Animal Models of the Neuromuscular Junction, Vitally Informative for Understanding Function and the Molecular Mechanisms of Congenital Myasthenic Syndromes

Abstract
1. Introduction
2. Methodology
3. Animal Models Used in the Study of CMS-Associated Proteins
3.1. Post-Synaptic
3.1.1. AChR Deficiency
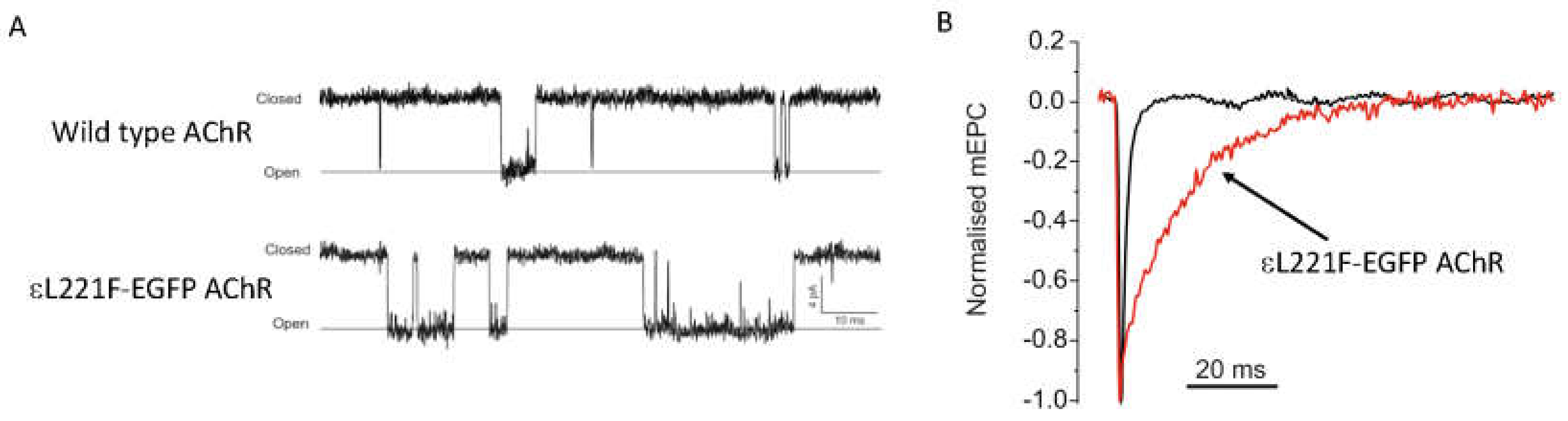
3.1.2. AChR Kinetic Mutations
3.1.3. MuSK/LRP4
3.1.4. Rapsyn
3.1.5. DOK7
3.2. Synaptic
3.2.1. ColQ
3.2.2. Agrin
3.2.3. The Extracellular Matrix (ECM)
3.3. Pre-Synaptic
4. Animal Models in Myasthenia Gravis
5. Conclusions
Acknowledgements
Conflicts of Interest
Abbreviations
| AAV | Adeno-Associated Virus |
| ACh | Acetylcholine |
| AChE | Acetylcholine Esterase |
| AChR | Acetylcholine Receptor |
| ChAT | Choline Acetyltransferase |
| CMAP | Compound Muscle Action Potential |
| CMS | Congenital Myasthenic Syndrome |
| Col13A1 | Collagen, Type XIII, α-1 |
| Col-Q | Collagen Like Tail Subunit of Asymmetric Acetylcholinesterase |
| DOK7 | Downstream of Tyrosine Kinase-7 |
| EPC | Endplate Current |
| EPP | Endplate Potential |
| LRP4 | Low Density Lipoprotein Receptor (LDLR)-related protein 4 |
| mEPP | Miniature Endplate Potential |
| MuSK | Muscle-Specific Kinase |
| NMJ | Neuromuscular Junction |
| P | Postnatal day |
| QC | Quantal Content |
| Rapsyn | Receptor-Associated Protein of the Synapse |
| SCCMS | Slow Channel Congenital Myasthenic Syndrome |
| SCL5A7 | Solute Carrier Family 5 (Choline Transporter), Member 7 |
| SLC18A3 | Solute Carrier Family 18 (Vesicular Acetylcholine), Member 3 |
| SNAP25 | Synaptosomal-Associated Protein, 25-KD |
| SYT2 | Synaptotagmin 2 |
| VAMP1 | Vesicle-Associated Membrane Protein 1 |
References
- Rodriguez Cruz, P.M.; Palace, J.; Beeson, D. Congenital myasthenic syndromes and the neuromuscular junction. Curr. Opin. Neurol. 2014, 27, 566–575. [Google Scholar] [CrossRef] [PubMed]
- Nicole, S.; Azuma, Y.; Bauche, S.; Eymard, B.; Lochmuller, H.; Slater, C. Congenital myasthenic syndromes or inherited disorders of neuromuscular transmission: Recent discoveries and open questions. J. Neuromuscul. Dis. 2017, 4, 269–284. [Google Scholar] [CrossRef] [PubMed]
- Cossins, J.; Webster, R.; Maxwell, S.; Burke, G.; Vincent, A.; Beeson, D. A mouse model of achr deficiency syndrome with a phenotype reflecting the human condition. Hum. Mol. Genet. 2004, 13, 2947–2957. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.M.; Bhattacharyya, B.B.; Charnet, P.; Day, J.W.; Labarca, C.; Wollmann, R.L.; Lambert, E.H. A transgenic mouse model of the slow-channel syndrome. Muscle Nerve 1996, 19, 79–87. [Google Scholar] [CrossRef]
- Gomez, C.M.; Maselli, R.; Gundeck, J.E.; Chao, M.; Day, J.W.; Tamamizu, S.; Lasalde, J.A.; McNamee, M.; Wollmann, R.L. Slow-channel transgenic mice: A model of postsynaptic organellar degeneration at the neuromuscular junction. J. Neurosci. 1997, 17, 4170–4179. [Google Scholar] [CrossRef] [PubMed]
- Gomez, C.M.; Maselli, R.A.; Groshong, J.; Zayas, R.; Wollmann, R.L.; Cens, T.; Charnet, P. Active calcium accumulation underlies severe weakness in a panel of mice with slow-channel syndrome. J. Neurosci. 2002, 22, 6447–6457. [Google Scholar] [CrossRef] [PubMed]
- Grajales-Reyes, J.G.; Garcia-Gonzalez, A.; Maria-Rios, J.C.; Grajales-Reyes, G.E.; Delgado-Velez, M.; Baez-Pagan, C.A.; Quesada, O.; Gomez, C.M.; Lasalde-Dominicci, J.A. A panel of slow-channel congenital myasthenic syndrome mice reveals a unique locomotor behavioral signature. J. Neuromuscul. Dis. 2017, 4, 341–347. [Google Scholar] [CrossRef] [PubMed]
- Chevessier, F.; Peter, C.; Mersdorf, U.; Girard, E.; Krejci, E.; McArdle, J.J.; Witzemann, V. A new mouse model for the slow-channel congenital myasthenic syndrome induced by the achr epsilonl221f mutation. Neurobiol. Dis. 2012, 45, 851–861. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.G.; Cossins, J.; Lashley, D.; Maxwell, S.; Liu, W.W.; Wickens, J.R.; Martinez-Martinez, P.; de Baets, M.; Beeson, D. A mouse model of the slow channel myasthenic syndrome: Neuromuscular physiology and effects of ephedrine treatment. Exp. Neurol. 2013, 248, 286–298. [Google Scholar] [CrossRef] [PubMed]
- Vohra, B.P.; Groshong, J.S.; Zayas, R.; Wollmann, R.L.; Gomez, C.M. Activation of apoptotic pathways at muscle fiber synapses is circumscribed and reversible in a slow-channel syndrome model. Neurobiol. Dis. 2006, 23, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Groshong, J.S.; Spencer, M.J.; Bhattacharyya, B.J.; Kudryashova, E.; Vohra, B.P.; Zayas, R.; Wollmann, R.L.; Miller, R.J.; Gomez, C.M. Calpain activation impairs neuromuscular transmission in a mouse model of the slow-channel myasthenic syndrome. J. Clin. Investig. 2007, 117, 2903–2912. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Grajales-Reyes, G.E.; Alicea-Vazquez, V.; Grajales-Reyes, J.G.; Robinson, K.; Pytel, P.; Baez-Pagan, C.A.; Lasalde-Dominicci, J.A.; Gomez, C.M. Fluoxetine is neuroprotective in slow-channel congenital myasthenic syndrome. Exp. Neurol. 2015, 270, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Chevessier, F.; Girard, E.; Molgo, J.; Bartling, S.; Koenig, J.; Hantai, D.; Witzemann, V. A mouse model for congenital myasthenic syndrome due to musk mutations reveals defects in structure and function of neuromuscular junctions. Hum. Mol. Genet. 2008, 17, 3577–3595. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Cao, Y.; Wu, H.; Ye, X.; Zhu, Z.; Xing, G.; Shen, C.; Barik, A.; Zhang, B.; Xie, X.; et al. Enzymatic activity of the scaffold protein rapsyn for synapse formation. Neuron 2016, 92, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Arimura, S.; Okada, T.; Tezuka, T.; Chiyo, T.; Kasahara, Y.; Yoshimura, T.; Motomura, M.; Yoshida, N.; Beeson, D.; Takeda, S.; et al. Neuromuscular disease. Dok7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science 2014, 345, 1505–1508. [Google Scholar] [CrossRef] [PubMed]
- Bogdanik, L.P.; Burgess, R.W. A valid mouse model of agrin-associated congenital myasthenic syndrome. Hum. Mol. Genet. 2011, 20, 4617–4633. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; Morsch, M.; Phillips, W.D.; Verschuuren, J.J. Electrophysiological analysis of neuromuscular synaptic function in myasthenia gravis patients and animal models. Exp. Neurol. 2015, 270, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Deacon, R.M. Measuring the strength of mice. J. Vis. Exp. 2013. [Google Scholar] [CrossRef] [PubMed]
- Willadt, S.; Nash, M.; Slater, C.R. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep. 2016, 6, 24849. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, B.J.; Day, J.W.; Gundeck, J.E.; Leonard, S.; Wollmann, R.L.; Gomez, C.M. Desensitization of mutant acetylcholine receptors in transgenic mice reduces the amplitude of neuromuscular synaptic currents. Synapse 1997, 27, 367–377. [Google Scholar] [CrossRef]
- Matthews-Bellinger, J.A.; Salpeter, M.M. Fine structural distribution of acetylcholine receptors at developing mouse neuromuscular junctions. J. Neurosci. 1983, 3, 644–657. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez Cruz, P.M.; Palace, J.; Beeson, D. Inherited disorders of the neuromuscular junction: An update. J. Neurol. 2014, 261, 2234–2243. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.G.; Shen, X.M.; Selcen, D.; Sine, S.M. Congenital myasthenic syndromes: Pathogenesis, diagnosis, and treatment. Lancet Neurol. 2015, 14, 461. [Google Scholar] [CrossRef]
- Witzemann, V.; Barg, B.; Nishikawa, Y.; Sakmann, B.; Numa, S. Differential regulation of muscle acetylcholine receptor gamma- and epsilon-subunit mrnas. FEBS Lett. 1987, 223, 104–112. [Google Scholar] [CrossRef]
- Brenner, H.R.; Witzemann, V.; Sakmann, B. Imprinting of acetylcholine receptor messenger rna accumulation in mammalian neuromuscular synapses. Nature 1990, 344, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Witzemann, V.; Schwarz, H.; Koenen, M.; Berberich, C.; Villarroel, A.; Wernig, A.; Brenner, H.R.; Sakmann, B. Acetylcholine receptor epsilon-subunit deletion causes muscle weakness and atrophy in juvenile and adult mice. Proc. Natl. Acad. Sci. USA 1996, 93, 13286–13291. [Google Scholar] [CrossRef] [PubMed]
- Missias, A.C.; Mudd, J.; Cunningham, J.M.; Steinbach, J.H.; Merlie, J.P.; Sanes, J.R. Deficient development and maintenance of postsynaptic specializations in mutant mice lacking an ‘adult’ acetylcholine receptor subunit. Development 1997, 124, 5075–5086. [Google Scholar] [PubMed]
- Croxen, R.; Young, C.; Slater, C.; Haslam, S.; Brydson, M.; Vincent, A.; Beeson, D. End-plate gamma- and epsilon-subunit mrna levels in achr deficiency syndrome due to epsilon-subunit null mutations. Brain 2001, 124, 1362–1372. [Google Scholar] [CrossRef] [PubMed]
- Webster, R.; Maxwell, S.; Spearman, H.; Tai, K.; Beckstein, O.; Sansom, M.; Beeson, D. A novel congenital myasthenic syndrome due to decreased acetylcholine receptor ion-channel conductance. Brain 2012, 135, 1070–1080. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Qian, L.; Yang, Z.H.; Huang, Y.; Ngo, S.T.; Ruan, N.J.; Wang, J.; Schneider, C.; Noakes, P.G.; Ding, Y.Q.; et al. Rapsyn interaction with calpain stabilizes achr clusters at the neuromuscular junction. Neuron 2007, 55, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Harper, C.M.; Fukodome, T.; Engel, A.G. Treatment of slow-channel congenital myasthenic syndrome with fluoxetine. Neurology 2003, 60, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Milone, M.; Wang, H.L.; Ohno, K.; Fukudome, T.; Pruitt, J.N.; Bren, N.; Sine, S.M.; Engel, A.G. Slow-channel myasthenic syndrome caused by enhanced activation, desensitization, and agonist binding affinity attributable to mutation in the m2 domain of the acetylcholine receptor alpha subunit. J. Neurosci. 1997, 17, 5651–5665. [Google Scholar] [CrossRef] [PubMed]
- DeChiara, T.M.; Bowen, D.C.; Valenzuela, D.M.; Simmons, M.V.; Poueymirou, W.T.; Thomas, S.; Kinetz, E.; Compton, D.L.; Rojas, E.; Park, J.S.; et al. The receptor tyrosine kinase musk is required for neuromuscular junction formation in vivo. Cell 1996, 85, 501–512. [Google Scholar] [CrossRef]
- Glass, D.J.; Bowen, D.C.; Stitt, T.N.; Radziejewski, C.; Bruno, J.; Ryan, T.E.; Gies, D.R.; Shah, S.; Mattsson, K.; Burden, S.J.; et al. Agrin acts via a musk receptor complex. Cell 1996, 85, 513–523. [Google Scholar] [CrossRef]
- Weatherbee, S.D.; Anderson, K.V.; Niswander, L.A. Ldl-receptor-related protein 4 is crucial for formation of the neuromuscular junction. Development 2006, 133, 4993–5000. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Stiegler, A.L.; Cameron, T.O.; Hallock, P.T.; Gomez, A.M.; Huang, J.H.; Hubbard, S.R.; Dustin, M.L.; Burden, S.J. Lrp4 is a receptor for agrin and forms a complex with musk. Cell 2008, 135, 334–342. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Luo, S.; Wang, Q.; Suzuki, T.; Xiong, W.C.; Mei, L. Lrp4 serves as a coreceptor of agrin. Neuron 2008, 60, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Burgess, R.W.; Dominguez, B.; Pfaff, S.L.; Sanes, J.R.; Lee, K.F. Distinct roles of nerve and muscle in postsynaptic differentiation of the neuromuscular synapse. Nature 2001, 410, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Arber, S.; William, C.; Li, L.; Tanabe, Y.; Jessell, T.M.; Birchmeier, C.; Burden, S.J. Patterning of muscle acetylcholine receptor gene expression in the absence of motor innervation. Neuron 2001, 30, 399–410. [Google Scholar] [CrossRef]
- Kim, N.; Burden, S.J. Musk controls where motor axons grow and form synapses. Nat. Neurosci. 2008, 11, 19–27. [Google Scholar] [CrossRef] [PubMed]
- Vock, V.M.; Ponomareva, O.N.; Rimer, M. Evidence for muscle-dependent neuromuscular synaptic site determination in mammals. J. Neurosci. 2008, 28, 3123–3130. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liang, C.; Bates, R.; Yin, Y.; Xiong, W.C.; Mei, L. Wnt proteins regulate acetylcholine receptor clustering in muscle cells. Mol. Brain 2012, 5, 7. [Google Scholar] [CrossRef] [PubMed]
- Barik, A.; Zhang, B.; Sohal, G.S.; Xiong, W.C.; Mei, L. Crosstalk between agrin and wnt signaling pathways in development of vertebrate neuromuscular junction. Dev. Neurobiol. 2014, 74, 828–838. [Google Scholar] [CrossRef] [PubMed]
- Messeant, J.; Ezan, J.; Delers, P.; Glebov, K.; Marchiol, C.; Lager, F.; Renault, G.; Tissir, F.; Montcouquiol, M.; Sans, N.; et al. Wnt proteins contribute to neuromuscular junction formation through distinct signaling pathways. Development 2017, 144, 1712–1724. [Google Scholar] [CrossRef] [PubMed]
- Froehner, S.C.; Luetje, C.W.; Scotland, P.B.; Patrick, J. The postsynaptic 43k protein clusters muscle nicotinic acetylcholine receptors in xenopus oocytes. Neuron 1990, 5, 403–410. [Google Scholar] [CrossRef]
- Phillips, W.D.; Maimone, M.M.; Merlie, J.P. Mutagenesis of the 43-kd postsynaptic protein defines domains involved in plasma membrane targeting and achr clustering. J. Cell Biol. 1991, 115, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Noakes, P.G.; Mudd, J.; Nichol, M.; Chu, G.C.; Sanes, J.R.; Merlie, J.P. Failure of postsynaptic specialization to develop at neuromuscular junctions of rapsyn-deficient mice. Nature 1995, 377, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Engel, A.G.; Shen, X.M.; Selcen, D.; Brengman, J.; Harper, C.M.; Tsujino, A.; Milone, M. Rapsyn mutations in humans cause endplate acetylcholine-receptor deficiency and myasthenic syndrome. Am. J. Hum. Genet. 2002, 70, 875–885. [Google Scholar] [CrossRef] [PubMed]
- Cossins, J.; Burke, G.; Maxwell, S.; Spearman, H.; Man, S.; Kuks, J.; Vincent, A.; Palace, J.; Fuhrer, C.; Beeson, D. Diverse molecular mechanisms involved in achr deficiency due to rapsyn mutations. Brain 2006, 129, 2773–2783. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.S.; Baumeister, S.K.; Schara, U.; Cossins, J.; Krause, S.; von der Hagen, M.; Huebner, A.; Webster, R.; Beeson, D.; Lochmüller, H.; et al. Chrnd mutation causes a congenital myasthenic syndrome by impairing co-clustering of the acetylcholine receptor with rapsyn. Brain 2006, 129, 2784–2793. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Inoue, A.; Okada, M.; Murata, Y.; Kakuta, S.; Jigami, T.; Kubo, S.; Shiraishi, H.; Eguchi, K.; Motomura, M.; et al. The muscle protein dok-7 is essential for neuromuscular synaptogenesis. Science 2006, 312, 1802–1805. [Google Scholar] [CrossRef] [PubMed]
- Beeson, D.; Higuchi, O.; Palace, J.; Cossins, J.; Spearman, H.; Maxwell, S.; Newsom-Davis, J.; Burke, G.; Fawcett, P.; Motomura, M.; et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science 2006, 313, 1975–1978. [Google Scholar] [CrossRef] [PubMed]
- Palace, J.; Lashley, D.; Newsom-Davis, J.; Cossins, J.; Maxwell, S.; Kennett, R.; Jayawant, S.; Yamanashi, Y.; Beeson, D. Clinical features of the dok7 neuromuscular junction synaptopathy. Brain 2007, 130, 1507–1515. [Google Scholar] [CrossRef] [PubMed]
- Miyoshi, S.; Tezuka, T.; Arimura, S.; Tomono, T.; Okada, T.; Yamanashi, Y. Dok7 gene therapy enhances motor activity and life span in als model mice. EMBO Mol. Med. 2017, 9, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Hallock, P.T.; Xu, C.F.; Park, T.J.; Neubert, T.A.; Curran, T.; Burden, S.J. Dok-7 regulates neuromuscular synapse formation by recruiting crk and crk-l. Genes Dev. 2010, 24, 2451–2461. [Google Scholar] [CrossRef] [PubMed]
- Massoulie, J.; Pezzementi, L.; Bon, S.; Krejci, E.; Vallette, F.M. Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 1993, 41, 31–91. [Google Scholar] [CrossRef]
- Arikawa-Hirasawa, E.; Rossi, S.G.; Rotundo, R.L.; Yamada, Y. Absence of acetylcholinesterase at the neuromuscular junctions of perlecan-null mice. Nat. Neurosci. 2002, 5, 119–123. [Google Scholar] [CrossRef] [PubMed]
- Bon, S.; Coussen, F.; Massoulie, J. Quaternary associations of acetylcholinesterase. Ii. The polyproline attachment domain of the collagen tail. J. Biol. Chem. 1997, 272, 3016–3021. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Brengman, J.; Tsujino, A.; Engel, A.G. Human endplate acetylcholinesterase deficiency caused by mutations in the collagen-like tail subunit (COLQ) of the asymmetric enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 9654–9659. [Google Scholar] [CrossRef] [PubMed]
- Donger, C.; Krejci, E.; Serradell, A.P.; Eymard, B.; Bon, S.; Nicole, S.; Chateau, D.; Gary, F.; Fardeau, M.; Massoulie, J.; et al. Mutation in the human acetylcholinesterase-associated collagen gene, colq, is responsible for congenital myasthenic syndrome with end-plate acetylcholinesterase deficiency (type ic). Am. J. Hum. Genet. 1998, 63, 967–975. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.; Krejci, E.; Molgo, J.; Cunningham, J.M.; Massoulie, J.; Sanes, J.R. Genetic analysis of collagen q: Roles in acetylcholinesterase and butyrylcholinesterase assembly and in synaptic structure and function. J. Cell Biol. 1999, 144, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Petrov, K.A.; Girard, E.; Nikitashina, A.D.; Colasante, C.; Bernard, V.; Nurullin, L.; Leroy, J.; Samigullin, D.; Colak, O.; Nikolsky, E.; et al. Schwann cells sense and control acetylcholine spillover at the neuromuscular junction by alpha7 nicotinic receptors and butyrylcholinesterase. J. Neurosci. 2014, 34, 11870–11883. [Google Scholar] [CrossRef] [PubMed]
- Sigoillot, S.M.; Bourgeois, F.; Karmouch, J.; Molgo, J.; Dobbertin, A.; Chevalier, C.; Houlgatte, R.; Leger, J.; Legay, C. Neuromuscular junction immaturity and muscle atrophy are hallmarks of the colq-deficient mouse, a model of congenital myasthenic syndrome with acetylcholinesterase deficiency. FASEB J. 2016, 30, 2382–2399. [Google Scholar] [CrossRef] [PubMed]
- Nitkin, R.M.; Smith, M.A.; Magill, C.; Fallon, J.R.; Yao, Y.M.; Wallace, B.G.; McMahan, U.J. Identification of agrin, a synaptic organizing protein from torpedo electric organ. J. Cell Biol. 1987, 105, 2471–2478. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, E.W.; Nitkin, R.M.; Wallace, B.G.; Rubin, L.L.; McMahan, U.J. Components of torpedo electric organ and muscle that cause aggregation of acetylcholine receptors on cultured muscle cells. J. Cell Biol. 1984, 99, 615–627. [Google Scholar] [CrossRef] [PubMed]
- Gautam, M.; Noakes, P.G.; Moscoso, L.; Rupp, F.; Scheller, R.H.; Merlie, J.P.; Sanes, J.R. Defective neuromuscular synaptogenesis in agrin-deficient mutant mice. Cell 1996, 85, 525–535. [Google Scholar] [CrossRef]
- Huze, C.; Bauche, S.; Richard, P.; Chevessier, F.; Goillot, E.; Gaudon, K.; Ben Ammar, A.; Chaboud, A.; Grosjean, I.; Lecuyer, H.A.; et al. Identification of an agrin mutation that causes congenital myasthenia and affects synapse function. Am. J. Hum. Genet. 2009, 85, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Fernandez, J.M.; Arredondo, J.; Navarro, C.; Ngo, M.; Beeson, D.; Cagney, O.; Williams, D.C.; Wollmann, R.L.; Yarov-Yarovoy, V.; et al. Lg2 agrin mutation causing severe congenital myasthenic syndrome mimics functional characteristics of non-neural (z-) agrin. Hum. Genet. 2012, 131, 1123–1135. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Martin, P.T. Role of extracellular matrix proteins and their receptors in the development of the vertebrate neuromuscular junction. Dev. Neurobiol. 2011, 71, 982–1005. [Google Scholar] [CrossRef] [PubMed]
- Zenker, M.; Aigner, T.; Wendler, O.; Tralau, T.; Muntefering, H.; Fenski, R.; Pitz, S.; Schumacher, V.; Royer-Pokora, B.; Wuhl, E.; et al. Human laminin beta2 deficiency causes congenital nephrosis with mesangial sclerosis and distinct eye abnormalities. Hum. Mol. Genet. 2004, 13, 2625–2632. [Google Scholar] [CrossRef] [PubMed]
- Noakes, P.G.; Gautam, M.; Mudd, J.; Sanes, J.R.; Merlie, J.P. Aberrant differentiation of neuromuscular junctions in mice lacking s-laminin/laminin beta 2. Nature 1995, 374, 258–262. [Google Scholar] [CrossRef] [PubMed]
- Maselli, R.A.; Ng, J.J.; Anderson, J.A.; Cagney, O.; Arredondo, J.; Williams, C.; Wessel, H.B.; Abdel-Hamid, H.; Wollmann, R.L. Mutations in lamb2 causing a severe form of synaptic congenital myasthenic syndrome. J. Med. Genet. 2009, 46, 203–208. [Google Scholar] [CrossRef] [PubMed]
- Latvanlehto, A.; Fox, M.A.; Sormunen, R.; Tu, H.; Oikarainen, T.; Koski, A.; Naumenko, N.; Shakirzyanova, A.; Kallio, M.; Ilves, M.; et al. Muscle-derived collagen xiii regulates maturation of the skeletal neuromuscular junction. J. Neurosci. 2010, 30, 12230–12241. [Google Scholar] [CrossRef] [PubMed]
- Haronen, H.; Zainul, Z.; Tu, H.; Naumenko, N.; Sormunen, R.; Miinalainen, I.; Shakirzyanova, A.; Oikarainen, T.; Abdullin, A.; Martin, P.; et al. Collagen xiii secures pre- and postsynaptic integrity of the neuromuscular synapse. Hum. Mol. Genet. 2017, 26, 2076–2090. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.V.; Cossins, J.; Rodriguez Cruz, P.M.; Parry, D.A.; Maxwell, S.; Martinez-Martinez, P.; Riepsaame, J.; Abdelhamed, Z.A.; Lake, A.V.; Moran, M.; et al. Congenital myasthenic syndrome type 19 is caused by mutations in col13a1, encoding the atypical non-fibrillar collagen type xiii alpha1 chain. Am. J. Hum. Genet. 2015, 97, 878–885. [Google Scholar] [CrossRef] [PubMed]
- Fox, M.A.; Sanes, J.R.; Borza, D.B.; Eswarakumar, V.P.; Fassler, R.; Hudson, B.G.; John, S.W.; Ninomiya, Y.; Pedchenko, V.; Pfaff, S.L.; et al. Distinct target-derived signals organize formation, maturation, and maintenance of motor nerve terminals. Cell 2007, 129, 179–193. [Google Scholar] [CrossRef] [PubMed]
- Ross, J.A.; Webster, R.G.; Lechertier, T.; Reynolds, L.E.; Turmaine, M.; Bencze, M.; Jamshidi, Y.; Cetin, H.; Muntoni, F.; Beeson, D.; et al. Multiple roles of integrin-alpha3 at the neuromuscular junction. J. Cell Sci. 2017, 130, 1772–1784. [Google Scholar] [CrossRef] [PubMed]
- Kreidberg, J.A.; Donovan, M.J.; Goldstein, S.L.; Rennke, H.; Shepherd, K.; Jones, R.C.; Jaenisch, R. Alpha 3 beta 1 integrin has a crucial role in kidney and lung organogenesis. Development 1996, 122, 3537–3547. [Google Scholar] [PubMed]
- Has, C.; Sparta, G.; Kiritsi, D.; Weibel, L.; Moeller, A.; Vega-Warner, V.; Waters, A.; He, Y.; Anikster, Y.; Esser, P.; et al. Integrin alpha3 mutations with kidney, lung, and skin disease. N. Engl. J. Med. 2012, 366, 1508–1514. [Google Scholar] [CrossRef] [PubMed]
- Ohno, K.; Tsujino, A.; Brengman, J.M.; Harper, C.M.; Bajzer, Z.; Udd, B.; Beyring, R.; Robb, S.; Kirkham, F.J.; Engel, A.G. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc. Natl. Acad. Sci. USA 2001, 98, 2017–2022. [Google Scholar] [CrossRef] [PubMed]
- Misgeld, T.; Burgess, R.W.; Lewis, R.M.; Cunningham, J.M.; Lichtman, J.W.; Sanes, J.R. Roles of neurotransmitter in synapse formation: Development of neuromuscular junctions lacking choline acetyltransferase. Neuron 2002, 36, 635–648. [Google Scholar] [CrossRef]
- Brandon, E.P.; Lin, W.; D’Amour, K.A.; Pizzo, D.P.; Dominguez, B.; Sugiura, Y.; Thode, S.; Ko, C.P.; Thal, L.J.; Gage, F.H.; et al. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J. Neurosci. 2003, 23, 539–549. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, D.N.; Horvath, R.; Sowden, J.E.; Gonzalez, M.; Sanchez-Mejias, A.; Guan, Z.; Whittaker, R.G.; Almodovar, J.L.; Lane, M.; Bansagi, B.; et al. Synaptotagmin 2 mutations cause an autosomal-dominant form of lambert-eaton myasthenic syndrome and nonprogressive motor neuropathy. Am. J. Hum. Genet. 2014, 95, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Whittaker, R.G.; Herrmann, D.N.; Bansagi, B.; Hasan, B.A.; Lofra, R.M.; Logigian, E.L.; Sowden, J.E.; Almodovar, J.L.; Littleton, J.T.; Zuchner, S.; et al. Electrophysiologic features of syt2 mutations causing a treatable neuromuscular syndrome. Neurology 2015, 85, 1964–1971. [Google Scholar] [CrossRef] [PubMed]
- Shields, M.C.; Bowers, M.R.; Fulcer, M.M.; Bollig, M.K.; Rock, P.J.; Sutton, B.R.; Vrailas-Mortimer, A.D.; Lochmuller, H.; Whittaker, R.G.; Horvath, R.; et al. Drosophila studies support a role for a presynaptic synaptotagmin mutation in a human congenital myasthenic syndrome. PLoS ONE 2017, 12, e0184817. [Google Scholar] [CrossRef] [PubMed]
- Salpietro, V.; Lin, W.; Delle Vedove, A.; Storbeck, M.; Liu, Y.; Efthymiou, S.; Manole, A.; Wiethoff, S.; Ye, Q.; Saggar, A.; et al. Homozygous mutations in vamp1 cause a presynaptic congenital myasthenic syndrome. Ann. Neurol. 2017, 81, 597–603. [Google Scholar] [CrossRef] [PubMed]
- Nystuen, A.M.; Schwendinger, J.K.; Sachs, A.J.; Yang, A.W.; Haider, N.B. A null mutation in vamp1/synaptobrevin is associated with neurological defects and prewean mortality in the lethal-wasting mouse mutant. Neurogenetics 2007, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Patrick, J.; Lindstrom, J. Autoimmune response to acetylcholine receptor. Science 1973, 180, 871–872. [Google Scholar] [CrossRef] [PubMed]
- Mantegazza, R.; Cordiglieri, C.; Consonni, A.; Baggi, F. Animal models of myasthenia gravis: Utility and limitations. Int. J. Gen. Med. 2016, 9, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Xiao, Y.; Zhang, K.; Luo, B.; Shen, C. Introducing autoimmunity at the synapse by a novel animal model of experimental autoimmune myasthenia gravis. Neuroscience 2018, 374, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Kusner, L.L.; Sengupta, M.; Kaminski, H.J. Acetylcholine receptor antibody-mediated animal models of myasthenia gravis and the role of complement. Ann. N. Y. Acad. Sci. 2018, 1413, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; Huijbers, M.G.M.; Verschuuren, J. Neuromuscular synapse electrophysiology in myasthenia gravis animal models. Ann. N. Y. Acad. Sci. 2018, 1412, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Viegas, S.; Jacobson, L.; Waters, P.; Cossins, J.; Jacob, S.; Leite, M.I.; Webster, R.; Vincent, A. Passive and active immunization models of musk-ab positive myasthenia: Electrophysiological evidence for pre and postsynaptic defects. Exp. Neurol. 2012, 234, 506–512. [Google Scholar] [CrossRef] [PubMed]
- Klooster, R.; Plomp, J.J.; Huijbers, M.G.; Niks, E.H.; Straasheijm, K.R.; Detmers, F.J.; Hermans, P.W.; Sleijpen, K.; Verrips, A.; Losen, M.; et al. Muscle-specific kinase myasthenia gravis igg4 autoantibodies cause severe neuromuscular junction dysfunction in mice. Brain 2012, 135, 1081–1101. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Lu, Y.; Zhang, B.; Figueiredo, D.; Bean, J.; Jung, J.; Wu, H.; Barik, A.; Yin, D.M.; Xiong, W.C.; et al. Antibodies against low-density lipoprotein receptor-related protein 4 induce myasthenia gravis. J. Clin. Investig. 2013, 123, 5190–5202. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, C.; Cavus, F.; Yilmaz, V.; Tuzun, E. Immunization with recombinantly expressed lrp4 induces experimental autoimmune myasthenia gravis in c57bl/6 mice. Immunol. Investig. 2017, 46, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Motohashi, N.; Takashima, R.; Kishi, M.; Nishimune, H.; Shigemoto, K. Immunization of mice with lrp4 induces myasthenia similar to musk-associated myasthenia gravis. Exp. Neurol. 2017, 297, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J.; van Kempen, G.T.; Molenaar, P.C. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J. Physiol. 1992, 458, 487–499. [Google Scholar] [CrossRef] [PubMed]
- Morsch, M.; Reddel, S.W.; Ghazanfari, N.; Toyka, K.V.; Phillips, W.D. Pyridostigmine but not 3,4-diaminopyridine exacerbates ach receptor loss and myasthenia induced in mice by muscle-specific kinase autoantibody. J. Physiol. 2013, 591, 2747–2762. [Google Scholar] [CrossRef] [PubMed]
- Plomp, J.J. Trans-synaptic homeostasis at the myasthenic neuromuscular junction. Front. Biosci. 2017, 22, 1033–1051. [Google Scholar] [CrossRef]
- Wang, X.; McIntosh, J.M.; Rich, M.M. Muscle nicotinic acetylcholine receptors may mediate trans-synaptic signaling at the mouse neuromuscular junction. J. Neurosci. 2018, 38, 1725–1736. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| CMS Disorder | CMS Subtype | Gene | Description | References |
|---|---|---|---|---|
| AChR-Deficiency | CMS4C | CHRNE, CHRNG | Expression of γ-AChR in ε-AChR knockout background generates weak mice with reduced endplate depolarisation but normal lifespan. | [3] |
| AChR-Slow channel kinetic | CMS4A, CMS1A, CMS3A | CHRNE, CHRNA1, CHRND | Expression of slow channel kinetic mutant AChR replicates prolongation of AChR current, muscle weakness, calcium overload and response to treatment. | [4,5,6,7,8,9,10,11,12] |
| MuSK | CMS9 | MUSK | Hemizygous expression of V789M mutant in knockout background generates overtly weak mouse with defects of NMJ structure and neurotransmission. | [13] |
| Rapsyn | CMS11 | RAPSN | Mutation within RING-domain of rapsyn inhibits E3-ligase activity, disrupts AChR cluster formation, motor nerve targeting and is perinatally lethal. | [14] |
| DOK7 | CMS10 | DOK7 | Duplication mutation (c.1124_1127dupTGCC) disrupts NMJ formation and is perinatally lethal.Overexpression of DOK7 rescues phenotype. | [15] |
| Agrin | CMS8 | AGRN | Chemically generated missense mutation causes NMJ degradation with decreased AChR density and reduced lifespan. | [16] |
© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webster, R.G. Animal Models of the Neuromuscular Junction, Vitally Informative for Understanding Function and the Molecular Mechanisms of Congenital Myasthenic Syndromes. Int. J. Mol. Sci. 2018, 19, 1326. https://doi.org/10.3390/ijms19051326
Webster RG. Animal Models of the Neuromuscular Junction, Vitally Informative for Understanding Function and the Molecular Mechanisms of Congenital Myasthenic Syndromes. International Journal of Molecular Sciences. 2018; 19(5):1326. https://doi.org/10.3390/ijms19051326
Chicago/Turabian StyleWebster, Richard G. 2018. "Animal Models of the Neuromuscular Junction, Vitally Informative for Understanding Function and the Molecular Mechanisms of Congenital Myasthenic Syndromes" International Journal of Molecular Sciences 19, no. 5: 1326. https://doi.org/10.3390/ijms19051326
APA StyleWebster, R. G. (2018). Animal Models of the Neuromuscular Junction, Vitally Informative for Understanding Function and the Molecular Mechanisms of Congenital Myasthenic Syndromes. International Journal of Molecular Sciences, 19(5), 1326. https://doi.org/10.3390/ijms19051326