Contribution of Tumor Endothelial Cells in Cancer Progression

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
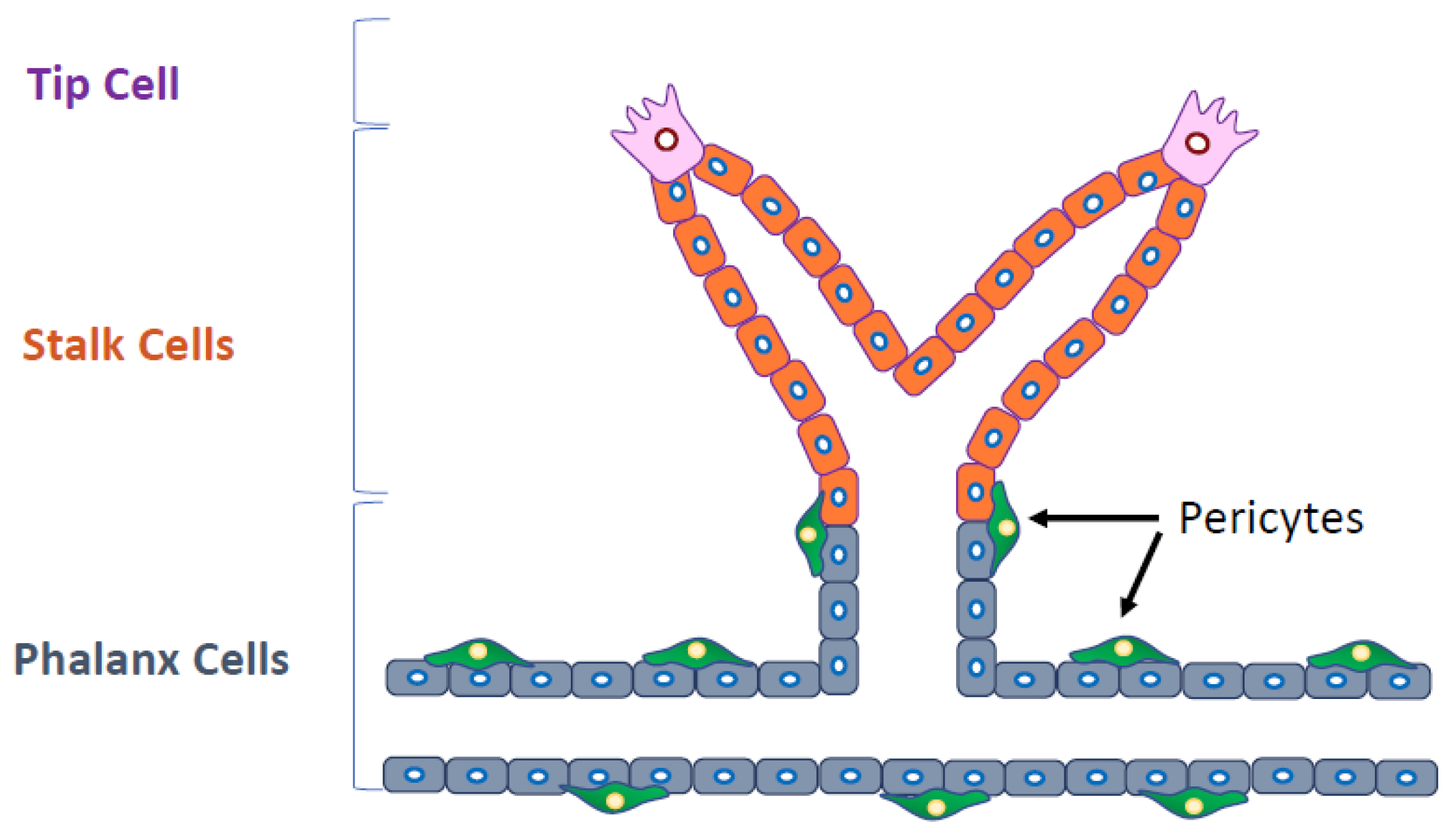
2. Molecules that Regulate Angiogenesis
3. Tumor Endothelial Cells
4. Drug Resistance and Cytogenetic Abnormalities in Tumor Endothelial Cells
5. Heterogeneity of Tumor Endothelial Cells
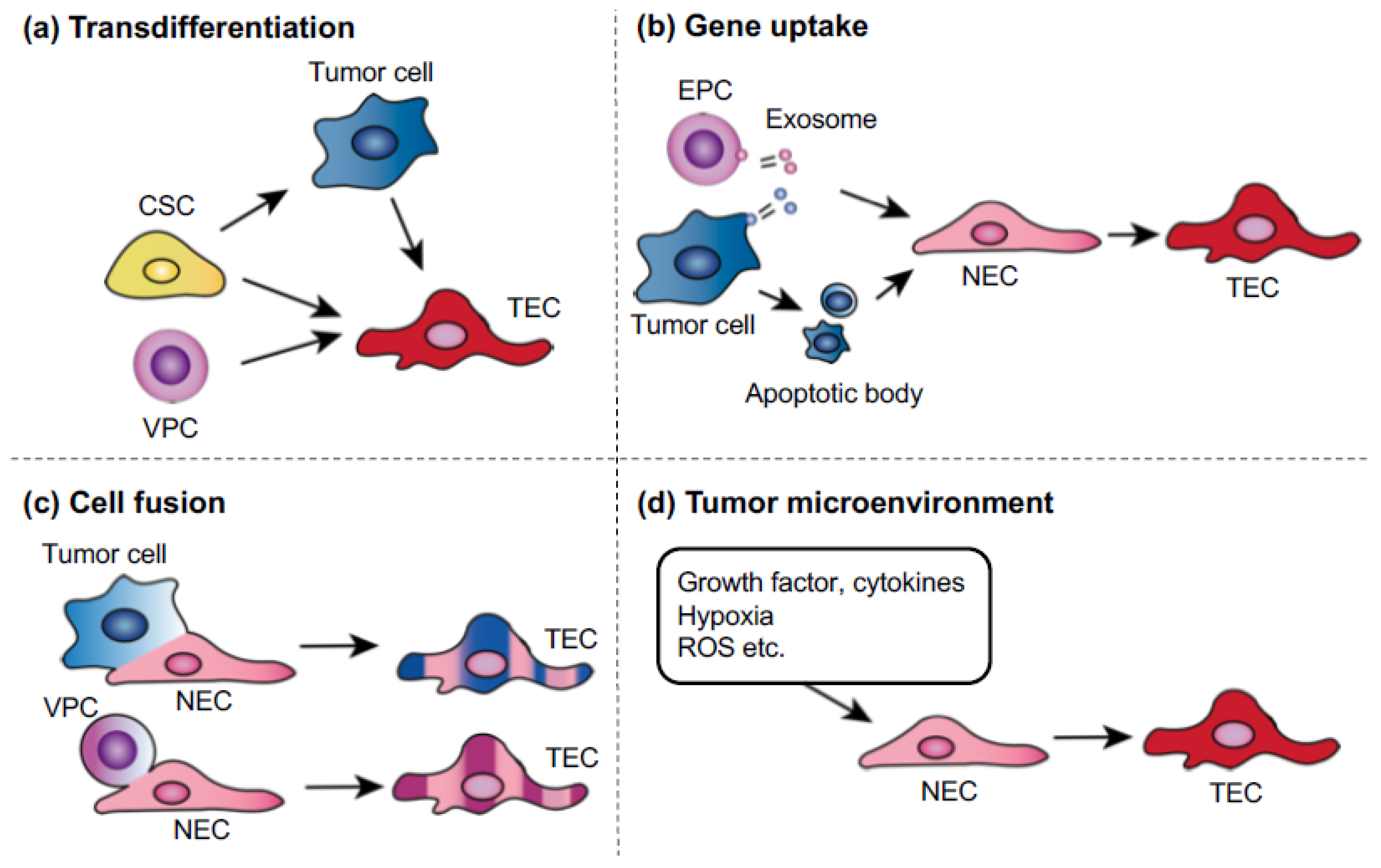
6. Mechanisms of Tumor Endothelial Cell Abnormality
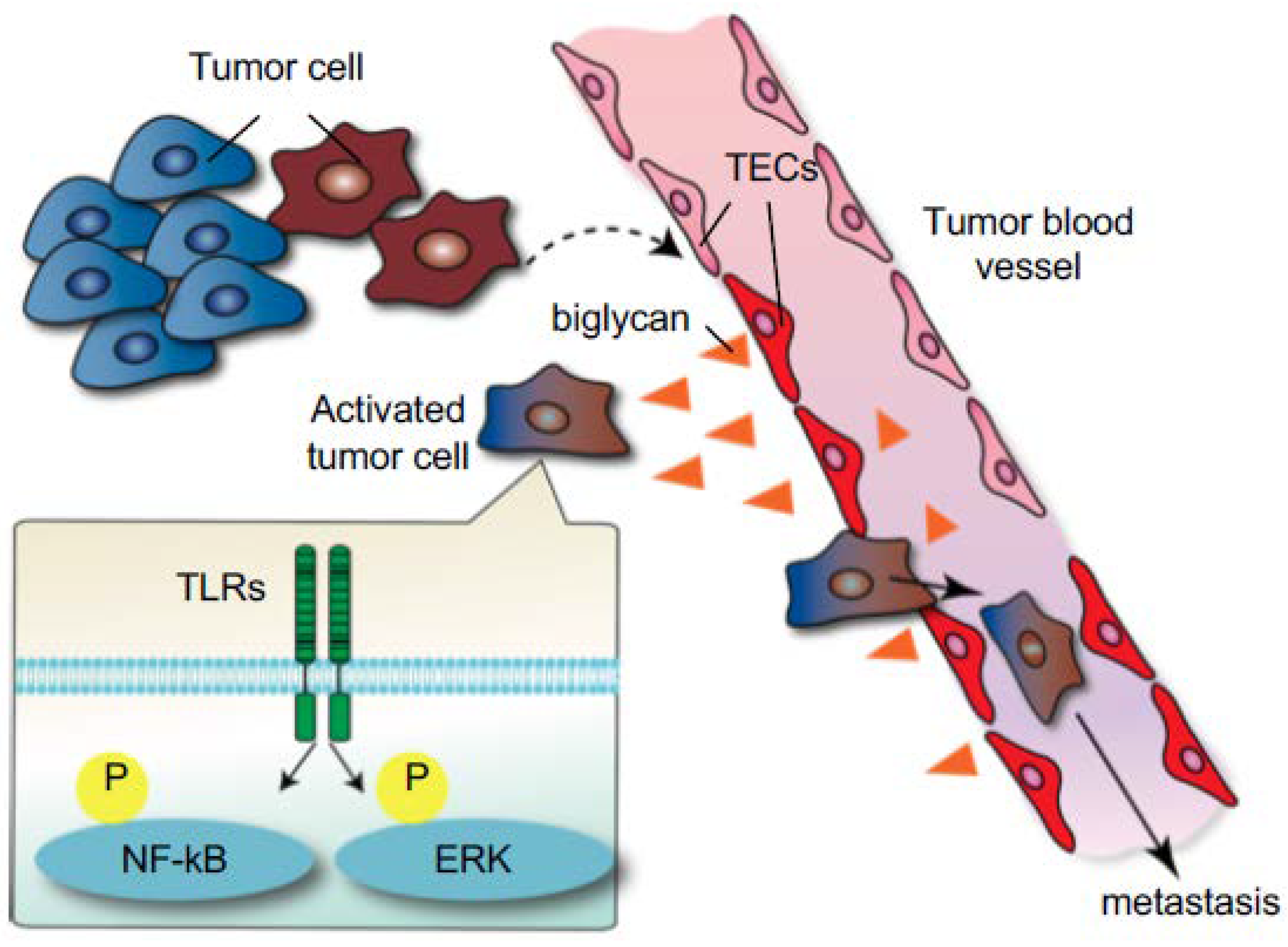
7. Tumor Endothelial Cells’ Roles in Cancer Progression
8. Conclusions
Acknowledgments
Conflicts of Interest
References
- Folkman, J. Tumor angiogenesis: Therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [PubMed]
- Folkman, J.; Kerbel, R. Role of angiogenesis in tumor growth and metastasis. Clinical translation of angiogenesis inhibitors. Semin. Oncol. 2002, 29, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.; Thomsen, J.L.; Primdahl, S.; Dyreborg, U.; Andersen, J.A. Breast cancer and atypia among young and middle-aged women: A study of 110 medicolegal autopsies. Br. J. Cancer 1987, 56, 814–819. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Galli, S.J.; Dvorak, A.M.; Perruzzi, C.A.; Harvey, V.S.; Dvorak, H.F. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 1983, 219, 983–985. [Google Scholar] [CrossRef] [PubMed]
- Ferrara, N.; Henzel, W.J. Pituitary follicular cells secrete a novel heparin-binding growth factor specific for vascular endothelial cells. Biochem. Biophys. Res. Commun. 1989, 161, 851–858. [Google Scholar] [CrossRef]
- Kim, K.J.; Li, B.; Winer, J.; Armanini, M.; Gillett, N.; Phillips, H.S.; Ferrara, N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature 1993, 362, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.H.; Fehrenbacher, L.; Novotny, W.F.; Herbst, R.S.; Nemunaitis, J.J.; Jablons, D.M.; Langer, C.J.; DeVore, R.F.; Gaudreault, J.; Damico, L.A.; et al. Randomized phase II trial comparing bevacizumab plus carboplatin and paclitaxel with carboplatin and paclitaxel alone in previously untreated locally advanced or metastatic non-small-cell lung cancer. J. Clin. Oncol. 2004, 22, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Keedy, V.L.; Sandler, A.B. Inhibition of angiogenesis in the treatment of non-small cell lung cancer. Cancer Sci. 2007, 98, 1825–1830. [Google Scholar] [CrossRef] [PubMed]
- Kindler, H.L.; Friberg, G.; Singh, D.A.; Locker, G.; Nattam, S.; Kozloff, M.; Taber, D.A.; Karrison, T.; Dachman, A.; Stadler, W.M.; et al. Phase II trial of bevacizumab plus gemcitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2005, 23, 8033–8040. [Google Scholar] [CrossRef] [PubMed]
- Saif, M.W.; Elfiky, A.; Salem, R.R. Gastrointestinal perforation due to bevacizumab in colorectal cancer. Ann. Surg. Oncol. 2007, 14, 1860–1869. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, M.; Claesson-Welsh, L. Signal transduction by VEGF receptors in regulation of angiogenesis and lymphangiogenesis. Exp. Cell Res. 2006, 312, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Dameron, K.M.; Volpert, O.V.; Tainsky, M.A.; Bouck, N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science 1994, 265, 1582–1584. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Troise, I.; Daly, C.; Papadopoulos, N.J.; Coetzee, S.; Boland, P.; Gale, N.W.; Lin, H.C.; Yancopoulos, G.D.; Thurston, G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 2006, 444, 1032–1037. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. The Vasohibin Family. Pharmaceuticals 2010, 3, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Minami, T.; Horiuchi, K.; Miura, M.; Abid, M.R.; Takabe, W.; Noguchi, N.; Kohro, T.; Ge, X.; Aburatani, H.; Hamakubo, T.; et al. Vascular endothelial growth factor- and thrombin-induced termination factor, Down syndrome critical region-1, attenuates endothelial cell proliferation and angiogenesis. J. Biol. Chem. 2004, 279, 50537–50554. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J. Antiangiogenesis in cancer therapy—Endostatin and its mechanisms of action. Exp. Cell Res. 2006, 312, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Maeshima, Y.; Manfredi, M.; Reimer, C.; Holthaus, K.A.; Hopfer, H.; Chandamuri, B.R.; Kharbanda, S.; Kalluri, R. Identification of the anti-angiogenic site within vascular basement membrane-derived tumstatin. J. Biol. Chem. 2001, 276, 15240–15248. [Google Scholar] [CrossRef] [PubMed]
- Ryeom, S.; Folkman, J. Role of endogenous angiogenesis inhibitors in Down syndrome. J. Craniofac. Surg. 2009, 20 (Suppl. 1), 595–596. [Google Scholar] [CrossRef] [PubMed]
- St Croix, B.; Rago, C.; Velculescu, V.; Traverso, G.; Romans, K.E.; Montgomery, E.; Lal, A.; Riggins, G.J.; Lengauer, C.; Vogelstein, B.; et al. Genes expressed in human tumor endothelium. Science 2000, 289, 1197–1202. [Google Scholar] [CrossRef] [PubMed]
- Nanda, A.; St Croix, B. Tumor endothelial markers: New targets for cancer therapy. Curr. Opin. Oncol. 2004, 16, 44–49. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Bonome, T.; Li, Y.; Kamat, A.A.; Han, L.Y.; Schmandt, R.; Coleman, R.L.; Gershenson, D.M.; Jaffe, R.B.; Birrer, M.J.; et al. Gene Alterations Identified by Expression Profiling in Tumor-Associated Endothelial Cells from Invasive Ovarian Carcinoma. Cancer Res. 2007, 67, 1757–1768. [Google Scholar] [CrossRef] [PubMed]
- Buckanovich, R.J.; Sasaroli, D.; O’Brien-Jenkins, A.; Botbyl, J.; Hammond, R.; Katsaros, D.; Sandaltzopoulos, R.; Liotta, L.A.; Gimotty, P.A.; Coukos, G. Tumor vascular proteins as biomarkers in ovarian cancer. J. Clin. Oncol. 2007, 25, 852–861. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R. Gene expression of tumor angiogenesis dissected: Specific targeting of colon cancer angiogenic vasculature. Blood 2006, 108, 2339–2348. [Google Scholar] [CrossRef] [PubMed]
- Seaman, S.; Stevens, J.; Yang, M.Y.; Logsdon, D.; Graff-Cherry, C.; St Croix, B. Genes that Distinguish Physiological and Pathological Angiogenesis. Cancer Cell 2007, 11, 539–554. [Google Scholar] [CrossRef] [PubMed]
- Van Beijnum, J.R.; Nowak-Sliwinska, P.; van den Boezem, E.; Hautvast, P.; Buurman, W.A.; Griffioen, A.W. Tumor angiogenesis is enforced by autocrine regulation of high-mobility group box 1. Oncogene 2013, 32, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Deambrosis, I.; Russo, S.; Deregibus, M.C.; Camussi, G. Altered angiogenesis and survival in human tumor-derived endothelial cells. FASEB J. 2003, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hida, K.; Hida, Y.; Amin, D.N.; Flint, A.F.; Panigrahy, D.; Morton, C.C.; Klagsbrun, M. Tumor-associated endothelial cells with cytogenetic abnormalities. Cancer Res. 2004, 64, 8249–8255. [Google Scholar] [CrossRef] [PubMed]
- Arbiser, J.L.; Raab, G.; Rohan, R.M.; Paul, S.; Hirschi, K.; Flynn, E.; Price, E.R.; Fisher, D.E.; Cohen, C.; Klagsbrun, M. Isolation of mouse stromal cells associated with a human tumor using differential diphtheria toxin sensitivity. Am. J. Pathol. 1999, 155, 723–729. [Google Scholar] [CrossRef]
- Matsuda, K.; Ohga, N.; Hida, Y.; Muraki, C.; Tsuchiya, K.; Kurosu, T.; Akino, T.; Shih, S.-C.; Totsuka, Y.; Klagsbrun, M.; et al. Isolated tumor endothelial cells maintain specific character during long-term culture. Biochem. Biophys. Res. Commun. 2010, 394, 947–954. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Ohga, N.; Hida, Y.; Akiyama, K.; Kitayama, K.; Osawa, T.; Onodera, Y.; Shinohara, N.; Nonomura, K.; Shindoh, M.; et al. CXCR7: A novel tumor endothelial marker in renal cell carcinoma. Pathol. Int. 2012, 62, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Osawa, T.; Ohga, N.; Akiyama, K.; Hida, Y.; Kitayama, K.; Kawamoto, T.; Yamamoto, K.; Maishi, N.; Kondoh, M.; Onodera, Y.; et al. Lysyl oxidase secreted by tumour endothelial cells promotes angiogenesis and metastasis. Br. J. Cancer 2013, 109, 2237–2247. [Google Scholar] [CrossRef] [PubMed]
- Amin, D.N.; Hida, K.; Bielenberg, D.R.; Klagsbrun, M. Tumor endothelial cells express epidermal growth factor receptor (EGFR) but not ErbB3 and are responsive to EGF and to EGFR kinase inhibitors. Cancer Res. 2006, 66, 2173–2180. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, K.; Hida, K.; Hida, Y.; Muraki, C.; Ohga, N.; Akino, T.; Kondo, T.; Miseki, T.; Nakagawa, K.; Shindoh, M.; et al. Adrenomedullin antagonist suppresses tumor formation in renal cell carcinoma through inhibitory effects on tumor endothelial cells and endothelial progenitor mobilization. Int. J. Oncol. 2010, 36, 1379–1386. [Google Scholar] [PubMed]
- Kurosu, T.; Ohga, N.; Hida, Y.; Maishi, N.; Akiyama, K.; Kakuguchi, W.; Kuroshima, T.; Kondo, M.; Akino, T.; Totsuka, Y.; et al. HuR keeps an angiogenic switch on by stabilising mRNA of VEGF and COX-2 in tumour endothelium. Br. J. Cancer 2011, 104, 819–829. [Google Scholar] [CrossRef] [PubMed]
- Bussolati, B.; Assenzio, B.; Deregibus, M.C.; Camussi, G. The proangiogenic phenotype of human tumor-derived endothelial cells depends on thrombospondin-1 downregulation via phosphatidylinositol 3-kinase/Akt pathway. J. Mol. Med. 2006, 84, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Muraki, C.; Ohga, N.; Hida, Y.; Nishihara, H.; Kato, Y.; Tsuchiya, K.; Matsuda, K.; Totsuka, Y.; Shindoh, M.; HIDA, K. Cyclooxygenase-2 inhibition causes antiangiogenic effects on tumor endothelial and vascular progenitor cells. Int. J. Cancer 2011, 130, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Akino, T.; Hida, K.; Hida, Y.; Tsuchiya, K.; Freedman, D.; Muraki, C.; Ohga, N.; Matsuda, K.; Akiyama, K.; Harabayashi, T.; et al. Cytogenetic abnormalities of tumor-associated endothelial cells in human malignant tumors. Am. J. Pathol. 2009, 175, 2657–2667. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.P.; Gires, O.; Wang, D.D.; Li, L.; Wang, H. Comprehensive in situ co-detection of aneuploid circulating endothelial and tumor cells. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Streubel, B.; Chott, A.; Huber, D.; Exner, M.; Jager, U.; Wagner, O.; Schwarzinger, I. Lymphoma-specific genetic aberrations in microvascular endothelial cells in B-cell lymphomas. N. Engl. J. Med. 2004, 351, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Chadalavada, K.; Wilshire, J.; Kowalik, U.; Hovinga, K.E.; Geber, A.; Fligelman, B.; Leversha, M.; Brennan, C.; Tabar, V. Glioblastoma stem-like cells give rise to tumour endothelium. Nature 2010, 468, 829–833. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.Q.; Sun, H.C.; Zhang, W.; Zhu, X.D.; Zhuang, P.Y.; Zhang, J.B.; Wang, L.; Wu, W.Z.; Qin, L.X.; Tang, Z.Y. Human Hepatocellular Carcinoma Tumor-derived Endothelial Cells Manifest Increased Angiogenesis Capability and Drug Resistance Compared with Normal Endothelial Cells. Clin. Cancer Res. 2009, 15, 4838–4846. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Ohga, N.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Ishikawa, S.; Maishi, N.; Akino, T.; Kondoh, M.; Matsuda, A.; et al. Tumor Endothelial Cells Acquire Drug Resistance by MDR1 Up-Regulation via VEGF Signaling in Tumor Microenvironment. Am. J. Pathol. 2012, 180, 1283–1293. [Google Scholar] [CrossRef] [PubMed]
- Duensing, S.; Munger, K. Human papillomaviruses and centrosome duplication errors: Modeling the origins of genomic instability. Oncogene 2002, 21, 6241–6248. [Google Scholar] [CrossRef] [PubMed]
- Ohga, N.; Ishikawa, S.; Maishi, N.; Akiyama, K.; Hida, Y.; Kawamoto, T.; Sadamoto, Y.; Osawa, T.; Yamamoto, K.; Kondoh, M.; et al. Heterogeneity of tumor endothelial cells: Comparison between tumor endothelial cells isolated from high- and low-metastatic tumors. Am. J. Pathol. 2012, 180, 1294–1307. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Ohba, Y.; Alam, M.T.; Kawamoto, T.; Ohmura, H.; Yamada, K.; Torii, C.; et al. Inhibition of Multidrug Transporter in Tumor Endothelial Cells Enhances Antiangiogenic Effects of Low-Dose Metronomic Paclitaxel. Am. J. Pathol. 2015, 185, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Mundhekar, A.N.; Bullard, D.C.; Kucik, D.F. Intracellular heterogeneity in adhesiveness of endothelium affects early steps in leukocyte adhesion. Am. J. Physiol. Cell Physiol. 2006, 291, C130–C137. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Molema, G. Heterogeneity in endothelial responsiveness to cytokines, molecular causes, and pharmacological consequences. Semin. Thromb. Hemost. 2010, 36, 246–264. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Kidoya, H.; Sakimoto, S.; Wakabayashi, T.; Takakura, N. Identification and characterization of a resident vascular stem/progenitor cell population in preexisting blood vessels. EMBO J. 2011, 31, 842–855. [Google Scholar] [CrossRef] [PubMed]
- Naito, H.; Wakabayashi, T.; Kidoya, H.; Muramatsu, F.; Takara, K.; Eino, D.; Yamane, K.; Iba, T.; Takakura, N. Endothelial Side Population Cells Contribute to Tumor Angiogenesis and Antiangiogenic Drug Resistance. Cancer Res. 2016, 76, 3200–3210. [Google Scholar] [CrossRef] [PubMed]
- Saubamea, B.; Cochois-Guegan, V.; Cisternino, S.; Scherrmann, J.M. Heterogeneity in the rat brain vasculature revealed by quantitative confocal analysis of endothelial barrier antigen and P-glycoprotein expression. J. Cereb. Blood Flow Metab. 2012, 32, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Ohmura-Kakutani, H.; Akiyama, K.; Maishi, N.; Ohga, N.; Hida, Y.; Kawamoto, T.; Iida, J.; Shindoh, M.; Tsuchiya, K.; Shinohara, N.; et al. Identification of tumor endothelial cells with high aldehyde dehydrogenase activity and a highly angiogenic phenotype. PLoS ONE 2014, 9, e113910. [Google Scholar] [CrossRef] [PubMed]
- Langenkamp, E.; Molema, G. Microvascular endothelial cell heterogeneity: General concepts and pharmacological consequences for anti-angiogenic therapy of cancer. Cell Tissue Res. 2009, 335, 205–222. [Google Scholar] [CrossRef] [PubMed]
- Nagy, J.A.; Dvorak, H.F. Heterogeneity of the tumor vasculature: The need for new tumor blood vessel type-specific targets. Clin. Exp. Metast. 2012, 29, 657–662. [Google Scholar] [CrossRef] [PubMed]
- Adya, R.; Tan, B.K.; Punn, A.; Chen, J.; Randeva, H.S. Visfatin induces human endothelial VEGF and MMP-2/9 production via MAPK and PI3K/Akt signalling pathways: Novel insights into visfatin-induced angiogenesis. Cardiovasc. Res. 2008, 78, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Peterson, T.E.; Kirkpatrick, N.D.; Huang, Y.; Farrar, C.T.; Marijt, K.A.; Kloepper, J.; Datta, M.; Amoozgar, Z.; Seano, G.; Jung, K.; et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc. Natl. Acad. Sci. USA 2016, 113, 4470–4475. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K. Normalizing tumor vasculature with anti-angiogenic therapy: A new paradigm for combination therapy. Nat. Med. 2001, 7, 987–989. [Google Scholar] [CrossRef] [PubMed]
- Winkler, F.; Kozin, S.V.; Tong, R.T.; Chae, S.S.; Booth, M.F.; Garkavtsev, I.; Xu, L.; Hicklin, D.J.; Fukumura, D.; di Tomaso, E.; et al. Kinetics of vascular normalization by VEGFR2 blockade governs brain tumor response to radiation: Role of oxygenation, angiopoietin-1, and matrix metalloproteinases. Cancer Cell 2004, 6, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Takara, K.; Yamakawa, D.; Kidoya, H.; Takakura, N. Apelin as a marker for monitoring the tumor vessel normalization window during antiangiogenic therapy. Cancer Sci. 2015, 107, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y. Persistent vascular normalization as an alternative goal of anti-angiogenic cancer therapy. Cancer Sci. 2011, 102, 1253–1256. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, I.; Scheffrahn, I.; Bartling, S.; Weis, J.; von Felbert, V.; Middleton, M.; Kato, M.; Ergun, S.; Schadendorf, D. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in malignant melanoma. J. Exp. Med. 2010, 207, 491–503. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Nolan, D.; McDonnell, K.; Vahdat, L.; Benezra, R.; Altorki, N.; Mittal, V. Bone marrow-derived endothelial progenitor cells contribute to the angiogenic switch in tumor growth and metastatic progression. Biochim. Biophys. Acta 2009, 1796, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Taylor, S.M.; Nevis, K.R.; Park, H.L.; Rogers, G.C.; Rogers, S.L.; Cook, J.G.; Bautch, V.L. Angiogenic factor signaling regulates centrosome duplication in endothelial cells of developing blood vessels. Blood 2010, 116, 3108–3117. [Google Scholar] [CrossRef] [PubMed]
- Kondoh, M.; Ohga, N.; Akiyama, K.; Hida, Y.; Maishi, N.; Towfik, A.M.; Inoue, N.; Shindoh, M.; Hida, K. Hypoxia-induced reactive oxygen species cause chromosomal abnormalities in endothelial cells in the tumor microenvironment. PLoS ONE 2013, 8, e80349. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Gutkind, J.S. VEGF controls endothelial-cell permeability by promoting the beta-arrestin-dependent endocytosis of VE-cadherin. Nat. Cell Biol. 2006, 8, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; di Tomaso, E.; McDonald, D.M.; Jones, R.; Jain, R.K.; Munn, L.L. Mosaic blood vessels in tumors: Frequency of cancer cells in contact with flowing blood. Proc. Natl. Acad. Sci. USA 2000, 97, 14608–14613. [Google Scholar] [CrossRef] [PubMed]
- Butler, J.M.; Kobayashi, H.; Rafii, S. Instructive role of the vascular niche in promoting tumour growth and tissue repair by angiocrine factors. Nat. Rev. Cancer 2010, 10, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Ding, B.S.; Guo, P.; Lee, S.B.; Butler, J.M.; Casey, S.C.; Simons, M.; Tam, W.; Felsher, D.W.; Shido, K.; et al. Angiocrine factors deployed by tumor vascular niche induce B cell lymphoma invasiveness and chemoresistance. Cancer Cell 2014, 25, 350–365. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Scandura, J.M.; Inghirami, G.G.; Shido, K.; Ding, B.S.; Rafii, S. Molecular Checkpoint Decisions Made by Subverted Vascular Niche Transform Indolent Tumor Cells into Chemoresistant Cancer Stem Cells. Cancer Cell 2017, 31, 110–126. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.S.; Costello, M.A.; Talsma, C.E.; Flack, C.G.; Crowley, J.G.; Hamm, L.L.; He, X.; Hervey-Jumper, S.L.; Heth, J.A.; Muraszko, K.M.; et al. Endothelial cells create a stem cell niche in glioblastoma by providing NOTCH ligands that nurture self-renewal of cancer stem-like cells. Cancer Res. 2011, 71, 6061–6072. [Google Scholar] [CrossRef] [PubMed]
- Wieland, E.; Rodriguez-Vita, J.; Liebler, S.S.; Mogler, C.; Moll, I.; Herberich, S.E.; Espinet, E.; Herpel, E.; Menuchin, A.; Chang-Claude, J.; et al. Endothelial Notch1 Activity Facilitates Metastasis. Cancer Cell 2017, 31, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Maishi, N.; Ohba, Y.; Akiyama, K.; Ohga, N.; Hamada, J.; Nagao-Kitamoto, H.; Alam, M.T.; Yamamoto, K.; Kawamoto, T.; Inoue, N.; et al. Tumour endothelial cells in high metastatic tumours promote metastasis via epigenetic dysregulation of biglycan. Sci. Rep. 2016, 6, 28039. [Google Scholar] [CrossRef] [PubMed]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hida, K.; Maishi, N.; Annan, D.A.; Hida, Y. Contribution of Tumor Endothelial Cells in Cancer Progression. Int. J. Mol. Sci. 2018, 19, 1272. https://doi.org/10.3390/ijms19051272
Hida K, Maishi N, Annan DA, Hida Y. Contribution of Tumor Endothelial Cells in Cancer Progression. International Journal of Molecular Sciences. 2018; 19(5):1272. https://doi.org/10.3390/ijms19051272
Chicago/Turabian StyleHida, Kyoko, Nako Maishi, Dorcas A. Annan, and Yasuhiro Hida. 2018. "Contribution of Tumor Endothelial Cells in Cancer Progression" International Journal of Molecular Sciences 19, no. 5: 1272. https://doi.org/10.3390/ijms19051272
APA StyleHida, K., Maishi, N., Annan, D. A., & Hida, Y. (2018). Contribution of Tumor Endothelial Cells in Cancer Progression. International Journal of Molecular Sciences, 19(5), 1272. https://doi.org/10.3390/ijms19051272