The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis



{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
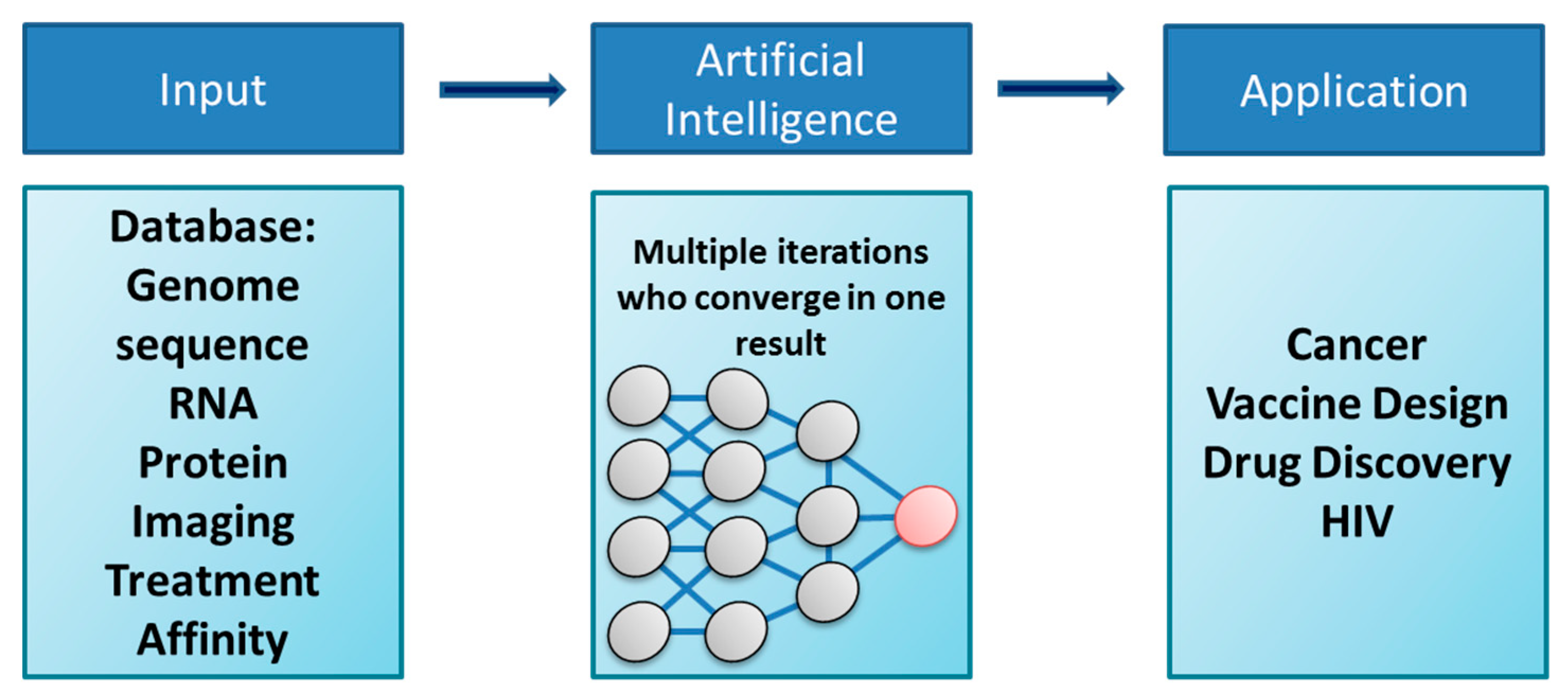
2. Artificial Intelligence and Machine Learning: New Tools to Identify Drug Targets and Common Pathways of Disease
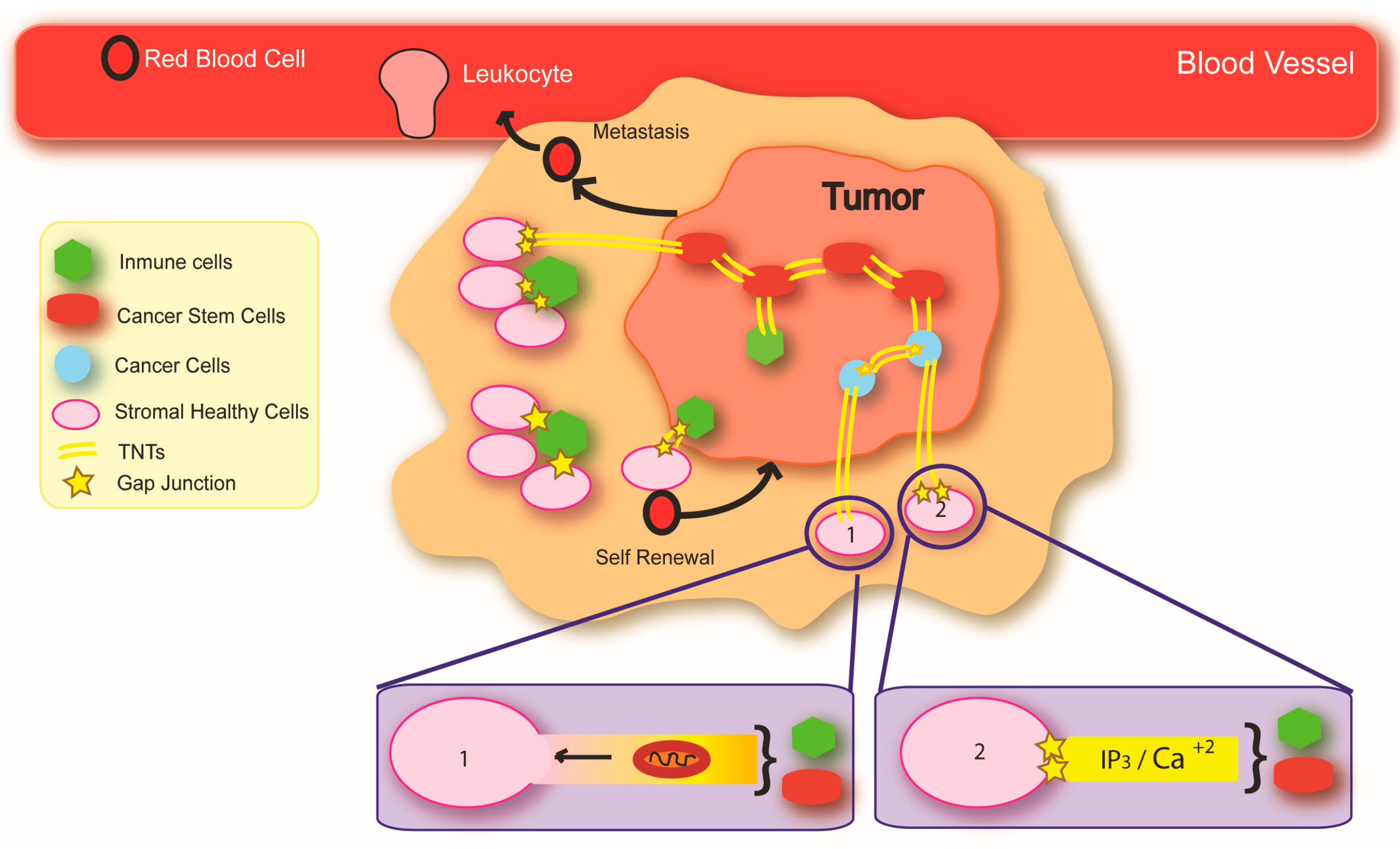
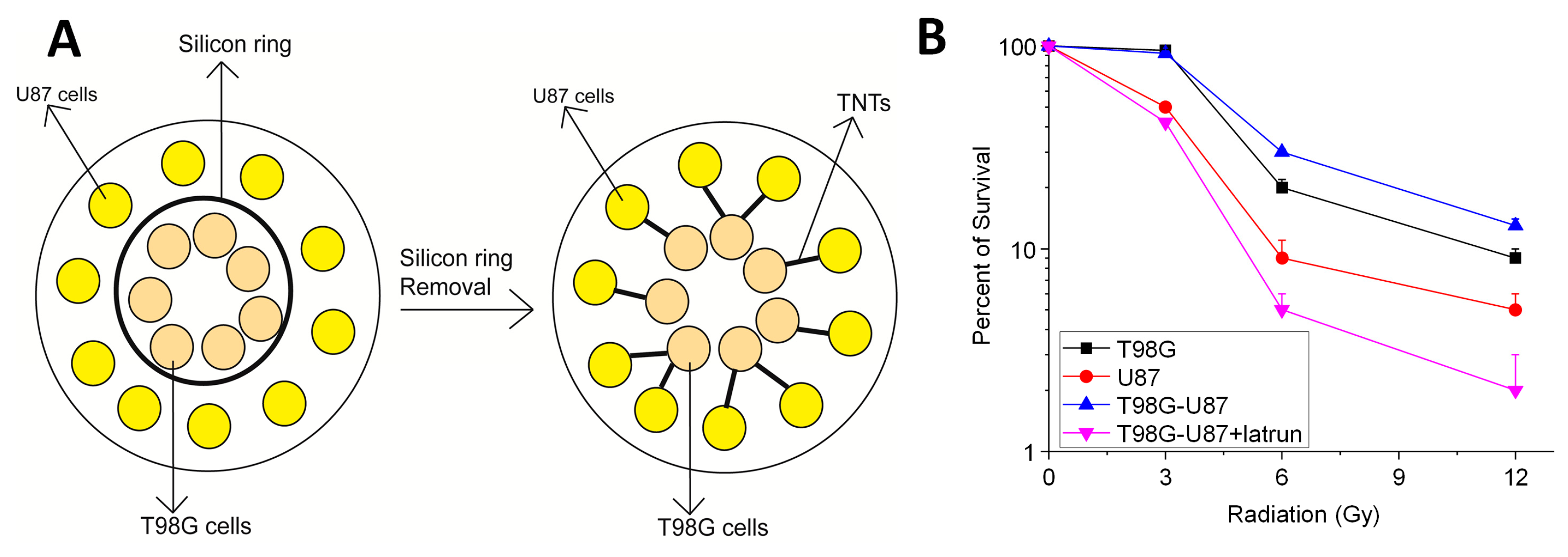
3. Connexins and Cancer
4. Mechanisms of Cancer Initiation and Spread: Potential Implications of the Intercellular Transfer of Genetic Alterations
5. Cancer and Metabolic Compromise: Focus on Central Nervous System Malignancies
6. Metabolism of Aggressive Glioblastoma
7. Connexin Channels: Novel Roles in Cancer
8. Future Directions and Conclusions
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Tomasetti, C.; Li, L.; Vogelstein, B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science 2017, 355, 1330–1334. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Heidt, D.G.; Dalerba, P.; Burant, C.F.; Zhang, L.; Adsay, V.; Wicha, M.; Clarke, M.F.; Simeone, D.M. Identification of pancreatic cancer stem cells. Cancer Res. 2007, 67, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Amanam, I.; Chung, V. Targeted Therapies for Pancreatic Cancer. Cancers 2018, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- McFaline-Figueroa, J.R.; Lee, E.Q. Brain Tumors. Am. J. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 17040. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.K.; di Tomaso, E.; Duda, D.G.; Loeffler, J.S.; Sorensen, A.G.; Batchelor, T.T. Angiogenesis in brain tumours. Nat. Rev. Neurosci. 2007, 8, 610–622. [Google Scholar] [CrossRef] [PubMed]
- Koshy, M.; Villano, J.L.; Dolecek, T.A.; Howard, A.; Mahmood, U.; Chmura, S.J.; Weichselbaum, R.R.; McCarthy, B.J. Improved survival time trends for glioblastoma using the SEER 17 population-based registries. J. Neurooncol. 2012, 107, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Crespin, S.; Fromont, G.; Wager, M.; Levillain, P.; Cronier, L.; Monvoisin, A.; Defamie, N.; Mesnil, M. Expression of a gap junction protein, connexin43, in a large panel of human gliomas: New insights. Cancer Med. 2016, 5, 1742–1752. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.M.; Liu, Y.; Regner, K.R.; Jotterand, F.; Liu, P.; Liang, M. Artificial Intelligence, Physiological Genomics, and Precision Medicine. Physiol. Genom. 2018. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Romero, M.; Vazquez-Naya, J.M.; Rabunal, J.R.; Pita-Fernandez, S.; Macenlle, R.; Castro-Alvarino, J.; Lopez-Roses, L.; Ulla, J.L.; Martinez-Calvo, A.V.; Vazquez, S.; et al. Artificial intelligence techniques for colorectal cancer drug metabolism: Ontology and complex network. Curr. Drug Metab. 2010, 11, 347–368. [Google Scholar] [CrossRef] [PubMed]
- Koscielny, G.; An, P.; Carvalho-Silva, D.; Cham, J.A.; Fumis, L.; Gasparyan, R.; Hasan, S.; Karamanis, N.; Maguire, M.; Papa, E.; et al. Open Targets: A platform for therapeutic target identification and validation. Nucleic Acids Res. 2017, 45, D985–D994. [Google Scholar] [CrossRef] [PubMed]
- Ching, T.; Himmelstein, D.S.; Beaulieu-Jones, B.K.; Kalinin, A.A.; Do, B.T.; Way, G.P.; Ferrero, E.; Agapow, P.-M.; Zietz, M.; Hoffman, M.M. Opportunities and obstacles for deep learning in biology and medicine. bioRxiv 2018, 142760. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, E.; Dunham, I.; Sanseau, P. In silico prediction of novel therapeutic targets using gene-disease association data. J. Transl. Med. 2017, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Ariazi, J.; Benowitz, A.; De Biasi, V.; Den Boer, M.L.; Cherqui, S.; Cui, H.; Douillet, N.; Eugenin, E.A.; Favre, D.; Goodman, S.; et al. Tunneling Nanotubes and Gap Junctions-Their Role in Long-Range Intercellular Communication during Development, Health, and Disease Conditions. Front. Mol. Neurosci. 2017, 10, 333. [Google Scholar] [CrossRef] [PubMed]
- Rios Velazquez, E.; Parmar, C.; Liu, Y.; Coroller, T.P.; Cruz, G.; Stringfield, O.; Ye, Z.; Makrigiorgos, M.; Fennessy, F.; Mak, R.H.; et al. Somatic Mutations Drive Distinct Imaging Phenotypes in Lung Cancer. Cancer Res. 2017, 77, 3922–3930. [Google Scholar] [CrossRef] [PubMed]
- Mullin, E. Stopping Breast Cancer with Help of AI. MIT Technol. Rev. 2016, 1, 1–5. [Google Scholar]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Senthebane, D.A.; Rowe, A.; Thomford, N.E.; Shipanga, H.; Munro, D.; Mazeedi, M.; Almazyadi, H.A.M.; Kallmeyer, K.; Dandara, C.; Pepper, M.S.; et al. The Role of Tumor Microenvironment in Chemoresistance: To Survive, Keep Your Enemies Closer. Int. J. Mol. Sci. 2017, 18, 1586. [Google Scholar] [CrossRef] [PubMed]
- Rahman, M.; Hasan, M.R. Cancer Metabolism and Drug Resistance. Metabolites 2015, 5, 571–600. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Solecki, G.; Wick, W.; Winkler, F. A malignant cellular network in gliomas: Potential clinical implications. Neuro Oncol. 2016, 18, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Desir, S.; Dickson, E.L.; Vogel, R.I.; Thayanithy, V.; Wong, P.; Teoh, D.; Geller, M.A.; Steer, C.J.; Subramanian, S.; Lou, E. Tunneling nanotube formation is stimulated by hypoxia in ovarian cancer cells. Oncotarget 2016, 7, 43150–43161. [Google Scholar] [CrossRef] [PubMed]
- Osswald, M.; Jung, E.; Sahm, F.; Solecki, G.; Venkataramani, V.; Blaes, J.; Weil, S.; Horstmann, H.; Wiestler, B.; Syed, M.; et al. Brain tumour cells interconnect to a functional and resistant network. Nature 2015, 528, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.V.; Contreras, J.E.; Bukauskas, F.F.; Saez, J.C. New roles for astrocytes: Gap junction hemichannels have something to communicate. Trends Neurosci. 2003, 26, 610–617. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.V.; Verselis, V.K. Biophysics of gap junctions. Semin. Cell Biol. 1992, 3, 29–47. [Google Scholar] [CrossRef]
- Saez, J.C.; Berthoud, V.M.; Branes, M.C.; Martinez, A.D.; Beyer, E.C. Plasma membrane channels formed by connexins: Their regulation and functions. Physiol. Rev. 2003, 83, 1359–1400. [Google Scholar] [CrossRef] [PubMed]
- Saez, J.C.; Contreras, J.E.; Bukauskas, F.F.; Retamal, M.A.; Bennett, M.V. Gap junction hemichannels in astrocytes of the CNS. Acta Physiol. Scand. 2003, 179, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Emerging issues of connexin channels: Biophysics fills the gap. Q. Rev. Biophys. 2001, 34, 325–472. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.L. Connexin channel permeability to cytoplasmic molecules. Prog. Biophys. Mol. Biol. 2007, 94, 120–143. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Monvoisin, A.; Vix, J.; Mesnil, M.; Thuringer, D.; Debiais, F.; Cronier, L. Connexins, important players in the dissemination of prostate cancer cells. Biochim. Biophys. Acta 2018, 1860, 202–215. [Google Scholar] [CrossRef] [PubMed]
- Aasen, T.; Mesnil, M.; Naus, C.C.; Lampe, P.D.; Laird, D.W. Gap junctions and cancer: Communicating for 50 years. Nat. Rev. Cancer 2016, 16, 775–788. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, D. Connexin’s Connection in Breast Cancer Growth and Progression. Int. J. Cell Biol. 2016, 2016, 9025905. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Crespin, S.; Mesnil, M. Opposing roles of connexin43 in glioma progression. Biochim. Biophys. Acta 2012, 1818, 2058–2067. [Google Scholar] [CrossRef] [PubMed]
- Sato, H.; Hagiwara, H.; Ohde, Y.; Senba, H.; Virgona, N.; Yano, T. Regulation of renal cell carcinoma cell proliferation, invasion and metastasis by connexin 32 gene. J. Membr. Biol. 2007, 216, 17–21. [Google Scholar] [CrossRef] [PubMed]
- Trosko, J.E.; Chang, C.C.; Upham, B.L.; Tai, M.H. Ignored hallmarks of carcinogenesis: Stem cells and cell-cell communication. Ann. N. Y. Acad. Sci. 2004, 1028, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Dahlberg, C.I.; Sarhan, D.; Chrobok, M.; Duru, A.D.; Alici, E. Natural Killer Cell-Based Therapies Targeting Cancer: Possible Strategies to Gain and Sustain Anti-Tumor Activity. Front. Immunol. 2015, 6, 605. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.B.; Rasmussen, M.; Rasmussen, L.J. Nuclear and mitochondrial DNA repair: Similar pathways? Mitochondrion 2005, 5, 89–108. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. Cancer cell cycles. Science 1996, 274, 1672–1677. [Google Scholar] [CrossRef] [PubMed]
- Branzei, D.; Foiani, M. Regulation of DNA repair throughout the cell cycle. Nat. Rev. Mol. Cell Biol. 2008, 9, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria and cancer. Cancer Metab. 2014, 2, 8. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Giampazolias, E.; Tait, S.W. Mitochondria and the hallmarks of cancer. FEBS J. 2016, 283, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Ahn, C.S.; Metallo, C.M. Mitochondria as biosynthetic factories for cancer proliferation. Cancer Metab. 2015, 3, 1. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.K. Mitochondria damage checkpoint, aging, and cancer. Ann. N. Y. Acad. Sci. 2006, 1067, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Rossignol, R.; Sonveaux, P. Mitochondria in cancer. Biochim. Biophys. Acta 2017, 1858, 553–555. [Google Scholar] [CrossRef] [PubMed]
- Bhandary, B.; Marahatta, A.; Kim, H.R.; Chae, H.J. Mitochondria in relation to cancer metastasis. J. Bioenerg. Biomembr. 2012, 44, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Gerbitz, K.D. Does the mitochondrial DNA play a role in the pathogenesis of diabetes? Diabetologia 1992, 35, 1181–1186. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell. Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef] [PubMed]
- Penta, J.S.; Johnson, F.M.; Wachsman, J.T.; Copeland, W.C. Mitochondrial DNA in human malignancy. Mutat. Res. 2001, 488, 119–133. [Google Scholar] [CrossRef]
- Frezza, C.; Gottlieb, E. Mitochondria in cancer: Not just innocent bystanders. Semin. Cancer Biol. 2009, 19, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Bogenhagen, D.F. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 2012, 1819, 914–920. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, F.; Schneider, P.M.; Metzger, R.; Warnecke-Eberz, U.; Baldus, S.E.; Dienes, H.P.; Aikou, T.; Hoelscher, A.H. Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett’s esophagus. Oncogene 2002, 21, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondrial genetics: A paradigm for aging and degenerative diseases? Science 1992, 256, 628–632. [Google Scholar] [CrossRef] [PubMed]
- Wei, Y.H. Oxidative stress and mitochondrial DNA mutations in human aging. Proc. Soc. Exp. Biol. Med. 1998, 217, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Han, H.; Hu, J.Q.; Yan, Q.; Zhu, J.Z.; Zhu, Z.B.; Chen, Y.J.; Sun, J.T.; Zhang, R.Y. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol. Med. Rep. 2016, 13, 1517–1524. [Google Scholar] [CrossRef] [PubMed]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Fujisawa, S.; Morozov, A.; Barlas, A.; Romin, Y.; Dogan, Y.; Gholami, S.; Moreira, A.L.; Manova-Todorova, K.; Moore, M.A. Tunneling nanotubes provide a unique conduit for intercellular transfer of cellular contents in human malignant pleural mesothelioma. PLoS ONE 2012, 7, e33093. [Google Scholar] [CrossRef] [PubMed]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.; Qiu, Y.; Shi, Y.; Cai, J.; Wang, B.; Wei, X.; Ke, Q.; Sui, X.; Wang, Y.; et al. Cell adhesion-mediated mitochondria transfer contributes to mesenchymal stem cell-induced chemoresistance on T cell acute lymphoblastic leukemia cells. J. Hematol. Oncol. 2018, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Guo, R.; Davis, D.; Fang, Y. Intercellular transfer of mitochondria rescues virus-induced cell death but facilitates cell-to-cell spreading of porcine reproductive and respiratory syndrome virus. Virology 2018. [Google Scholar] [CrossRef] [PubMed]
- Da Cunha, F.M.; Torelli, N.Q.; Kowaltowski, A.J. Mitochondrial Retrograde Signaling: Triggers, Pathways, and Outcomes. Oxid. Med. Cell. Longev. 2015, 2015, 482582. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C. Mitochondria and cancer. Nat. Rev. Cancer 2012, 12, 685–698. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.A.; Picard, M.; Sondheimer, N. Mitochondrial DNA, nuclear context, and the risk for carcinogenesis. Environ. Mol. Mutagen. 2018. [Google Scholar] [CrossRef] [PubMed]
- Lightowlers, R.N.; Chinnery, P.F.; Turnbull, D.M.; Howell, N. Mammalian mitochondrial genetics: Heredity, heteroplasmy and disease. Trends Genet. 1997, 13, 450–455. [Google Scholar] [CrossRef]
- Zong, W.X.; Rabinowitz, J.D.; White, E. Mitochondria and Cancer. Mol. Cell 2016, 61, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Bardella, C.; Al-Dalahmah, O.; Krell, D.; Brazauskas, P.; Al-Qahtani, K.; Tomkova, M.; Adam, J.; Serres, S.; Lockstone, H.; Freeman-Mills, L.; et al. Expression of Idh1(R132H) in the Murine Subventricular Zone Stem Cell Niche Recapitulates Features of Early Gliomagenesis. Cancer Cell 2016, 30, 578–594. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Liang, X.; Sun, X.; Zhang, L.; Fu, X.; Rogers, C.J.; Berim, A.; Zhang, S.; Wang, S.; Wang, B.; et al. AMPK/α-Ketoglutarate Axis Dynamically Mediates DNA Demethylation in the Prdm16 Promoter and Brown Adipogenesis. Cell Metab. 2016, 24, 542–554. [Google Scholar] [CrossRef] [PubMed]
- Sciacovelli, M.; Frezza, C. Oncometabolites: Unconventional triggers of oncogenic signalling cascades. Free Radic. Biol. Med. 2016, 100, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Krebs, H.A.; Johnson, W.A. The role of citric acid in intermediate metabolism in animal tissues. FEBS Lett. 1980, 117, K2–K10. [Google Scholar] [CrossRef]
- McDonough, M.A.; Loenarz, C.; Chowdhury, R.; Clifton, I.J.; Schofield, C.J. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr. Opin. Struct. Biol. 2010, 20, 659–672. [Google Scholar] [CrossRef] [PubMed]
- Schofield, C.J.; Zhang, Z. Structural and mechanistic studies on 2-oxoglutarate-dependent oxygenases and related enzymes. Curr. Opin. Struct. Biol. 1999, 9, 722–731. [Google Scholar] [CrossRef]
- Bruick, R.K.; McKnight, S.L. A conserved family of prolyl-4-hydroxylases that modify HIF. Science 2001, 294, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef]
- Hirsila, M.; Koivunen, P.; Gunzler, V.; Kivirikko, K.I.; Myllyharju, J. Characterization of the human prolyl 4-hydroxylases that modify the hypoxia-inducible factor. J. Biol. Chem. 2003, 278, 30772–30780. [Google Scholar] [CrossRef] [PubMed]
- Kivirikko, K.I.; Pihlajaniemi, T. Collagen hydroxylases and the protein disulfide isomerase subunit of prolyl 4-hydroxylases. Adv. Enzymol. Relat. Areas Mol. Biol. 1998, 72, 325–398. [Google Scholar] [PubMed]
- He, W.; Miao, F.J.; Lin, D.C.; Schwandner, R.T.; Wang, Z.; Gao, J.; Chen, J.L.; Tian, H.; Ling, L. Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 2004, 429, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, N.; Itoh, H. Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals 2009, 17, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Gasparre, G.; Porcelli, A.M.; Bonora, E.; Pennisi, L.F.; Toller, M.; Iommarini, L.; Ghelli, A.; Moretti, M.; Betts, C.M.; Martinelli, G.N.; et al. Disruptive mitochondrial DNA mutations in complex I subunits are markers of oncocytic phenotype in thyroid tumors. Proc. Natl. Acad. Sci. USA 2007, 104, 9001–9006. [Google Scholar] [CrossRef] [PubMed]
- Park, J.S.; Sharma, L.K.; Li, H.; Xiang, R.; Holstein, D.; Wu, J.; Lechleiter, J.; Naylor, S.L.; Deng, J.J.; Lu, J.; et al. A heteroplasmic, not homoplasmic, mitochondrial DNA mutation promotes tumorigenesis via alteration in reactive oxygen species generation and apoptosis. Hum. Mol. Genet. 2009, 18, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Larman, T.C.; DePalma, S.R.; Hadjipanayis, A.G.; Protopopov, A.; Zhang, J.; Gabriel, S.B.; Chin, L.; Seidman, C.E.; Kucherlapati, R.; Seidman, J.G. Spectrum of somatic mitochondrial mutations in five cancers. Proc. Natl. Acad. Sci. USA 2012, 109, 14087–14091. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, K.; Takenaga, K.; Akimoto, M.; Koshikawa, N.; Yamaguchi, A.; Imanishi, H.; Nakada, K.; Honma, Y.; Hayashi, J. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008, 320, 661–664. [Google Scholar] [CrossRef] [PubMed]
- Tipirisetti, N.R.; Govatati, S.; Pullari, P.; Malempati, S.; Thupurani, M.K.; Perugu, S.; Guruvaiah, P.; Rao, K.L.; Digumarti, R.R.; Nallanchakravarthula, V.; et al. Mitochondrial control region alterations and breast cancer risk: A study in South Indian population. PLoS ONE 2014, 9, e85363. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.B.; Ye, Y.; Huang, M.; Chang, D.W.; Kamat, A.M.; Pu, X.; Dinney, C.P.; Wu, X. Mitochondrial DNA Content as Risk Factor for Bladder Cancer and Its Association with Mitochondrial DNA Polymorphisms. Cancer Prev. Res. 2015, 8, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Guney, A.I.; Ergec, D.S.; Tavukcu, H.H.; Koc, G.; Kirac, D.; Ulucan, K.; Javadova, D.; Turkeri, L. Detection of mitochondrial DNA mutations in nonmuscle invasive bladder cancer. Genet. Test. Mol. Biomark. 2012, 16, 672–678. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Peng, F.; Wang, L.; Peng, L.; Lan, G.; Yu, S. Roles of mitochondrial transcription factor A and microRNA-590-3p in the development of bladder cancer. Oncol. Lett. 2013, 6, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, S.; Favia, P.; Weigl, S.; Bianco, A.; Pilato, B.; Russo, L.; Paradiso, A.; Petruzzella, V. Mitochondrial DNA variants and risk of familial breast cancer: An exploratory study. Int. J. Oncol. 2014, 44, 1691–1698. [Google Scholar] [CrossRef] [PubMed]
- Li, L.H.; Kang, T.; Chen, L.; Zhang, W.; Liao, Y.; Chen, J.; Shi, Y. Detection of mitochondrial DNA mutations by high-throughput sequencing in the blood of breast cancer patients. Int. J. Mol. Med. 2014, 33, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Wan, J.; Song, R.; Zhao, H. Peripheral blood mitochondrial DNA copy number, length heteroplasmy and breast cancer risk: A replication study. Carcinogenesis 2015, 36, 1307–1313. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Gao, Y.T.; Shu, X.O.; Wen, W.; Yang, G.; Li, G.; Courtney, R.; Ji, B.T.; Li, H.L.; Purdue, M.P.; et al. Association of leukocyte mitochondrial DNA copy number with colorectal cancer risk: Results from the Shanghai Women’s Health Study. Cancer Epidemiol. Biomark. Prev. 2014, 23, 2357–2365. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.; Zhao, Z.; Xiao, Y.; Peng, J.; Chen, J.; He, Y. Roles of mitochondrial transcription factor A and microRNA5903p in the development of colon cancer. Mol. Med. Rep. 2016, 14, 5475–5480. [Google Scholar] [CrossRef] [PubMed]
- Namslauer, I.; Brzezinski, P. A mitochondrial DNA mutation linked to colon cancer results in proton leaks in cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2009, 106, 3402–3407. [Google Scholar] [CrossRef] [PubMed]
- Allegra, E.; Garozzo, A.; Lombardo, N.; De Clemente, M.; Carey, T.E. Mutations and polymorphisms in mitochondrial DNA in head and neck cancer cell lines. Acta Otorhinolaryngol. Ital. 2006, 26, 185–190. [Google Scholar] [PubMed]
- Challen, C.; Brown, H.; Cai, C.; Betts, G.; Paterson, I.; Sloan, P.; West, C.; Birch-Machin, M.; Robinson, M. Mitochondrial DNA mutations in head and neck cancer are infrequent and lack prognostic utility. Br. J. Cancer 2011, 104, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.A.; Jiang, R.S.; Wang, W.Y.; Lin, J.C. Somatic mutations in the D-loop of mitochondrial DNA in head and neck squamous cell carcinoma. Head Neck 2015, 37, 878–883. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.M.; Glazer, C.A.; Mambo, E.; Chatterjee, A.; Zhao, M.; Sidransky, D.; Califano, J.A. Head and neck cancer cell lines exhibit differential mitochondrial repair deficiency in response to 4NQO. Oral Oncol. 2006, 42, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Vidone, M.; Clima, R.; Santorsola, M.; Calabrese, C.; Girolimetti, G.; Kurelac, I.; Amato, L.B.; Iommarini, L.; Trevisan, E.; Leone, M.; et al. A comprehensive characterization of mitochondrial DNA mutations in glioblastoma multiforme. Int. J. Biochem. Cell Biol. 2015, 63, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Yeung, K.Y.; Dickinson, A.; Donoghue, J.F.; Polekhina, G.; White, S.J.; Grammatopoulos, D.K.; McKenzie, M.; Johns, T.G.; St John, J.C. The identification of mitochondrial DNA variants in glioblastoma multiforme. Acta Neuropathol. Commun. 2014, 2, 1. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, E.; Scorrano, L. When numbers matters: Mitochondrial DNA and gliomagenesis. Cell Death Differ. 2013, 20, 1601–1602. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lohrer, H.D.; Hieber, L.; Zitzelsberger, H. Differential mutation frequency in mitochondrial DNA from thyroid tumours. Carcinogenesis 2002, 23, 1577–1582. [Google Scholar] [CrossRef] [PubMed]
- Lang, M.; Vocke, C.D.; Merino, M.J.; Schmidt, L.S.; Linehan, W.M. Mitochondrial DNA mutations distinguish bilateral multifocal renal oncocytomas from familial Birt-Hogg-Dube tumors. Mod. Pathol. 2015, 28, 1458–1469. [Google Scholar] [CrossRef] [PubMed]
- Purdue, M.P.; Hofmann, J.N.; Colt, J.S.; Hoxha, M.; Ruterbusch, J.J.; Davis, F.G.; Rothman, N.; Wacholder, S.; Schwartz, K.L.; Baccarelli, A.; et al. A case-control study of peripheral blood mitochondrial DNA copy number and risk of renal cell carcinoma. PLoS ONE 2012, 7, e43149. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Hann, H.W.; Hann, R.S.; Wan, S.; Myers, R.E.; Ye, Z.; Xing, J.; Yang, H. Circulating mitochondrial DNA content associated with the risk of liver cirrhosis: A nested case-control study. Dig. Dis. Sci. 2015, 60, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Sanin, D.E.; Wang, X. Mitochondrial DNA in Lung Cancer. Adv. Exp. Med. Biol. 2017, 1038, 9–22. [Google Scholar] [PubMed]
- Lee, H.C.; Huang, K.H.; Yeh, T.S.; Chi, C.W. Somatic alterations in mitochondrial DNA and mitochondrial dysfunction in gastric cancer progression. World J. Gastroenterol. 2014, 20, 3950–3959. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Bassig, B.A.; Seow, W.J.; Hu, W.; Purdue, M.P.; Huang, W.Y.; Liu, C.S.; Cheng, W.L.; Mannisto, S.; Vermeulen, R.; et al. Mitochondrial DNA copy number and chronic lymphocytic leukemia/small lymphocytic lymphoma risk in two prospective studies. Cancer Epidemiol. Biomark. Prev. 2015, 24, 148–153. [Google Scholar] [CrossRef] [PubMed]
- Payne, B.A.; Gardner, K.; Chinnery, P.F. Mitochondrial DNA mutations in ageing and disease: Implications for HIV? Antivir. Ther. 2015, 20, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Chattopadhyay, K.; Aldous, C. A brief review on human mtDNA mutations and NRTI-associated mtDNA toxicity and mutations. Mitochondrial DNA A DNA Mapp. Seq. Anal. 2016, 27, 1685–1687. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.Y.; Kao, M.C. Therapeutic applications of the TAT-mediated protein transduction system for complex I deficiency and other mitochondrial diseases. Ann. N. Y. Acad. Sci. 2015, 1350, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Lindl, K.A.; Marks, D.R.; Kolson, D.L.; Jordan-Sciutto, K.L. HIV-associated neurocognitive disorder: Pathogenesis and therapeutic opportunities. J. Neuroimmune Pharmacol. 2010, 5, 294–309. [Google Scholar] [CrossRef] [PubMed]
- Goda, J.S.; Pachpor, T.; Basu, T.; Chopra, S.; Gota, V. Targeting the AKT pathway: Repositioning HIV protease inhibitors as radiosensitizers. Indian J. Med. Res. 2016, 143, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Petrovas, C.; Mueller, Y.M.; Katsikis, P.D. Apoptosis of HIV-specific CD8+ T cells: An HIV evasion strategy. Cell Death Differ. 2005, 12, 859–870. [Google Scholar] [CrossRef] [PubMed]
- Widlak, P.; Garrard, W.T. Roles of the major apoptotic nuclease-DNA fragmentation factor-in biology and disease. Cell. Mol. Life Sci. 2009, 66, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Zur Hausen, H. Papillomaviruses and cancer: From basic studies to clinical application. Nat. Rev. Cancer 2002, 2, 342–350. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Eugenin, E.A. Mechanisms of HIV Neuropathogenesis: Role of Cellular Communication Systems. Curr. HIV Res. 2016, 14, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Vannemreddy, P.S.; Fowler, M.; Polin, R.S.; Todd, J.R.; Nanda, A. Glioblastoma multiforme in a case of acquired immunodeficiency syndrome: Investigation a possible oncogenic influence of human immunodeficiency virus on glial cells. Case report and review of the literature. J. Neurosurg. 2000, 92, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.R.; Short, S.C. Management of glioblastoma multiforme in HIV patients: A case series and review of published studies. Clin. Oncol. 2009, 21, 591–597. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, B.T.; Davis, T.P. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol. Rev. 2005, 57, 173–185. [Google Scholar] [CrossRef] [PubMed]
- Castellano, P.; Nwagbo, C.; Martinez, L.R.; Eugenin, E.A. Methamphetamine compromises gap junctional communication in astrocytes and neurons. J. Neurochem. 2016, 137, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol. 2017, 7, 306. [Google Scholar] [CrossRef] [PubMed]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Lu, Y.; Yang, J.; Chen, G.; Kim, S.; Feng, L.; Ogasawara, M.; Hammoudi, N.; Lu, W.; Zhang, H.; et al. Metabolic activation of mitochondria in glioma stem cells promotes cancer development through a reactive oxygen species-mediated mechanism. Stem Cell Res. Ther. 2015, 6, 198. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Poppleton, H.; Kocak, M.; Hogg, T.L.; Fuller, C.; Hamner, B.; Oh, E.Y.; Gaber, M.W.; Finklestein, D.; Allen, M.; et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007, 11, 69–82. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation. Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef] [PubMed]
- Dovmark, T.H.; Saccomano, M.; Hulikova, A.; Alves, F.; Swietach, P. Connexin-43 channels are a pathway for discharging lactate from glycolytic pancreatic ductal adenocarcinoma cells. Oncogene 2017, 36, 4538–4550. [Google Scholar] [CrossRef] [PubMed]
- Weil, S.; Osswald, M.; Solecki, G.; Grosch, J.; Jung, E.; Lemke, D.; Ratliff, M.; Hanggi, D.; Wick, W.; Winkler, F. Tumor microtubes convey resistance to surgical lesions and chemotherapy in gliomas. Neuro Oncol. 2017, 19, 1316–1326. [Google Scholar] [CrossRef] [PubMed]
- Lou, E. Intercellular conduits in tumours: The new social network. Trends Cancer 2016, 2, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Okafo, G.; Prevedel, L.; Eugenin, E. Tunneling nanotubes (TNT) mediate long-range gap junctional communication: Implications for HIV cell to cell spread. Sci. Rep. 2017, 7, 16660. [Google Scholar] [CrossRef] [PubMed]
- Hitomi, M.; Deleyrolle, L.P.; Mulkearns-Hubert, E.E.; Jarrar, A.; Li, M.; Sinyuk, M.; Otvos, B.; Brunet, S.; Flavahan, W.A.; Hubert, C.G.; et al. Differential connexin function enhances self-renewal in glioblastoma. Cell Rep. 2015, 11, 1031–1042. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.T.; Parker, I.; Smith, I.F. Communication of Ca2+ signals via tunneling membrane nanotubes is mediated by transmission of inositol trisphosphate through gap junctions. Cell Calcium 2016, 60, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Kirsten, R.; Heintz, B.; Nelson, K.; Oremek, G. Reduction of hyperlipidemia with 3-sn-polyenyl-phosphatidylcholine in dialysis patients. Int. J. Clin. Pharmacol. Ther. Toxicol. 1989, 27, 129–134. [Google Scholar] [PubMed]
- Ady, J.W.; Desir, S.; Thayanithy, V.; Vogel, R.I.; Moreira, A.L.; Downey, R.J.; Fong, Y.; Manova-Todorova, K.; Moore, M.A.; Lou, E. Intercellular communication in malignant pleural mesothelioma: Properties of tunneling nanotubes. Front. Physiol. 2014, 5, 400. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.H.; Bukoreshtliev, N.V.; Barroso, J.F. Tunneling nanotubes: A new route for the exchange of components between animal cells. FEBS Lett. 2007, 581, 2194–2201. [Google Scholar] [CrossRef] [PubMed]
- Hsiung, F.; Ramirez-Weber, F.A.; Iwaki, D.D.; Kornberg, T.B. Dependence of Drosophila wing imaginal disc cytonemes on Decapentaplegic. Nature 2005, 437, 560–563. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, T. Pictures in cell biology. Cytonemes. Trends Cell Biol. 1999, 9, 434. [Google Scholar] [CrossRef]
- Rupp, I.; Sologub, L.; Williamson, K.C.; Scheuermayer, M.; Reininger, L.; Doerig, C.; Eksi, S.; Kombila, D.U.; Frank, M.; Pradel, G. Malaria parasites form filamentous cell-to-cell connections during reproduction in the mosquito midgut. Cell Res. 2011, 21, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Pontes, B.; Viana, N.B.; Campanati, L.; Farina, M.; Neto, V.M.; Nussenzveig, H.M. Structure and elastic properties of tunneling nanotubes. Eur. Biophys. J. 2008, 37, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Thayanithy, V.; Dickson, E.L.; Steer, C.; Subramanian, S.; Lou, E. Tumor-stromal cross talk: Direct cell-to-cell transfer of oncogenic microRNAs via tunneling nanotubes. Transl. Res. 2014, 164, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Guerrouahen, B.S.; Al Thawadi, H.; Ghiabi, P.; Maleki, M.; Abu-Kaoud, N.; Jacob, A.; Mirshahi, M.; Galas, L.; Rafii, S.; et al. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J. Transl. Med. 2013, 11, 94. [Google Scholar] [CrossRef] [PubMed]
- Antanaviciute, I.; Rysevaite, K.; Liutkevicius, V.; Marandykina, A.; Rimkute, L.; Sveikatiene, R.; Uloza, V.; Skeberdis, V.A. Long-distance communication between laryngeal carcinoma cells. PLoS ONE 2014, 9, e99196. [Google Scholar] [CrossRef] [PubMed]
- Polak, R.; de Rooij, B.; Pieters, R.; den Boer, M.L. B-cell precursor acute lymphoblastic leukemia cells use tunneling nanotubes to orchestrate their microenvironment. Blood 2015, 126, 2404–2414. [Google Scholar] [CrossRef] [PubMed]
- Quinn, T.A.; Camelliti, P.; Rog-Zielinska, E.A.; Siedlecka, U.; Poggioli, T.; O’Toole, E.T.; Knopfel, T.; Kohl, P. Electrotonic coupling of excitable and nonexcitable cells in the heart revealed by optogenetics. Proc. Natl. Acad. Sci. USA 2016. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Sinovas, A.; Ruiz-Meana, M.; Denuc, A.; García-Dorado, D. Mitochondrial Cx43, an important component of cardiac preconditioning. Biochim. Biophys. Acta (BBA) Biomembr. 2015, 1860, 182–183. [Google Scholar] [CrossRef] [PubMed]
- Kirca, M.; Kleinbongard, P.; Soetkamp, D.; Heger, J.; Csonka, C.; Ferdinandy, P.; Schulz, R. Interaction between connexin 43 and nitric oxide synthase in mice heart mitochondria. J. Cell. Mol. Med. 2015, 19, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Trudeau, K.; Muto, T.; Roy, S. Downregulation of mitochondrial connexin 43 by high glucose triggers mitochondrial shape change and cytochrome C release in retinal endothelial cells. Investig. Ophthalmol. Vis. Sci. 2012, 53, 6675–6681. [Google Scholar] [CrossRef] [PubMed]
- Shan, H.; Wei, J.; Zhang, M.; Lin, L.; Yan, R.; Zhang, R.; Zhu, Y.H. Suppression of PKCε-mediated mitochondrial connexin 43 phosphorylation at serine 368 is involved in myocardial mitochondrial dysfunction in a rat model of dilated cardiomyopathy. Mol. Med. Rep. 2015, 11, 4720–4726. [Google Scholar] [CrossRef] [PubMed]
- Malik, S.; Theis, M.; Eugenin, E.A. Connexin43 Containing Gap Junction Channels Facilitate HIV Bystander Toxicity: Implications in NeuroHIV. Front. Mol. Neurosci. 2017, 10, 404. [Google Scholar] [CrossRef] [PubMed]
- Ady, J.; Thayanithy, V.; Mojica, K.; Wong, P.; Carson, J.; Rao, P.; Fong, Y.; Lou, E. Tunneling nanotubes: An alternate route for propagation of the bystander effect following oncolytic viral infection. Mol. Ther. Oncolytics 2016, 3, 16029. [Google Scholar] [CrossRef] [PubMed]
- Datta, P.K.; Deshmane, S.; Khalili, K.; Merali, S.; Gordon, J.C.; Fecchio, C.; Barrero, C.A. Glutamate metabolism in HIV-1 infected macrophages: Role of HIV-1 Vpr. Cell Cycle 2016, 15, 2288–2298. [Google Scholar] [CrossRef] [PubMed]
- Van Lith, S.A.; Navis, A.C.; Verrijp, K.; Niclou, S.P.; Bjerkvig, R.; Wesseling, P.; Tops, B.; Molenaar, R.; van Noorden, C.J.; Leenders, W.P. Glutamate as chemotactic fuel for diffuse glioma cells: Are they glutamate suckers? Biochim. Biophys. Acta 2014, 1846, 66–74. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Sasayama, T.; Irino, Y.; Takata, K.; Nagashima, H.; Satoh, N.; Kyotani, K.; Mizowaki, T.; Imahori, T.; Ejima, Y.; et al. Compensatory glutamine metabolism promotes glioblastoma resistance to mTOR inhibitor treatment. J. Clin. Investig. 2015, 125, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Muller, F.L.; Liu, Y.; Van Remmen, H. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J. Biol. Chem. 2004, 279, 49064–49073. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Ralph, S.J.; Rodriguez-Enriquez, S.; Neuzil, J.; Saavedra, E.; Moreno-Sanchez, R. The causes of cancer revisited: “mitochondrial malignancy” and ROS-induced oncogenic Transformation—Why mitochondria are targets for cancer therapy. Mol. Aspects Med. 2010, 31, 145–170. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, L.B.; Chandel, N.S. Mitochondrial reactive oxygen species and cancer. Cancer Metab. 2014, 2, 17. [Google Scholar] [CrossRef] [PubMed]
- Draganov, D.; Gopalakrishna-Pillai, S.; Chen, Y.R.; Zuckerman, N.; Moeller, S.; Wang, C.; Ann, D.; Lee, P.P. Modulation of P2X4/P2X7/Pannexin-1 sensitivity to extracellular ATP via Ivermectin induces a non-apoptotic and inflammatory form of cancer cell death. Sci. Rep. 2015, 5, 16222. [Google Scholar] [CrossRef] [PubMed]
- Rayfield, C.A.; Grady, F.; De Leon, G.; Rockne, R.; Carrasco, E.; Jackson, P.; Vora, M.; Johnston, S.K.; Hawkins-Daarud, A.; Clark-Swanson, K.R. Distinct Phenotypic Clusters of Glioblastoma Growth and Response Kinetics Predict Survival. JCO Clin. Cancer Inform. 2018, 2, 1–14. [Google Scholar] [CrossRef]
- Carette, D.; Gilleron, J.; Chevallier, D.; Segretain, D.; Pointis, G. Connexin a check-point component of cell apoptosis in normal and physiopathological conditions. Biochimie 2014, 101, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Amoedo, N.D.; Valencia, J.P.; Rodrigues, M.F.; Galina, A.; Rumjanek, F.D. How does the metabolism of tumour cells differ from that of normal cells. Biosci. Rep. 2013, 33, e00080. [Google Scholar] [CrossRef] [PubMed]
- De Vitto, H.; Perez-Valencia, J.; Radosevich, J.A. Glutamine at focus: Versatile roles in cancer. Tumour Biol. 2016, 37, 1541–1558. [Google Scholar] [CrossRef] [PubMed]
- Land, H.; Chen, A.C.; Morgenstern, J.P.; Parada, L.F.; Weinberg, R.A. Behavior of myc and ras oncogenes in transformation of rat embryo fibroblasts. Mol. Cell. Biol. 1986, 6, 1917–1925. [Google Scholar] [CrossRef] [PubMed]
- De Feijter, A.W.; Matesic, D.F.; Ruch, R.J.; Guan, X.; Chang, C.C.; Trosko, J.E. Localization and function of the connexin 43 gap-junction protein in normal and various oncogene-expressing rat liver epithelial cells. Mol. Carcinog. 1996, 16, 203–212. [Google Scholar] [CrossRef]
- Maqbool, R.; Rashid, R.; Ismail, R.; Niaz, S.; Chowdri, N.A.; Hussain, M.U. The carboxy-terminal domain of connexin 43 (CT-Cx43) modulates the expression of p53 by altering miR-125b expression in low-grade human breast cancers. Cell Oncol. 2015, 38, 443–451. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.; Zhao, W.; Wang, Y.; Xu, Y.; Xu, J.; Tang, K.; Zhang, S.; Yin, Z.; Wu, Q.; Wang, X. Connexin32 regulates hepatoma cell metastasis and proliferation via the p53 and Akt pathways. Oncotarget 2015, 6, 10116–10133. [Google Scholar] [CrossRef] [PubMed]
- Moennikes, O.; Stahl, S.; Bannasch, P.; Buchmann, A.; Schwarz, M. WY-14,643-mediated promotion of hepatocarcinogenesis in connexin32-wild-type and connexin32-null mice. Carcinogenesis 2003, 24, 1561–1565. [Google Scholar] [CrossRef] [PubMed]
- Moennikes, O.; Buchmann, A.; Romualdi, A.; Ott, T.; Werringloer, J.; Willecke, K.; Schwarz, M. Lack of phenobarbital-mediated promotion of hepatocarcinogenesis in connexin32-null mice. Cancer Res. 2000, 60, 5087–5091. [Google Scholar] [PubMed]
- Murphy, S.F.; Varghese, R.T.; Lamouille, S.; Guo, S.; Pridham, K.J.; Kanabur, P.; Osimani, A.M.; Sharma, S.; Jourdan, J.; Rodgers, C.M.; et al. Connexin 43 Inhibition Sensitizes Chemoresistant Glioblastoma Cells to Temozolomide. Cancer Res. 2016, 76, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wang, D.; Du, X.; He, Y.; Chen, S.; Shao, Q.; Ma, C.; Huang, B.; Chen, A.; Zhao, P.; et al. Glioma cells escaped from cytotoxicity of temozolomide and vincristine by communicating with human astrocytes. Med. Oncol. 2015, 32, 43. [Google Scholar] [CrossRef] [PubMed]
- Sin, W.C.; Aftab, Q.; Bechberger, J.F.; Leung, J.H.; Chen, H.; Naus, C.C. Astrocytes promote glioma invasion via the gap junction protein connexin43. Oncogene 2016, 35, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Cui, J.; Sun, X.; Zhang, Y. Tunneling-nanotube development in astrocytes depends on p53 activation. Cell Death Differ. 2011, 18, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, Y. Tunneling nanotubes between rat primary astrocytes and C6 glioma cells alter proliferation potential of glioma cells. Neurosci. Bull. 2015, 31, 371–378. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Bhuyan, F.; Hiyoshi, M.; Noyori, O.; Nasser, H.; Miyazaki, M.; Saito, T.; Kondoh, Y.; Osada, H.; Kimura, S.; et al. Potential Role of the Formation of Tunneling Nanotubes in HIV-1 Spread in Macrophages. J. Immunol. 2016, 196, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- Mann, J.; Ramakrishna, R.; Magge, R.; Wernicke, A.G. Advances in Radiotherapy for Glioblastoma. Front. Neurol. 2017, 8, 748. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, F. Glioblastoma update: Molecular biology, diagnosis, treatment, response assessment, and translational clinical trials. F1000Res 2017, 6, 1892. [Google Scholar] [CrossRef] [PubMed]
- Tamimi, A.F.; Juweid, M. Epidemiology and Outcome of Glioblastoma. In Glioblastoma; De Vleeschouwer, S., Ed.; Codon Publications: Brisbane, Australia, 2017. [Google Scholar] [CrossRef]
- Sisakhtnezhad, S.; Khosravi, L. Emerging physiological and pathological implications of tunneling nanotubes formation between cells. Eur. J. Cell Biol. 2015, 94, 429–443. [Google Scholar] [CrossRef] [PubMed]
- Saenz-de-Santa-Maria, I.; Bernardo-Castineira, C.; Enciso, E.; Garcia-Moreno, I.; Chiara, J.L.; Suarez, C.; Chiara, M.D. Control of long-distance cell-to-cell communication and autophagosome transfer in squamous cell carcinoma via tunneling nanotubes. Oncotarget 2017, 8, 20939–20960. [Google Scholar] [CrossRef] [PubMed]
- Teleki, I.; Szasz, A.M.; Maros, M.E.; Gyorffy, B.; Kulka, J.; Meggyeshazi, N.; Kiszner, G.; Balla, P.; Samu, A.; Krenacs, T. Correlations of differentially expressed gap junction connexins Cx26, Cx30, Cx32, Cx43 and Cx46 with breast cancer progression and prognosis. PLoS ONE 2014, 9, e112541. [Google Scholar] [CrossRef] [PubMed]
- Kotini, M.; Mayor, R. Connexins in migration during development and cancer. Dev. Biol. 2015, 401, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Tabernero, A.; Gangoso, E.; Jaraiz-Rodriguez, M.; Medina, J.M. The role of connexin43-Src interaction in astrocytomas: A molecular puzzle. Neuroscience 2016, 323, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Leithe, E.; Mesnil, M.; Aasen, T. The connexin 43 C-terminus: A tail of many tales. Biochim. Biophys. Acta 2018, 1860, 48–64. [Google Scholar] [CrossRef] [PubMed]
- Grossman, H.B.; Liebert, M.; Lee, I.W.; Lee, S.W. Decreased connexin expression and intercellular communication in human bladder cancer cells. Cancer Res. 1994, 54, 3062–3065. [Google Scholar] [PubMed]
- Naus, C.C.; Laird, D.W. Implications and challenges of connexin connections to cancer. Nat. Rev. Cancer 2010, 10, 435–441. [Google Scholar] [CrossRef] [PubMed]
- Tsai, C.F.; Cheng, Y.K.; Lu, D.Y.; Wang, S.L.; Chang, C.N.; Chang, P.C.; Yeh, W.L. Inhibition of estrogen receptor reduces connexin 43 expression in breast cancers. Toxicol. Appl. Pharmacol. 2018, 338, 182–190. [Google Scholar] [CrossRef] [PubMed]
- Mandel, K.; Yang, Y.; Schambach, A.; Glage, S.; Otte, A.; Hass, R. Mesenchymal stem cells directly interact with breast cancer cells and promote tumor cell growth in vitro and in vivo. Stem Cells Dev. 2013, 22, 3114–3127. [Google Scholar] [CrossRef] [PubMed]
- Lim, P.K.; Bliss, S.A.; Patel, S.A.; Taborga, M.; Dave, M.A.; Gregory, L.A.; Greco, S.J.; Bryan, M.; Patel, P.S.; Rameshwar, P. Gap junction-mediated import of microRNA from bone marrow stromal cells can elicit cell cycle quiescence in breast cancer cells. Cancer Res. 2011, 71, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Hong, X.; Sin, W.C.; Harris, A.L.; Naus, C.C. Gap junctions modulate glioma invasion by direct transfer of microRNA. Oncotarget 2015, 6, 15566–15577. [Google Scholar] [CrossRef] [PubMed]
- Suzhi, Z.; Liang, T.; Yuexia, P.; Lucy, L.; Xiaoting, H.; Yuan, Z.; Qin, W. Gap Junctions Enhance the Antiproliferative Effect of MicroRNA-124-3p in Glioblastoma Cells. J. Cell. Physiol. 2015, 230, 2476–2488. [Google Scholar] [CrossRef] [PubMed]
- Menachem, A.; Makovski, V.; Bodner, O.; Pasmanik-Chor, M.; Stein, R.; Shomron, N.; Kloog, Y. Intercellular transfer of small RNAs from astrocytes to lung tumor cells induces resistance to chemotherapy. Oncotarget 2016, 7, 12489–12504. [Google Scholar] [CrossRef] [PubMed]
- Zong, L.; Zhu, Y.; Liang, R.; Zhao, H.B. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci. Rep. 2016, 6, 19884. [Google Scholar] [CrossRef] [PubMed]
- Lou, E. Can you hear them now? Tumor microtubes form cellular communication networks that protect gliomas from surgical lesions and chemotherapy treatments. Neuro Oncol. 2017, 19, 1289–1291. [Google Scholar] [CrossRef] [PubMed]
- Connor, Y.; Tekleab, S.; Nandakumar, S.; Walls, C.; Tekleab, Y.; Husain, A.; Gadish, O.; Sabbisetti, V.; Kaushik, S.; Sehrawat, S.; et al. Physical nanoscale conduit-mediated communication between tumour cells and the endothelium modulates endothelial phenotype. Nat. Commun. 2015, 6, 8671. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Shaw, R.M. Trafficking highways to the intercalated disc: New insights unlocking the specificity of connexin 43 localization. Cell Commun. Adhes. 2014, 21, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Schalper, K.A.; Carvajal-Hausdorf, D.; Oyarzo, M.P. Possible role of hemichannels in cancer. Front. Physiol. 2014, 5, 237. [Google Scholar] [CrossRef] [PubMed]
- Naderi, E.H.; Skah, S.; Ugland, H.; Myklebost, O.; Sandnes, D.L.; Torgersen, M.L.; Josefsen, D.; Ruud, E.; Naderi, S.; Blomhoff, H.K. Bone marrow stroma-derived PGE2 protects BCP-ALL cells from DNA damage-induced p53 accumulation and cell death. Mol. Cancer 2015, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Shehzad, A.; Islam, S.U.; Ahn, E.M.; Lee, Y.M.; Lee, Y.S. Decursinol angelate inhibits PGE2-induced survival of the human leukemia HL-60 cell line via regulation of the EP2 receptor and NFκB pathway. Cancer Biol. Ther. 2016, 17, 985–993. [Google Scholar] [CrossRef] [PubMed]
- Lou, E.; Gholami, S.; Romin, Y.; Thayanithy, V.; Fujisawa, S.; Desir, S.; Steer, C.J.; Subramanian, S.; Fong, Y.; Manova-Todorova, K.; et al. Imaging Tunneling Membrane Tubes Elucidates Cell Communication in Tumors. Trends Cancer 2017, 3, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.S.; Baty, J.W.; Dong, L.F.; Bezawork-Geleta, A.; Endaya, B.; Goodwin, J.; Bajzikova, M.; Kovarova, J.; Peterka, M.; Yan, B.; et al. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab. 2015, 21, 81–94. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, B.; Polak, R.; Stalpers, F.; Pieters, R.; den Boer, M.L. Tunneling nanotubes facilitate autophagosome transfer in the leukemic niche. Leukemia 2017, 31, 1651–1654. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.; Osswald, M.; Blaes, J.; Wiestler, B.; Sahm, F.; Schmenger, T.; Solecki, G.; Deumelandt, K.; Kurz, F.T.; Xie, R.F.; et al. Tweety-Homolog 1 Drives Brain Colonization of Gliomas. J. Neurosci. 2017, 37, 6837–6850. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.S.; Ganesan, P.; Choi, D.K. Cx43 Mediates Resistance against MPP+-Induced Apoptosis in SH-SY5Y Neuroblastoma Cells via Modulating the Mitochondrial Apoptosis Pathway. Int. J. Mol. Sci. 2016, 17, 1819. [Google Scholar] [CrossRef] [PubMed]
- Asklund, T.; Appelskog, I.B.; Ammerpohl, O.; Langmoen, I.A.; Dilber, M.S.; Aints, A.; Ekstrom, T.J.; Almqvist, P.M. Gap junction-mediated bystander effect in primary cultures of human malignant gliomas with recombinant expression of the HSVtk gene. Exp. Cell Res. 2003, 284, 185–195. [Google Scholar] [CrossRef]
- Cirenei, N.; Colombo, B.M.; Mesnil, M.; Benedetti, S.; Yamasaki, H.; Finocchiaro, G. In vitro and in vivo effects of retrovirus-mediated transfer of the connexin 43 gene in malignant gliomas: Consequences for HSVtk/GCV anticancer gene therapy. Gene Ther. 1998, 5, 1221–1226. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Grignet-Debrus, C.; Cool, V.; Baudson, N.; Velu, T.; Calberg-Bacq, C.M. The role of cellular- and prodrug-associated factors in the bystander effect induced by the Varicella zoster and Herpes simplex viral thymidine kinases in suicide gene therapy. Cancer Gene Ther. 2000, 7, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Liu, X.Z.; Kang, C.S.; Wang, G.X.; Zhong, Y.; Pu, P.Y. The anti-glioma effect of suicide gene therapy using BMSC expressing HSV/TK combined with overexpression of Cx43 in glioma cells. Cancer Gene Ther. 2010, 17, 192–202. [Google Scholar] [CrossRef] [PubMed]
- Marconi, P.; Tamura, M.; Moriuchi, S.; Krisky, D.M.; Niranjan, A.; Goins, W.F.; Cohen, J.B.; Glorioso, J.C. Connexin 43-enhanced suicide gene therapy using herpesviral vectors. Mol. Ther. 2000, 1, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Namba, H.; Iwadate, Y.; Kawamura, K.; Sakiyama, S.; Tagawa, M. Efficacy of the bystander effect in the herpes simplex virus thymidine kinase-mediated gene therapy is influenced by the expression of connexin43 in the target cells. Cancer Gene Ther. 2001, 8, 414–420. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shinoura, N.; Chen, L.; Wani, M.A.; Kim, Y.G.; Larson, J.J.; Warnick, R.E.; Simon, M.; Menon, A.G.; Bi, W.L.; Stambrook, P.J. Protein and messenger RNA expression of connexin43 in astrocytomas: Implications in brain tumor gene therapy. J. Neurosurg. 1996, 84, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Dilber, M.S.; Abedi, M.R.; Christensson, B.; Bjorkstrand, B.; Kidder, G.M.; Naus, C.C.; Gahrton, G.; Smith, C.I. Gap junctions promote the bystander effect of herpes simplex virus thymidine kinase in vivo. Cancer Res. 1997, 57, 1523–1528. [Google Scholar] [PubMed]
- Andrade-Rozental, A.F.; Rozental, R.; Hopperstad, M.G.; Wu, J.K.; Vrionis, F.D.; Spray, D.C. Gap junctions: The “kiss of death” and the “kiss of life”. Brain Res. Brain Res. Rev. 2000, 32, 308–315. [Google Scholar] [CrossRef]
- Mesnil, M.; Piccoli, C.; Tiraby, G.; Willecke, K.; Yamasaki, H. Bystander killing of cancer cells by herpes simplex virus thymidine kinase gene is mediated by connexins. Proc. Natl. Acad. Sci. USA 1996, 93, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Fischer, N.O.; Mbuy, G.N.; Woodruff, R.I. HSV-2 disrupts gap junctional intercellular communication between mammalian cells in vitro. J. Virol. Methods 2001, 91, 157–166. [Google Scholar] [CrossRef]
- Musee, J.; Mbuy, G.N.; Woodruff, R.I. Antiviral agents alter ability of HSV-2 to disrupt gap junctional intercellular communication between mammalian cells in vitro. Antivir. Res. 2002, 56, 143–151. [Google Scholar] [CrossRef]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdebenito, S.; Lou, E.; Baldoni, J.; Okafo, G.; Eugenin, E. The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis. Int. J. Mol. Sci. 2018, 19, 1270. https://doi.org/10.3390/ijms19051270
Valdebenito S, Lou E, Baldoni J, Okafo G, Eugenin E. The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis. International Journal of Molecular Sciences. 2018; 19(5):1270. https://doi.org/10.3390/ijms19051270
Chicago/Turabian StyleValdebenito, Silvana, Emil Lou, John Baldoni, George Okafo, and Eliseo Eugenin. 2018. "The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis" International Journal of Molecular Sciences 19, no. 5: 1270. https://doi.org/10.3390/ijms19051270
APA StyleValdebenito, S., Lou, E., Baldoni, J., Okafo, G., & Eugenin, E. (2018). The Novel Roles of Connexin Channels and Tunneling Nanotubes in Cancer Pathogenesis. International Journal of Molecular Sciences, 19(5), 1270. https://doi.org/10.3390/ijms19051270