Cell Rep. 2015, 13, 1194–1205. [Google Scholar] [CrossRef] [PubMed]
- Kafri, T.; Ariel, M.; Brandeis, M.; Shemer, R.; Urven, L.; McCarrey, J.; Cedar, H.; Razin, A. Developmental pattern of gene-specific DNA methylation in the mouse embryo and germ line. Genes Dev. 1992, 6, 705–714. [Google Scholar] [CrossRef] [PubMed]
- Bao, J.; Bedford, M.T. Epigenetic regulation of the histone-to-protamine transition during spermiogenesis. Reproduction 2016, 151, R55–R70. [Google Scholar] [CrossRef] [PubMed]
- Takada, S.; Paulsen, M.; Tevendale, M.; Tsai, C.E.; Kelsey, G.; Cattanach, B.M.; Ferguson-Smith, A.C. Epigenetic analysis of the Dlk1-Gtl2 imprinted domain on mouse chromosome 12: Implications for imprinting control from comparison with Igf2-H19. Hum. Mol. Genet. 2002, 11, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cheunsuchon, P.; Nakayama, Y.; Lawlor, M.W.; Zhong, Y.; Rice, K.A.; Zhang, L.; Zhang, X.; Gordon, F.E.; Lidov, H.G.; et al. Activation of paternally expressed genes and perinatal death caused by deletion of the Gtl2 gene. Development 2010, 137, 2643–2652. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, S.B.; Schwartz, S.; Miller, J.L.; Driscoll, D.J. Prader-Willi syndrome. Genet. Med. 2012, 14, 10–26. [Google Scholar] [CrossRef] [PubMed]
- Kantor, B.; Kaufman, Y.; Makedonski, K.; Razin, A.; Shemer, R. Establishing the epigenetic status of the Prader-Willi/Angelman imprinting center in the gametes and embryo. Hum. Mol. Genet. 2004, 13, 2767–2779. [Google Scholar] [CrossRef] [PubMed]
- Brant, J.O.; Riva, A.; Resnick, J.L.; Yang, T.P. Influence of the Prader-Willi syndrome imprinting center on the DNA methylation landscape in the mouse brain. Epigenetics 2014, 9, 1540–1556. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Richetto, J.; Massart, R.; Weber-Stadlbauer, U.; Szyf, M.; Riva, M.A.; Meyer, U. Genome-wide DNA Methylation Changes in a Mouse Model of Infection-Mediated Neurodevelopmental Disorders. Biol. Psychiatry 2017, 81, 265–276. [Google Scholar] [CrossRef] [PubMed]
- Joehanes, R.; Just, A.C.; Marioni, R.E.; Pilling, L.C.; Reynolds, L.M.; Mandaviya, P.R.; Guan, W.; Xu, T.; Elks, C.E.; Aslibekyan, S.; et al. Epigenetic Signatures of Cigarette Smoking. Circ. Cardiovasc. Genet. 2016, 9, 436–447. [Google Scholar] [CrossRef] [PubMed]
- Ornoy, A.; Reece, E.A.; Pavlinkova, G.; Kappen, C.; Miller, R.K. Effect of maternal diabetes on the embryo, fetus, and children: Congenital anomalies, genetic and epigenetic changes and developmental outcomes. Birth Defects Res. C Embryo Today 2015, 105, 53–72. [Google Scholar] [CrossRef] [PubMed]
- Serpeloni, F.; Radtke, K.; de Assis, S.G.; Henning, F.; Natt, D.; Elbert, T. Grandmaternal stress during pregnancy and DNA methylation of the third generation: An epigenome-wide association study. Transl. Psychiatry 2017, 7, e1202. [Google Scholar] [CrossRef] [PubMed]
- Januar, V.; Desoye, G.; Novakovic, B.; Cvitic, S.; Saffery, R. Epigenetic regulation of human placental function and pregnancy outcome: Considerations for causal inference. Am. J. Obstet. Gynecol. 2015, 213, S182–S196. [Google Scholar] [CrossRef] [PubMed]
- Paquette, A.G.; Houseman, E.A.; Green, B.B.; Lesseur, C.; Armstrong, D.A.; Lester, B.; Marsit, C.J. Regions of variable DNA methylation in human placenta associated with newborn neurobehavior. Epigenetics 2016, 11, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Monk, C.; Feng, T.; Lee, S.; Krupska, I.; Champagne, F.A.; Tycko, B. Distress During Pregnancy: Epigenetic Regulation of Placenta Glucocorticoid-Related Genes and Fetal Neurobehavior. Am. J. Psychiatry 2016, 173, 705–713. [Google Scholar] [CrossRef] [PubMed]
- Filiberto, A.C.; Maccani, M.A.; Koestler, D.; Wilhelm-Benartzi, C.; Avissar-Whiting, M.; Banister, C.E.; Gagne, L.A.; Marsit, C.J. Birthweight is associated with DNA promoter methylation of the glucocorticoid receptor in human placenta. Epigenetics 2011, 6, 566–572. [Google Scholar] [CrossRef] [PubMed]
- Bromer, C.; Marsit, C.J.; Armstrong, D.A.; Padbury, J.F.; Lester, B. Genetic and epigenetic variation of the glucocorticoid receptor (NR3C1) in placenta and infant neurobehavior. Dev. Psychobiol. 2013, 55, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Turecki, G.; Meaney, M.J. Effects of the Social Environment and Stress on Glucocorticoid Receptor Gene Methylation: A Systematic Review. Biol. Psychiatry 2016, 79, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Tyrka, A.R.; Parade, S.H.; Welch, E.S.; Ridout, K.K.; Price, L.H.; Marsit, C.; Philip, N.S.; Carpenter, L.L. Methylation of the leukocyte glucocorticoid receptor gene promoter in adults: Associations with early adversity and depressive, anxiety and substance-use disorders. Transl. Psychiatry 2016, 6, e848. [Google Scholar] [CrossRef] [PubMed]
- Roseboom, T.J.; van der Meulen, J.H.; Ravelli, A.C.; Osmond, C.; Barker, D.J.; Bleker, O.P. Effects of prenatal exposure to the Dutch famine on adult disease in later life: An overview. Mol. Cell. Endocrinol. 2001, 185, 93–98. [Google Scholar] [CrossRef]
- Heijmans, B.T.; Tobi, E.W.; Stein, A.D.; Putter, H.; Blauw, G.J.; Susser, E.S.; Slagboom, P.E.; Lumey, L.H. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. USA 2008, 105, 17046–17049. [Google Scholar] [CrossRef] [PubMed]
- Tobi, E.W.; Goeman, J.J.; Monajemi, R.; Gu, H.; Putter, H.; Zhang, Y.; Slieker, R.C.; Stok, A.P.; Thijssen, P.E.; Muller, F.; et al. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat. Commun. 2014, 5, 5592. [Google Scholar] [CrossRef] [PubMed]
- Vukojevic, V.; Kolassa, I.T.; Fastenrath, M.; Gschwind, L.; Spalek, K.; Milnik, A.; Heck, A.; Vogler, C.; Wilker, S.; Demougin, P.; et al. Epigenetic modification of the glucocorticoid receptor gene is linked to traumatic memory and post-traumatic stress disorder risk in genocide survivors. J. Neurosci. 2014, 34, 10274–10284. [Google Scholar] [CrossRef] [PubMed]
- Yehuda, R.; Flory, J.D.; Bierer, L.M.; Henn-Haase, C.; Lehrner, A.; Desarnaud, F.; Makotkine, I.; Daskalakis, N.P.; Marmar, C.R.; Meaney, M.J. Lower methylation of glucocorticoid receptor gene promoter 1F in peripheral blood of veterans with posttraumatic stress disorder. Biol. Psychiatry 2015, 77, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Davies, M.N.; Volta, M.; Pidsley, R.; Lunnon, K.; Dixit, A.; Lovestone, S.; Coarfa, C.; Harris, R.A.; Milosavljevic, A.; Troakes, C.; et al. Functional annotation of the human brain methylome identifies tissue-specific epigenetic variation across brain and blood. Genome Biol. 2012, 13, R43. [Google Scholar] [CrossRef] [PubMed]
- Bell, J.T.; Tsai, P.C.; Yang, T.P.; Pidsley, R.; Nisbet, J.; Glass, D.; Mangino, M.; Zhai, G.; Zhang, F.; Valdes, A.; et al. Epigenome-wide scans identify differentially methylated regions for age and age-related phenotypes in a healthy ageing population. PLoS Genet. 2012, 8, e1002629. [Google Scholar] [CrossRef] [PubMed]
- Ladd-Acosta, C. Epigenetic Signatures as Biomarkers of Exposure. Curr. Environ. Health Rep. 2015, 2, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.D.; Jones, M.J.; Meaney, M.J.; Turecki, G.; Kobor, M.S. BECon: A tool for interpreting DNA methylation findings from blood in the context of brain. Transl. Psychiatry 2017, 7, e1187. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Teh, A.L.; Chen, L.; Lim, I.Y.; Tan, P.F.; MacIsaac, J.L.; Morin, A.M.; Yap, F.; Tan, K.H.; Saw, S.M.; et al. Choice of surrogate tissue influences neonatal EWAS findings. BMC Med. 2017, 15, 211. [Google Scholar] [CrossRef] [PubMed]
- Williamson, P.R.; Gamble, C.; Altman, D.G.; Hutton, J.L. Outcome selection bias in meta-analysis. Stat. Methods Med. Res. 2005, 14, 515–524. [Google Scholar] [CrossRef] [PubMed]
- Blewitt, M.; Whitelaw, E. The use of mouse models to study epigenetics. Cold Spring Harb. Perspect. Biol. 2013, 5, a017939. [Google Scholar] [CrossRef] [PubMed]
- Nagy, C.; Turecki, G. Transgenerational epigenetic inheritance: An open discussion. Epigenomics 2015, 7, 781–790. [Google Scholar] [CrossRef] [PubMed]
- Klamt, S.; Stelling, J. Combinatorial complexity of pathway analysis in metabolic networks. Mol. Biol. Rep. 2002, 29, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Mills, J.L. Data torturing. N. Engl. J. Med. 1993, 329, 1196–1199. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| miRNA | Known Function | Reference |
|---|---|---|
| miRNA-9 | Neural progenitor proliferation and maintaining of self-renewal state. | [64] |
| Determination of neuron fate. | ||
| miRNA-124 | Promotes neuronal differentiation and maturation. | [65,66] |
| miRNA-128 | Neuronal migration and plasticity. | [67] |
| miRNA-137 | Promotes differentiation in neural stem cells from ventricular zones (embryonic mice brains) and subventricular zones (adult mice brains). | [68,69] |
| miRNA-132 | Roles in brain plasticity and memory. | [70] |
| MiRNA-145 | Inhibits expression of developmental factors (as Oct4). | [71] |
| miRNA-200 | Inhibit differentiation of neurodermal precursors. | [72] |
| miRNA-219 | Promotes oligodendrocyte differentiation. | [73] |
| miRNA-290/295 cluster | Promoter pluripotency and cell cycle phase distribution of ES cells. | [74] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cariaga-Martínez, A.E.; Gutiérrez, K.J.; Alelú-Paz, R. The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function. Int. J. Mol. Sci. 2018, 19, 1333. https://doi.org/10.3390/ijms19051333
Cariaga-Martínez AE, Gutiérrez KJ, Alelú-Paz R. The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function. International Journal of Molecular Sciences. 2018; 19(5):1333. https://doi.org/10.3390/ijms19051333
Chicago/Turabian StyleCariaga-Martínez, Ariel Ernesto, Kilian Jesús Gutiérrez, and Raúl Alelú-Paz. 2018. "The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function" International Journal of Molecular Sciences 19, no. 5: 1333. https://doi.org/10.3390/ijms19051333
APA StyleCariaga-Martínez, A. E., Gutiérrez, K. J., & Alelú-Paz, R. (2018). The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function. International Journal of Molecular Sciences, 19(5), 1333. https://doi.org/10.3390/ijms19051333