Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
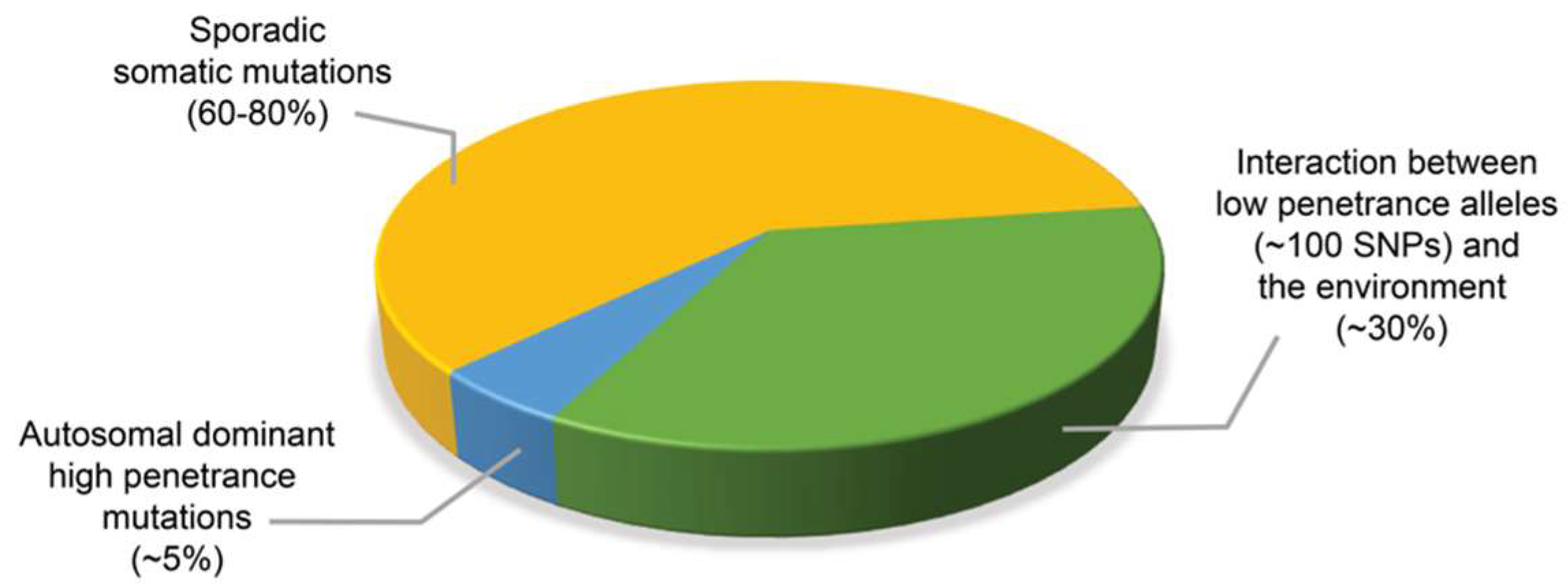
2. Assessing the Contribution of Mendelian Inheritance to Prostate Cancer Risk by Segregation and Linkage Analyses Studies
3. Identification of Prostate Cancer Susceptibility Loci by Genome-Wide Association Studies (GWAS)
3.1. The 8q24 Locus and Other Prostate Cancer Risk Alleles Associated with African Americans
3.2. Potential Mechanisms Contributing to the Association with Prostate Cancer Risk
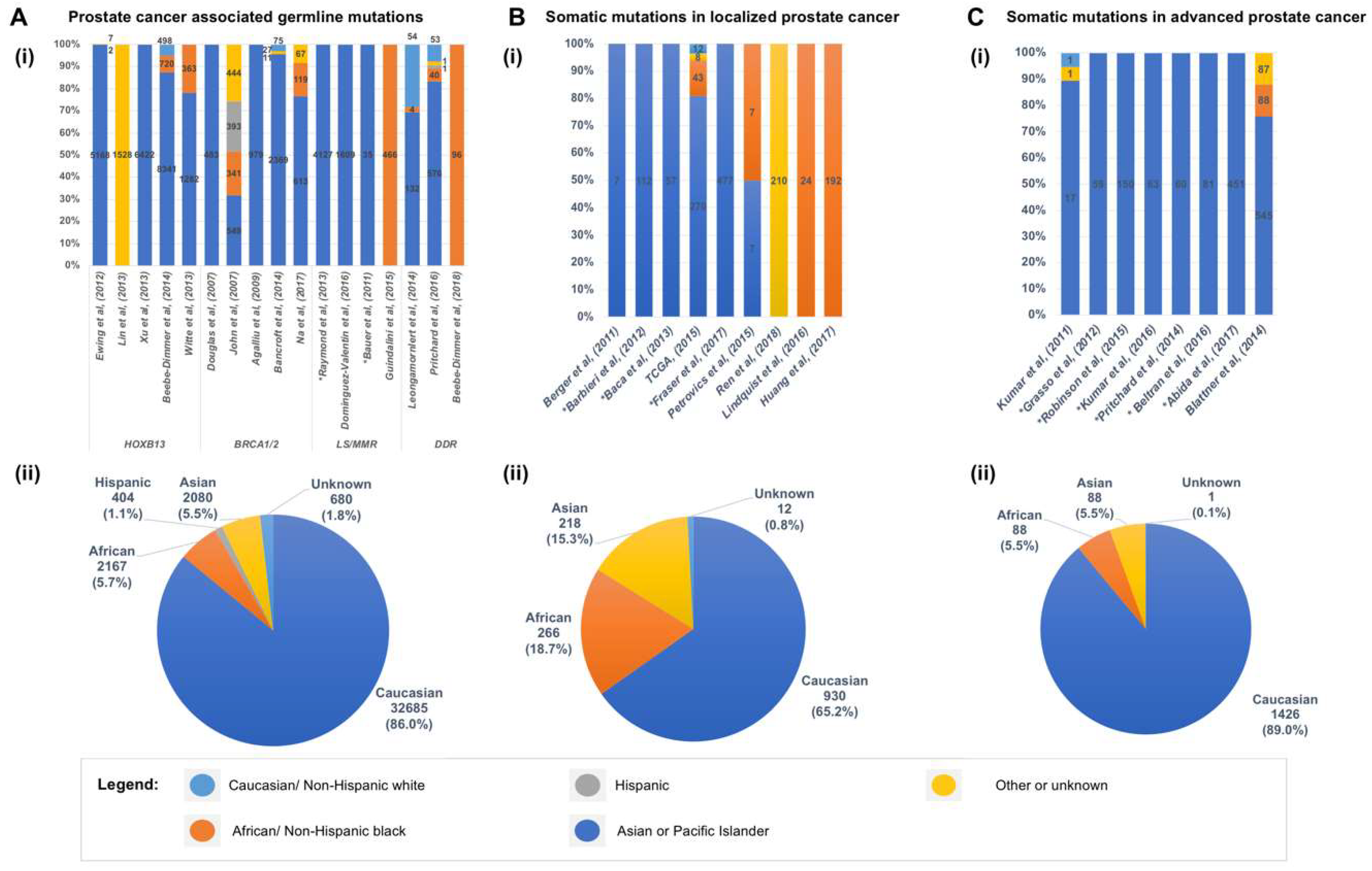
4. Germline Mutations Associated with Hereditary Prostate Cancer
4.1. ELAC2, RNASEL and MSR1
4.2. HOXB13
4.3. BRCA1 and BRCA2
4.4. DNA Mismatch Repair (MMR) Genes
4.5. Germline Alteration in DNA Damage Repair Pathways
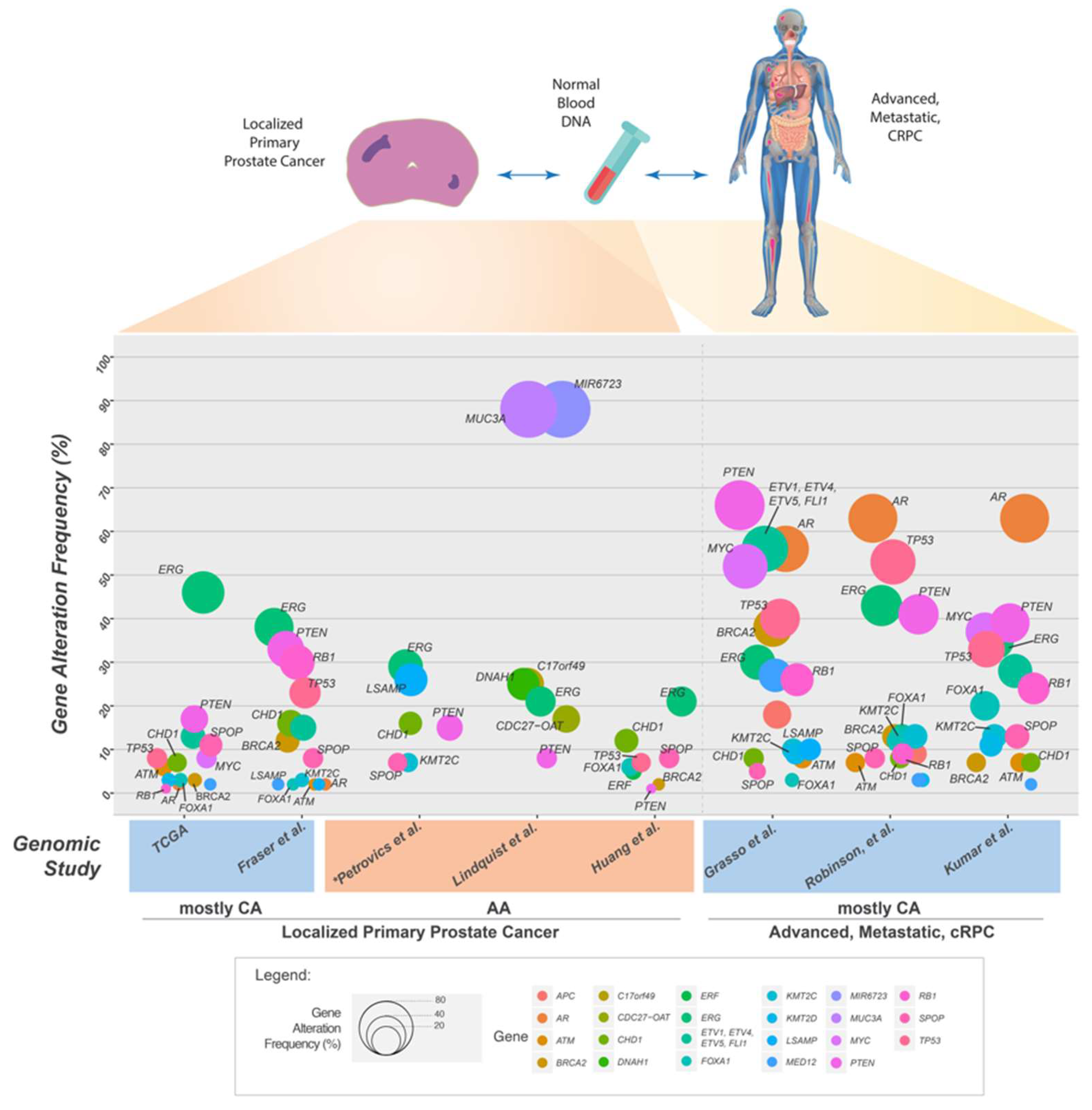
5. Somatic Mutations in the Genome of Localized Primary Prostate Cancer
6. Somatic Mutations in the Genome of Prostate Cancers from African American Men
7. Genomic Landscape of Advanced and Metastatic Castrate Resistant Prostate Cancer (mCRPC)
8. Impact of Prostate Cancer Genomics on the Prognosis, Treatment, and Ethnic Disparity of Prostate Cancer
9. Addressing the Under-Representation of Minority Populations in CaP Genomic Studies
Conflicts of Interest
Disclaimer
Abbreviations
| 3C | chromosome conformation capture |
| 4C | circularized chromosome conformation capture |
| AR | Androgen Receptor |
| CNAs | Copy Number Alterations |
| CPC-GENE | Canadian CaP Genome Network |
| CR-NEPC | castration resistant neuroendocrine prostate cancer |
| CRPC | castration resistant prostate adenocarcinoma |
| DDR | Damage Repair |
| DSB | double-strand break |
| EGFR | epidermal growth factor receptor |
| FA | Fanconi anemia |
| FISH | fluorescent in-situ hybridization |
| HBOC | hereditary breast and ovarian cancer |
| HBOC | ovarian cancer |
| HPC | hereditary Prostate Cancer |
| HR | homologous recombination |
| HR | homologous recombination |
| ICGC | International Cancer Genome Consortium |
| LS MAPK | Lynch Syndrome Mitogen-activated protein kinase |
| MATCH | Molecular Analysis for Therapy Choice |
| mCRPC | metastatic castrate resistant prostate cancer |
| MMR | Mismatch Repair |
| MSI | microsatellite instability |
| NCI | National Cancer Institute |
| NGS NIH | Next-Gen sequencing National Institutes of Health |
| NHEJ | Non-homologous end joining |
| NSCLC | non-small-cell lung cancer |
| OMB PARP PI3K | Office of Management and Budget Poly (ADP-ribose) polymerase Phosphatidylinositol 3- kinase |
| PCF | Prostate Cancer Foundation |
| SEER SNPs | Surveillance, Epidemiology, and End Results single-nucleotide polymorphisms |
| SSA | single-strand annealing |
| SU2C | Stand Up to Cancer |
| TCGA | The Cancer Genome Atlas |
| TSG | tumor suppressor genes |
| WES | Whole exome sequencing |
References
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global Cancer Statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2018. CA Cancer J. Clin. 2018, 68, 7–30. [Google Scholar] [CrossRef] [PubMed]
- DeSantis, C.E.; Siegel, R.L.; Sauer, A.G.; Miller, K.D.; Fedewa, S.A.; Alcaraz, K.I.; Jemal, A. Cancer Statistics for African Americans, 2016: Progress and Opportunities in Reducing Racial Disparities. CA Cancer J. Clin. 2016, 66, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Chornokur, G.; Dalton, K.; Borysova, M.E.; Kumar, N.B. Disparities at Presentation, Diagnosis, Treatment, and Survival in African American Men, Affected by Prostate Cancer. Prostate 2011, 71, 985–997. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, K.; Powell, I.J.; Underwood, W., 3rd; George, J.; Yee, C.; Banerjee, M. Interplay of Race, Socioeconomic Status, and Treatment on Survival of Patients with Prostate Cancer. Urology 2009, 74, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.; Witte, J.S.; McClure, L.A.; Shema, S.J.; Cockburn, M.G.; John, E.M.; Clarke, C.A. Socioeconomic Status and Prostate Cancer Incidence and Mortality Rates among the Diverse Population of California. Cancer Causes Control 2009, 20, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Farrell, J.; Petrovics, G.; McLeod, D.G.; Srivastava, S. Genetic and Molecular Differences in Prostate Carcinogenesis between African American and Caucasian American Men. Int. J. Mol. Sci. 2013, 14, 15510–15531. [Google Scholar] [CrossRef] [PubMed]
- Hanna, M.C.; Go, C.; Roden, C.; Jones, R.T.; Pochanard, P.; Javed, A.Y.; Javed, A.; Mondal, C.; Palescandolo, E.; Van Hummelen, P.; et al. Colorectal Cancers from Distinct Ancestral Populations Show Variations in Braf Mutation Frequency. PLoS ONE 2013, 8, e74950. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Ren, Y.; Fang, Z.; Li, C.; Fang, R.; Gao, B.; Han, X.; Tian, W.; Pao, W.; Chen, H.; et al. Lung Adenocarcinoma from East Asian Never-Smokers Is a Disease Largely Defined by Targetable Oncogenic Mutant Kinases. J. Clin. Oncol. 2010, 28, 4616–4620. [Google Scholar] [CrossRef] [PubMed]
- Guda, K.; Veigl, M.L.; Varadan, V.; Nosrati, A.; Ravi, L.; Lutterbaugh, J.; Beard, L.; Willson, J.K.; Sedwick, W.D.; Wang, Z.J.; et al. Novel Recurrently Mutated Genes in African American Colon Cancers. Proc. Natl. Acad. Sci. USA 2015, 112, 1149–1154. [Google Scholar] [CrossRef] [PubMed]
- Haber, D.A.; Bell, D.W.; Sordella, R.; Kwak, E.L.; Godin-Heymann, N.; Sharma, S.V.; Lynch, T.J.; Settleman, J. Molecular Targeted Therapy of Lung Cancer: EGFR Mutations and Response to Egfr Inhibitors. Cold Spring Harb. Symp. Quant. Biol. 2005, 70, 419–426. [Google Scholar] [CrossRef] [PubMed]
- Sanger, F.; Nicklen, S.; Coulson, A.R. DNA Sequencing with Chain-Terminating Inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The Sequence of the Human Genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [PubMed]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial Sequencing and Analysis of the Human Genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed]
- Thorisson, G.A.; Smith, A.V.; Krishnan, L.; Stein, L.D. The International Hapmap Project Web Site. Genome Res. 2005, 15, 1592–1593. [Google Scholar] [CrossRef] [PubMed]
- Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; Abecasis, G.R. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Stranger, B.E.; Stahl, E.A.; Raj, T. Progress and Promise of Genome-Wide Association Studies for Human Complex Trait Genetics. Genetics 2011, 187, 367–383. [Google Scholar] [CrossRef] [PubMed]
- Spratt, D.E.; Chan, T.; Waldron, L.; Speers, C.; Feng, F.Y.; Ogunwobi, O.O.; Osborne, J.R. Racial/Ethnic Disparities in Genomic Sequencing. JAMA Oncol. 2016, 2, 1070–1074. [Google Scholar] [CrossRef] [PubMed]
- Page, W.F.; Braun, M.M.; Partin, A.W.; Caporaso, N.; Walsh, P. Heredity and Prostate Cancer: A Study of World War Ii Veteran Twins. Prostate 1997, 33, 240–245. [Google Scholar] [CrossRef]
- Ahlbom, A.; Lichtenstein, P.; Malmstrom, H.; Feychting, M.; Hemminki, K.; Pedersen, N.L. Cancer in Twins: Genetic and Nongenetic Familial Risk Factors. J. Natl. Cancer Inst. 1997, 89, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Mucci, L.A.; Hjelmborg, J.B.; Harris, J.R.; Czene, K.; Havelick, D.J.; Scheike, T.; Graff, R.E.; Holst, K.; Moller, S.; Unger, R.H.; et al. Familial Risk and Heritability of Cancer among Twins in Nordic Countries. JAMA 2016, 315, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Albright, F.S.; Stephenson, R.A.; Agarwal, N.; Cannon-Albright, L.A. Relative Risks for Lethal Prostate Cancer Based on Complete Family History of Prostate Cancer Death. Prostate 2017, 77, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Whittemore, A.S.; Wu, A.H.; Kolonel, L.N.; John, E.M.; Gallagher, R.P.; Howe, G.R.; West, D.W.; Teh, C.Z.; Stamey, T. Family History and Prostate Cancer Risk in Black, White, and Asian Men in the United States and Canada. Am. J. Epidemiol. 1995, 141, 732–740. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.S.; Beaty, T.H.; Steinberg, G.D.; Childs, B.; Walsh, P.C. Mendelian Inheritance of Familial Prostate Cancer. Proc. Natl. Acad. Sci. USA 1992, 89, 3367–3371. [Google Scholar] [CrossRef] [PubMed]
- Gronberg, H.; Damber, L.; Damber, J.E.; Iselius, L. Segregation Analysis of Prostate Cancer in Sweden: Support for Dominant Inheritance. Am. J. Epidemiol. 1997, 146, 552–557. [Google Scholar] [CrossRef] [PubMed]
- MacInnis, R.J.; Antoniou, A.C.; Eeles, R.A.; Severi, G.; Guy, M.; McGuffog, L.; Hall, A.L.; O’Brien, L.T.; Wilkinson, R.A.; Dearnaley, D.P.; et al. Prostate Cancer Segregation Analyses Using 4390 Families from UK and Australian Population-Based Studies. Genet. Epidemiol. 2010, 34, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Pakkanen, S.; Baffoe-Bonnie, A.B.; Matikainen, M.P.; Koivisto, P.A.; Tammela, T.L.; Deshmukh, S.; Ou, L.; Bailey-Wilson, J.E.; Schleutker, J. Segregation Analysis of 1546 Prostate Cancer Families in Finland Shows Recessive Inheritance. Hum. Genet. 2007, 121, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Pilie, P.G.; Giri, V.N.; Cooney, K.A. Hoxb13 and Other High Penetrant Genes for Prostate Cancer. Asian J. Androl. 2016, 18, 530–532. [Google Scholar] [PubMed]
- Ewing, C.M.; Ray, A.M.; Lange, E.M.; Zuhlke, K.A.; Robbins, C.M.; Tembe, W.D.; Wiley, K.E.; Isaacs, S.D.; Johng, D.; Wang, Y.; et al. Germline Mutations in Hoxb13 and Prostate-Cancer Risk. N. Engl. J. Med. 2012, 366, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Nagy, R.; Sweet, K.; Eng, C. Highly Penetrant Hereditary Cancer Syndromes. Oncogene 2004, 23, 6445–6470. [Google Scholar] [CrossRef] [PubMed]
- Simard, J.; Dumont, M.; Labuda, D.; Sinnett, D.; Meloche, C.; El-Alfy, M.; Berger, L.; Lees, E.; Labrie, F.; Tavtigian, S.V. Prostate Cancer Susceptibility Genes: Lessons Learned and Challenges Posed. Endocr. Relat. Cancer 2003, 10, 225–259. [Google Scholar] [CrossRef] [PubMed]
- Sfanos, K.S.; De Marzo, A.M. Prostate Cancer and Inflammation: The Evidence. Histopathology 2012, 60, 199–215. [Google Scholar] [CrossRef] [PubMed]
- Witte, J.S. Prostate Cancer Genomics: Towards a New Understanding. Nat. Rev. Genet. 2009, 10, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Freedman, M.L. Genetics of Prostate Cancer Risk. Mt. Sinai J. Med. N. Y. 2010, 77, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Hormozdiari, F.; Kichaev, G.; Yang, W.Y.; Pasaniuc, B.; Eskin, E. Identification of Causal Genes for Complex Traits. Bioinformatics 2015, 31, i206–i213. [Google Scholar] [CrossRef] [PubMed]
- Kote-Jarai, Z.; Easton, D.F.; Stanford, J.L.; Ostrander, E.A.; Schleutker, J.; Ingles, S.A.; Schaid, D.; Thibodeau, S.; Dork, T.; Neal, D.; et al. Multiple Novel Prostate Cancer Predisposition Loci Confirmed by an International Study: The Practical Consortium. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2052–2061. [Google Scholar] [CrossRef] [PubMed]
- Al Olama, A.A.; Dadaev, T.; Hazelett, D.J.; Li, Q.; Leongamornlert, D.; Saunders, E.J.; Stephens, S.; Cieza-Borrella, C.; Whitmore, I.; Benlloch Garcia, S.; et al. Multiple Novel Prostate Cancer Susceptibility Signals Identified by Fine-Mapping of Known Risk Loci among Europeans. Hum. Mol. Genet. 2015, 24, 5589–5602. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, N.; Rohland, N.; Rand, K.A.; Tandon, A.; Allen, A.; Quinque, D.; Mallick, S.; Li, H.; Stram, A.; Sheng, X.; et al. The Contribution of Rare Variation to Prostate Cancer Heritability. Nat. Genet. 2016, 48, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Auer, P.L.; Lettre, G. Rare Variant Association Studies: Considerations, Challenges and Opportunities. Genome Med. 2015, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, S.; Das, S.; Kretzschmar, W.; Delaneau, O.; Wood, A.R.; Teumer, A.; Kang, H.M.; Fuchsberger, C.; Danecek, P.; Sharp, K.; et al. A Reference Panel of 64,976 Haplotypes for Genotype Imputation. Nat. Genet. 2016, 48, 1279–1283. [Google Scholar] [PubMed]
- Huang, J.; Howie, B.; McCarthy, S.; Memari, Y.; Walter, K.; Min, J.L.; Danecek, P.; Malerba, G.; Trabetti, E.; Zheng, H.F.; et al. Improved Imputation of Low-Frequency and Rare Variants Using the UK10K Haplotype Reference Panel. Nat. Commun. 2015, 6, 8111. [Google Scholar] [CrossRef] [PubMed]
- Sud, A.; Kinnersley, B.; Houlston, R.S. Genome-Wide Association Studies of Cancer: Current Insights and Future Perspectives. Nat. Rev. Cancer 2017, 17, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Howie, B.N.; Donnelly, P.; Marchini, J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009, 5, e1000529. [Google Scholar] [CrossRef] [PubMed]
- Amundadottir, L.T.; Sulem, P.; Gudmundsson, J.; Helgason, A.; Baker, A.; Agnarsson, B.A.; Sigurdsson, A.; Benediktsdottir, K.R.; Cazier, J.B.; Sainz, J.; et al. A Common Variant Associated with Prostate Cancer in European and African Populations. Nat. Genet. 2006, 38, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Sulem, P.; Manolescu, A.; Amundadottir, L.T.; Gudbjartsson, D.; Helgason, A.; Rafnar, T.; Bergthorsson, J.T.; Agnarsson, B.A.; Baker, A.; et al. Genome-Wide Association Study Identifies a Second Prostate Cancer Susceptibility Variant at 8q24. Nat. Genet. 2007, 39, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Yeager, M.; Orr, N.; Hayes, R.B.; Jacobs, K.B.; Kraft, P.; Wacholder, S.; Minichiello, M.J.; Fearnhead, P.; Yu, K.; Chatterjee, N.; et al. Genome-Wide Association Study of Prostate Cancer Identifies a Second Risk Locus at 8q24. Nat. Genet. 2007, 39, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Freedman, M.L.; Haiman, C.A.; Patterson, N.; McDonald, G.J.; Tandon, A.; Waliszewska, A.; Penney, K.; Steen, R.G.; Ardlie, K.; John, E.M.; et al. Admixture Mapping Identifies 8q24 as a Prostate Cancer Risk Locus in African-American Men. Proc. Natl. Acad. Sci. USA 2006, 103, 14068–14073. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, F.R.; Feigelson, H.S.; Cox, D.G.; Haiman, C.A.; Albanes, D.; Buring, J.; Calle, E.E.; Chanock, S.J.; Colditz, G.A.; Diver, W.R.; et al. A Common 8q24 Variant in Prostate and Breast Cancer from a Large Nested Case-Control Study. Cancer Res. 2007, 67, 2951–2956. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Patterson, N.; Freedman, M.L.; Myers, S.R.; Pike, M.C.; Waliszewska, A.; Neubauer, J.; Tandon, A.; Schirmer, C.; McDonald, G.J.; et al. Multiple Regions within 8q24 Independently Affect Risk for Prostate Cancer. Nat. Genet. 2007, 39, 638–644. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Chen, G.K.; Blot, W.J.; Strom, S.S.; Berndt, S.I.; Kittles, R.A.; Rybicki, B.A.; Isaacs, W.B.; Ingles, S.A.; Stanford, J.L.; et al. Characterizing Genetic Risk at Known Prostate Cancer Susceptibility Loci in African Americans. PLoS Genet. 2011, 7, e1001387. [Google Scholar] [CrossRef] [PubMed]
- Troutman, S.M.; Sissung, T.M.; Cropp, C.D.; Venzon, D.J.; Spencer, S.D.; Adesunloye, B.A.; Huang, X.; Karzai, F.H.; Price, D.K.; Figg, W.D. Racial Disparities in the Association between Variants on 8q24 and Prostate Cancer: A Systematic Review and Meta-Analysis. Oncologist 2012, 17, 312–320. [Google Scholar] [CrossRef] [PubMed]
- Haiman, C.A.; Chen, G.K.; Blot, W.J.; Strom, S.S.; Berndt, S.I.; Kittles, R.A.; Rybicki, B.A.; Isaacs, W.B.; Ingles, S.A.; Stanford, J.L.; et al. Genome-Wide Association Study of Prostate Cancer in Men of African Ancestry Identifies a Susceptibility Locus at 17q21. Nat. Genet. 2011, 43, 570–573. [Google Scholar] [CrossRef] [PubMed]
- Taioli, E.; Sears, V.; Watson, A.; Flores-Obando, R.E.; Jackson, M.D.; Ukoli, F.A.; de Syllos Colus, I.M.; Fernandez, P.; McFarlane-Anderson, N.; Ostrander, E.A.; et al. Polymorphisms in Cyp17 and Cyp3a4 and Prostate Cancer in Men of African Descent. Prostate 2013, 73, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Whitman, E.J.; Pomerantz, M.; Chen, Y.; Chamberlin, M.M.; Furusato, B.; Gao, C.; Ali, A.; Ravindranath, L.; Dobi, A.; Sesterhenn, I.A.; et al. Prostate Cancer Risk Allele Specific for African Descent Associates with Pathologic Stage at Prostatectomy. Cancer Epidemiol. Biomark. Prev. 2010, 19, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Koboldt, D.C.; Kanchi, K.L.; Gui, B.; Larson, D.E.; Fulton, R.S.; Isaacs, W.B.; Kraja, A.; Borecki, I.B.; Jia, L.; Wilson, R.K.; et al. Rare Variation in Tet2 Is Associated with Clinically Relevant Prostate Carcinoma in African Americans. Cancer Epidemiol. Biomark. Prev. 2016, 25, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Olama, A.A.; Benlloch, S.; Saunders, E.J.; Leongamornlert, D.A.; Tymrakiewicz, M.; Ghoussaini, M.; Luccarini, C.; Dennis, J.; Jugurnauth-Little, S.; et al. Identification of 23 New Prostate Cancer Susceptibility Loci Using the Icogs Custom Genotyping Array. Nat. Genet. 2013, 45, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Sampson, J.N.; Wheeler, W.A.; Yeager, M.; Panagiotou, O.; Wang, Z.; Berndt, S.I.; Lan, Q.; Abnet, C.C.; Amundadottir, L.T.; Figueroa, J.D.; et al. Analysis of Heritability and Shared Heritability Based on Genome-Wide Association Studies for Thirteen Cancer Types. J. Natl. Cancer Inst. 2015, 107, djv279. [Google Scholar] [CrossRef] [PubMed]
- Al Olama, A.A.; Kote-Jarai, Z.; Berndt, S.I.; Conti, D.V.; Schumacher, F.; Han, Y.; Benlloch, S.; Hazelett, D.J.; Wang, Z.; Saunders, E.; et al. A Meta-Analysis of 87,040 Individuals Identifies 23 New Susceptibility Loci for Prostate Cancer. Nat. Genet. 2014, 46, 1103–1109. [Google Scholar] [CrossRef] [PubMed]
- Cheng, I.; Chen, G.K.; Nakagawa, H.; He, J.; Wan, P.; Laurie, C.C.; Shen, J.; Sheng, X.; Pooler, L.C.; Crenshaw, A.T.; et al. Evaluating Genetic Risk for Prostate Cancer among Japanese and Latinos. Cancer Epidemiol. Biomark. Prev. 2012, 21, 2048–2058. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Sakoda, L.C.; Shen, L.; Jorgenson, E.; Habel, L.A.; Liu, J.; Kvale, M.N.; Asgari, M.M.; Banda, Y.; Corley, D.; et al. Imputation of the Rare Hoxb13 G84E Mutation and Cancer Risk in a Large Population-Based Cohort. PLoS Genet. 2015, 11, e1004930. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Kote-Jarai, Z.; Al Olama, A.A.; Giles, G.G.; Guy, M.; Severi, G.; Muir, K.; Hopper, J.L.; Henderson, B.E.; Haiman, C.A.; et al. Identification of Seven New Prostate Cancer Susceptibility Loci through a Genome-Wide Association Study. Nat. Genet. 2009, 41, 1116–1121. [Google Scholar] [CrossRef] [PubMed]
- Nam, R.K.; Zhang, W.; Siminovitch, K.; Shlien, A.; Kattan, M.W.; Klotz, L.H.; Trachtenberg, J.; Lu, Y.; Zhang, J.; Yu, C.; et al. New Variants at 10q26 and 15q21 Are Associated with Aggressive Prostate Cancer in a Genome-Wide Association Study from a Prostate Biopsy Screening Cohort. Cancer Biol. Ther. 2011, 12, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Kote-Jarai, Z.; Olama, A.A.; Giles, G.G.; Severi, G.; Schleutker, J.; Weischer, M.; Campa, D.; Riboli, E.; Key, T.; Gronberg, H.; et al. Seven Prostate Cancer Susceptibility Loci Identified by a Multi-Stage Genome-Wide Association Study. Nat. Genet. 2011, 43, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Kibel, A.S.; Hu, J.J.; Turner, A.R.; Pruett, K.; Zheng, S.L.; Sun, J.; Isaacs, S.D.; Wiley, K.E.; Kim, S.T.; et al. Prostate Cancer Risk Associated Loci in African Americans. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2145–2149. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, T.J.; Van Den Eeden, S.K.; Sakoda, L.C.; Jorgenson, E.; Habel, L.A.; Graff, R.E.; Passarelli, M.N.; Cario, C.L.; Emami, N.C.; Chao, C.R.; et al. A Large Multiethnic Genome-Wide Association Study of Prostate Cancer Identifies Novel Risk Variants and Substantial Ethnic Differences. Cancer Discov. 2015, 5, 878–891. [Google Scholar] [CrossRef] [PubMed]
- Gusev, A.; Shi, H.; Kichaev, G.; Pomerantz, M.; Li, F.; Long, H.W.; Ingles, S.A.; Kittles, R.A.; Strom, S.S.; Rybicki, B.A.; et al. Atlas of Prostate Cancer Heritability in European and African-American Men Pinpoints Tissue-Specific Regulation. Nat. Commun. 2016, 7, 10979. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Signorello, L.B.; Strom, S.S.; Kittles, R.A.; Rybicki, B.A.; Stanford, J.L.; Goodman, P.J.; Berndt, S.I.; Carpten, J.; Casey, G.; et al. Generalizability of Established Prostate Cancer Risk Variants in Men of African Ancestry. Int. J. Cancer 2015, 136, 1210–1217. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Rand, K.A.; Hazelett, D.J.; Ingles, S.A.; Kittles, R.A.; Strom, S.S.; Rybicki, B.A.; Nemesure, B.; Isaacs, W.B.; Stanford, J.L.; et al. Prostate Cancer Susceptibility in Men of African Ancestry at 8q24. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.A.; Rohland, N.; Tandon, A.; Stram, A.; Sheng, X.; Do, R.; Pasaniuc, B.; Allen, A.; Quinque, D.; Mallick, S.; et al. Whole-Exome Sequencing of over 4100 Men of African Ancestry and Prostate Cancer Risk. Hum. Mol. Genet. 2016, 25, 371–381. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.C.; Zeigler-Johnson, C.; Mittal, R.D.; Mandhani, A.; Mital, B.; Rebbeck, T.R.; Rennert, H. Common 8q24 Sequence Variations Are Associated with Asian Indian Advanced Prostate Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2008, 17, 2431–2435. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Batra, J.; Lose, F.; Chambers, S.; Gardiner, R.A.; Aitken, J.; Yaxley, J.; Clements, J.A.; Spurdle, A.B.; Australian Prostate Cancer, B. A Replication Study Examining Novel Common Single Nucleotide Polymorphisms Identified through a Prostate Cancer Genome-Wide Association Study in a Japanese Population. Am. J. Epidemiol. 2011, 174, 1391–1395. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Mo, Z.; Ye, D.; Wang, M.; Liu, F.; Jin, G.; Xu, C.; Wang, X.; Shao, Q.; Chen, Z.; et al. Genome-Wide Association Study in Chinese Men Identifies Two New Prostate Cancer Risk Loci at 9q31.2 and 19q13.4. Nat. Genet. 2012, 44, 1231–1235. [Google Scholar] [CrossRef] [PubMed]
- Na, R.; Liu, F.; Zhang, P.; Ye, D.; Xu, C.; Shao, Q.; Qi, J.; Wang, X.; Chen, Z.; Wang, M.; et al. Evaluation of Reported Prostate Cancer Risk-Associated Snps from Genome-Wide Association Studies of Various Racial Populations in Chinese Men. Prostate 2013, 73, 1623–1635. [Google Scholar] [CrossRef] [PubMed]
- Marzec, J.; Mao, X.; Li, M.; Wang, M.; Feng, N.; Gou, X.; Wang, G.; Sun, Z.; Xu, J.; Xu, H.; et al. A Genetic Study and Meta-Analysis of the Genetic Predisposition of Prostate Cancer in a Chinese Population. Oncotarget 2016, 7, 21393–21403. [Google Scholar] [CrossRef] [PubMed]
- MacArthur, D.G.; Manolio, T.A.; Dimmock, D.P.; Rehm, H.L.; Shendure, J.; Abecasis, G.R.; Adams, D.R.; Altman, R.B.; Antonarakis, S.E.; Ashley, E.A.; et al. Guidelines for Investigating Causality of Sequence Variants in Human Disease. Nature 2014, 508, 469–476. [Google Scholar] [CrossRef] [PubMed]
- FitzGerald, L.M.; Raspin, K.; Marthick, J.R.; Field, M.A.; Malley, R.C.; Thomson, R.J.; Blackburn, N.B.; Banks, A.; Charlesworth, J.C.; Donovan, S.; et al. Impact of the G84e Variant on Hoxb13 Gene and Protein Expression in Formalin-Fixed, Paraffin-Embedded Prostate Tumours. Sci. Rep. 2017, 7, 17778. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.D.; Eeles, R.; Freedland, S.J.; Isaacs, W.B.; Pomerantz, M.M.; Schalken, J.A.; Tammela, T.L.; Visakorpi, T. The Role of Genetic Markers in the Management of Prostate Cancer. Eur. Urol. 2012, 62, 577–587. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ewing, C.M.; Zheng, S.; Grindedaal, E.M.; Cooney, K.A.; Wiley, K.; Djurovic, S.; Andreassen, O.A.; Axcrona, K.; Mills, I.G.; et al. Genetic Factors Influencing Prostate Cancer Risk in Norwegian Men. Prostate 2018, 78, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Giri, V.N.; Knudsen, K.E.; Kelly, W.K.; Abida, W.; Andriole, G.L.; Bangma, C.H.; Bekelman, J.E.; Benson, M.C.; Blanco, A.; Burnett, A.; et al. Role of Genetic Testing for Inherited Prostate Cancer Risk: Philadelphia Prostate Cancer Consensus Conference 2017. J. Clin. Oncol. 2018, 36, 414–424. [Google Scholar] [CrossRef] [PubMed]
- Chang, B.L.; Cramer, S.D.; Wiklund, F.; Isaacs, S.D.; Stevens, V.L.; Sun, J.; Smith, S.; Pruett, K.; Romero, L.M.; Wiley, K.E.; et al. Fine Mapping Association Study and Functional Analysis Implicate a Snp in Msmb at 10q11 as a Causal Variant for Prostate Cancer Risk. Hum. Mol. Genet. 2009, 18, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Shrestha, Y.; Flavin, R.J.; Regan, M.M.; Penney, K.L.; Mucci, L.A.; Stampfer, M.J.; Hunter, D.J.; Chanock, S.J.; Schafer, E.J.; et al. Analysis of the 10q11 Cancer Risk Locus Implicates Msmb and Ncoa4 in Human Prostate Tumorigenesis. PLoS Genet. 2010, 6, e1001204. [Google Scholar] [CrossRef] [PubMed]
- Dias, A.; Kote-Jarai, Z.; Mikropoulos, C.; Eeles, R. Prostate Cancer Germline Variations and Implications for Screening and Treatment. Cold Spring Harb. Perspect. Med. 2017. pii: A030379. [Google Scholar] [CrossRef] [PubMed]
- Stelloo, S.; Nevedomskaya, E.; Kim, Y.; Hoekman, L.; Bleijerveld, O.B.; Mirza, T.; Wessels, L.F.A.; van Weerden, W.M.; Altelaar, A.F.M.; Bergman, A.M.; et al. Endogenous Androgen Receptor Proteomic Profiling Reveals Genomic Subcomplex Involved in Prostate Tumorigenesis. Oncogene 2018, 37, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Rinckleb, A.E.; Surowy, H.M.; Luedeke, M.; Varga, D.; Schrader, M.; Hoegel, J.; Vogel, W.; Maier, C. The Prostate Cancer Risk Locus at 10q11 Is Associated with DNA Repair Capacity. DNA Repair 2012, 11, 693–701. [Google Scholar] [CrossRef] [PubMed]
- El Gammal, A.T.; Bruchmann, M.; Zustin, J.; Isbarn, H.; Hellwinkel, O.J.; Kollermann, J.; Sauter, G.; Simon, R.; Wilczak, W.; Schwarz, J.; et al. Chromosome 8p Deletions and 8q Gains Are Associated with Tumor Progression and Poor Prognosis in Prostate Cancer. Clin. Cancer Res. 2010, 16, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Gurel, B.; Iwata, T.; C, M.K.; Jenkins, R.B.; Lan, F.; Van Dang, C.; Hicks, J.L.; Morgan, J.; Cornish, T.C.; Sutcliffe, S.; et al. Nuclear Myc Protein Overexpression Is an Early Alteration in Human Prostate Carcinogenesis. Mod. Pathol. 2008, 21, 1156–1167. [Google Scholar] [CrossRef] [PubMed]
- Fromont, G.; Godet, J.; Peyret, A.; Irani, J.; Celhay, O.; Rozet, F.; Cathelineau, X.; Cussenot, O. 8q24 Amplification Is Associated with Myc Expression and Prostate Cancer Progression and Is an Independent Predictor of Recurrence after Radical Prostatectomy. Hum. Pathol. 2013, 44, 1617–1623. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Beckwith, C.A.; Regan, M.M.; Wyman, S.K.; Petrovics, G.; Chen, Y.; Hawksworth, D.J.; Schumacher, F.R.; Mucci, L.; Penney, K.L.; et al. Evaluation of the 8q24 Prostate Cancer Risk Locus and Myc Expression. Cancer Res. 2009, 69, 5568–5574. [Google Scholar] [CrossRef] [PubMed]
- Wasserman, N.F.; Aneas, I.; Nobrega, M.A. An 8q24 Gene Desert Variant Associated with Prostate Cancer Risk Confers Differential in Vivo Activity to a Myc Enhancer. Genome Res. 2010, 20, 1191–1197. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Ahmadiyeh, N.; Jia, L.; Herman, P.; Verzi, M.P.; Doddapaneni, H.; Beckwith, C.A.; Chan, J.A.; Hills, A.; Davis, M.; et al. The 8q24 Cancer Risk Variant Rs6983267 Shows Long-Range Interaction with Myc in Colorectal Cancer. Nat. Genet. 2009, 41, 882–884. [Google Scholar] [CrossRef] [PubMed]
- Ahmadiyeh, N.; Pomerantz, M.M.; Grisanzio, C.; Herman, P.; Jia, L.; Almendro, V.; He, H.H.; Brown, M.; Liu, X.S.; Davis, M.; et al. 8q24 Prostate, Breast, and Colon Cancer Risk Loci Show Tissue-Specific Long-Range Interaction with Myc. Proc. Natl. Acad. Sci. USA 2010, 107, 9742–9746. [Google Scholar] [CrossRef] [PubMed]
- Cai, M.; Kim, S.; Wang, K.; Farnham, P.J.; Coetzee, G.A.; Lu, W. 4c-Seq Revealed Long-Range Interactions of a Functional Enhancer at the 8q24 Prostate Cancer Risk Locus. Sci. Rep. 2016, 6, 22462. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Tillmans, L.; Gao, J.; Gao, P.; Yuan, T.; Dittmar, R.L.; Song, W.; Yang, Y.; Sahr, N.; Wang, T.; et al. Chromatin Interactions and Candidate Genes at Ten Prostate Cancer Risk Loci. Sci. Rep. 2016, 6, 23202. [Google Scholar] [CrossRef] [PubMed]
- Ott, J. Analysis of Human Genetic Linkage; Johns Hopkins University Press: Baltimore, MD, USA, 1991; p. 302. [Google Scholar]
- Carpten, J.; Nupponen, N.; Isaacs, S.; Sood, R.; Robbins, C.; Xu, J.; Faruque, M.; Moses, T.; Ewing, C.; Gillanders, E.; et al. Germline Mutations in the Ribonuclease L Gene in Families Showing Linkage with Hpc1. Nat. Genet. 2002, 30, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Tavtigian, S.V.; Simard, J.; Teng, D.H.; Abtin, V.; Baumgard, M.; Beck, A.; Camp, N.J.; Carillo, A.R.; Chen, Y.; Dayananth, P.; et al. A Candidate Prostate Cancer Susceptibility Gene at Chromosome 17p. Nat. Genet. 2001, 27, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Zheng, S.L.; Komiya, A.; Mychaleckyj, J.C.; Isaacs, S.D.; Hu, J.J.; Sterling, D.; Lange, E.M.; Hawkins, G.A.; Turner, A.; et al. Germline Mutations and Sequence Variants of the Macrophage Scavenger Receptor 1 Gene Are Associated with Prostate Cancer Risk. Nat. Genet. 2002, 32, 321–325. [Google Scholar] [CrossRef] [PubMed]
- Thibodeau, S.N.; French, A.J.; Roche, P.C.; Cunningham, J.M.; Tester, D.J.; Lindor, N.M.; Moslein, G.; Baker, S.M.; Liskay, R.M.; Burgart, L.J.; et al. Altered Expression of Hmsh2 and Hmlh1 in Tumors with Microsatellite Instability and Genetic Alterations in Mismatch Repair Genes. Cancer Res. 1996, 56, 4836–4840. [Google Scholar] [PubMed]
- Leongamornlert, D.; Saunders, E.; Dadaev, T.; Tymrakiewicz, M.; Goh, C.; Jugurnauth-Little, S.; Kozarewa, I.; Fenwick, K.; Assiotis, I.; Barrowdale, D.; et al. Frequent Germline Deleterious Mutations in DNA Repair Genes in Familial Prostate Cancer Cases Are Associated with Advanced Disease. Br. J. Cancer 2014, 110, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Beebe-Dimmer, J.L.; Zuhlke, K.A.; Johnson, A.M.; Liesman, D.; Cooney, K.A. Rare Germline Mutations in African American Men Diagnosed with Early-Onset Prostate Cancer. Prostate 2018, 78, 321–326. [Google Scholar] [CrossRef] [PubMed]
- Beebe-Dimmer, J.L.; Isaacs, W.B.; Zuhlke, K.A.; Yee, C.; Walsh, P.C.; Isaacs, S.D.; Johnson, A.M.; Ewing, C.E.; Humphreys, E.B.; Chowdhury, W.H.; et al. Prevalence of the Hoxb13 G84e Prostate Cancer Risk Allele in Men Treated with Radical Prostatectomy. BJU Int. 2014, 113, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Na, R.; Zheng, S.L.; Han, M.; Yu, H.; Jiang, D.; Shah, S.; Ewing, C.M.; Zhang, L.; Novakovic, K.; Petkewicz, J.; et al. Germline Mutations in Atm and Brca1/2 Distinguish Risk for Lethal and Indolent Prostate Cancer and Are Associated with Early Age at Death. Eur. Urol. 2017, 71, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Raymond, V.M.; Mukherjee, B.; Wang, F.; Huang, S.C.; Stoffel, E.M.; Kastrinos, F.; Syngal, S.; Cooney, K.A.; Gruber, S.B. Elevated Risk of Prostate Cancer among Men with Lynch Syndrome. J. Clin. Oncol. 2013, 31, 1713–1718. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Valentin, M.; Joost, P.; Therkildsen, C.; Jonsson, M.; Rambech, E.; Nilbert, M. Frequent Mismatch-Repair Defects Link Prostate Cancer to Lynch Syndrome. BMC Urol. 2016, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Guindalini, R.S.; Win, A.K.; Gulden, C.; Lindor, N.M.; Newcomb, P.A.; Haile, R.W.; Raymond, V.; Stoffel, E.; Hall, M.; Llor, X.; et al. Mutation Spectrum and Risk of Colorectal Cancer in African American Families with Lynch Syndrome. Gastroenterology 2015, 149, 1446–1453. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Walker, A.H.; Zeigler-Johnson, C.; Weisburg, S.; Martin, A.M.; Nathanson, K.L.; Wein, A.J.; Malkowicz, S.B. Association of Hpc2/Elac2 Genotypes and Prostate Cancer. Am. J. Hum. Genet. 2000, 67, 1014–1019. [Google Scholar] [CrossRef] [PubMed]
- Severi, G.; Giles, G.G.; Southey, M.C.; Tesoriero, A.; Tilley, W.; Neufing, P.; Morris, H.; English, D.R.; McCredie, M.R.; Boyle, P.; et al. Elac2/Hpc2 Polymorphisms, Prostate-Specific Antigen Levels, and Prostate Cancer. J. Natl. Cancer Inst. 2003, 95, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Tong, N.; Li, J.M.; Zhang, Z.D.; Wu, H.F. Elac2 Polymorphisms and Prostate Cancer Risk: A Meta-Analysis Based on 18 Case-Control Studies. Prostate Cancer Prostatic Dis. 2010, 13, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.M.; Hernandez, W.; Ahaghotu, C.; Bennett, J.; Hoke, G.; Mason, T.; Pettaway, C.A.; Vijayakumar, S.; Weinrich, S.; Furbert-Harris, P.; et al. Association of Hpc2/Elac2 and Rnasel Non-Synonymous Variants with Prostate Cancer Risk in African American Familial and Sporadic Cases. Prostate 2008, 68, 1790–1797. [Google Scholar] [CrossRef] [PubMed]
- Casey, G.; Neville, P.J.; Plummer, S.J.; Xiang, Y.; Krumroy, L.M.; Klein, E.A.; Catalona, W.J.; Nupponen, N.; Carpten, J.D.; Trent, J.M.; et al. Rnasel Arg462gln Variant Is Implicated in up to 13% of Prostate Cancer Cases. Nat. Genet. 2002, 32, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Shook, S.J.; Beuten, J.; Torkko, K.C.; Johnson-Pais, T.L.; Troyer, D.A.; Thompson, I.M.; Leach, R.J. Association of Rnasel Variants with Prostate Cancer Risk in Hispanic Caucasians and African Americans. Clin. Cancer Res. 2007, 13, 5959–5964. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Tai, B.C. Rnasel Gene Polymorphisms and the Risk of Prostate Cancer: A Meta-Analysis. Clin. Cancer Res. 2006, 12, 5713–5719. [Google Scholar] [CrossRef] [PubMed]
- Eeles, R.A.; Durocher, F.; Edwards, S.; Teare, D.; Badzioch, M.; Hamoudi, R.; Gill, S.; Biggs, P.; Dearnaley, D.; Ardern-Jones, A.; et al. Linkage Analysis of Chromosome 1q Markers in 136 Prostate Cancer Families. The Cancer Research Campaign/British Prostate Group U.K. Familial Prostate Cancer Study Collaborators. Am. J. Hum. Genet. 1998, 62, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, F.; Jonsson, B.A.; Brookes, A.J.; Stromqvist, L.; Adolfsson, J.; Emanuelsson, M.; Adami, H.O.; Augustsson-Balter, K.; Gronberg, H. Genetic Analysis of the Rnasel Gene in Hereditary, Familial, and Sporadic Prostate Cancer. Clin. Cancer Res. 2004, 10, 7150–7156. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.M.; Lange, E.M.; Chen, H.; Zheng, S.L.; Chang, B.; Wiley, K.E.; Isaacs, S.D.; Walsh, P.C.; Isaacs, W.B.; Xu, J.; et al. Hereditary Prostate Cancer in African American Families: Linkage Analysis Using Markers That Map to Five Candidate Susceptibility Loci. Br. J. Cancer 2004, 90, 510–514. [Google Scholar] [CrossRef] [PubMed]
- Powell, I.J.; Carpten, J.; Dunston, G.; Kittles, R.; Bennett, J.; Hoke, G.; Pettaway, C.; Weinrich, S.; Vijayakumar, S.; Ahaghotu, C.A.; et al. African-American Heredity Prostate Cancer Study: A Model for Genetic Research. J. Natl. Med. Assoc. 2001, 93, 120–123. [Google Scholar] [PubMed]
- Xu, J.; Zheng, S.L.; Hawkins, G.A.; Faith, D.A.; Kelly, B.; Isaacs, S.D.; Wiley, K.E.; Chang, B.; Ewing, C.M.; Bujnovszky, P.; et al. Linkage and Association Studies of Prostate Cancer Susceptibility: Evidence for Linkage at 8p22-23. Am. J. Hum. Genet. 2001, 69, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Wiklund, F.; Jonsson, B.A.; Goransson, I.; Bergh, A.; Gronberg, H. Linkage Analysis of Prostate Cancer Susceptibility: Confirmation of Linkage at 8p22-23. Hum. Genet. 2003, 112, 414–418. [Google Scholar] [PubMed]
- Sun, J.; Hsu, F.C.; Turner, A.R.; Zheng, S.L.; Chang, B.L.; Liu, W.; Isaacs, W.B.; Xu, J. Meta-Analysis of Association of Rare Mutations and Common Sequence Variants in the Msr1 Gene and Prostate Cancer Risk. Prostate 2006, 66, 728–737. [Google Scholar] [CrossRef] [PubMed]
- Beuten, J.; Gelfond, J.A.; Franke, J.L.; Shook, S.; Johnson-Pais, T.L.; Thompson, I.M.; Leach, R.J. Single and Multivariate Associations of Msr1, Elac2, and Rnasel with Prostate Cancer in an Ethnic Diverse Cohort of Men. Cancer Epidemiol. Biomark. Prev. 2010, 19, 588–599. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, S.M.; Barrette, T.R.; Ghosh, D.; Shah, R.; Varambally, S.; Kurachi, K.; Pienta, K.J.; Rubin, M.A.; Chinnaiyan, A.M. Delineation of Prognostic Biomarkers in Prostate Cancer. Nature 2001, 412, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Norris, J.D.; Chang, C.Y.; Wittmann, B.M.; Kunder, R.S.; Cui, H.; Fan, D.; Joseph, J.D.; McDonnell, D.P. The Homeodomain Protein Hoxb13 Regulates the Cellular Response to Androgens. Mol. Cell 2009, 36, 405–416. [Google Scholar] [CrossRef] [PubMed]
- Pomerantz, M.M.; Li, F.; Takeda, D.Y.; Lenci, R.; Chonkar, A.; Chabot, M.; Cejas, P.; Vazquez, F.; Cook, J.; Shivdasani, R.A.; et al. The Androgen Receptor Cistrome Is Extensively Reprogrammed in Human Prostate Tumorigenesis. Nat. Genet. 2015, 47, 1346–1351. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Lange, E.M.; Lu, L.; Zheng, S.L.; Wang, Z.; Thibodeau, S.N.; Cannon-Albright, L.A.; Teerlink, C.C.; Camp, N.J.; Johnson, A.M.; et al. Hoxb13 Is a Susceptibility Gene for Prostate Cancer: Results from the International Consortium for Prostate Cancer Genetics (Icpcg). Hum. Genet. 2013, 132, 5–14. [Google Scholar] [CrossRef] [PubMed]
- Witte, J.S.; Mefford, J.; Plummer, S.J.; Liu, J.; Cheng, I.; Klein, E.A.; Rybicki, B.A.; Casey, G. Hoxb13 Mutation and Prostate Cancer: Studies of Siblings and Aggressive Disease. Cancer Epidemiol. Biomark. Prev. 2013, 22, 675–680. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cai, Q.; Wang, X.; Li, X.; Gong, R.; Guo, X.; Tang, Y.; Yang, K.; Niu, Y.; Zhao, Y. Germline Hoxb13 P.Gly84glu Mutation and Cancer Susceptibility: A Pooled Analysis of 25 Epidemiological Studies with 145,257 Participates. Oncotarget 2015, 6, 42312–42321. [Google Scholar] [CrossRef] [PubMed]
- Maia, S.; Cardoso, M.; Pinto, P.; Pinheiro, M.; Santos, C.; Peixoto, A.; Bento, M.J.; Oliveira, J.; Henrique, R.; Jeronimo, C.; et al. Identification of Two Novel Hoxb13 Germline Mutations in Portuguese Prostate Cancer Patients. PLoS ONE 2015, 10, e0132728. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, V.H.; Wahlfors, T.; Saaristo, L.; Rantapero, T.; Pelttari, L.M.; Kilpivaara, O.; Laasanen, S.L.; Kallioniemi, A.; Nevanlinna, H.; Aaltonen, L.; et al. Hoxb13 G84e Mutation in Finland: Population-Based Analysis of Prostate, Breast, and Colorectal Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2013, 22, 452–460. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Qu, L.; Chen, Z.; Xu, C.; Ye, D.; Shao, Q.; Wang, X.; Qi, J.; Chen, Z.; Zhou, F.; et al. A Novel Germline Mutation in Hoxb13 Is Associated with Prostate Cancer Risk in Chinese Men. Prostate 2013, 73, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, N.; Daly, M.B.; Feldman, G.L. Hereditary Breast and Ovarian Cancer Due to Mutations in Brca1 and Brca2. Genet. Med. 2010, 12, 245–259. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Chun, J.; Powell, S.N. Brca1 and Brca2: Different Roles in a Common Pathway of Genome Protection. Nat. Rev. Cancer 2011, 12, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.X. Brca1: Cell Cycle Checkpoint, Genetic Instability, DNA Damage Response and Cancer Evolution. Nucleic Acids Res. 2006, 34, 1416–1426. [Google Scholar] [CrossRef] [PubMed]
- Moynahan, M.E.; Pierce, A.J.; Jasin, M. Brca2 Is Required for Homology-Directed Repair of Chromosomal Breaks. Mol. Cell 2001, 7, 263–272. [Google Scholar] [CrossRef]
- Eeles, R.; Goh, C.; Castro, E.; Bancroft, E.; Guy, M.; Al Olama, A.A.; Easton, D.; Kote-Jarai, Z. The Genetic Epidemiology of Prostate Cancer and Its Clinical Implications. Nat. Rev. Urol. 2014, 11, 18–31. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Kumaraswamy, E.; Harlan-Williams, L.M.; Jensen, R.A. The Role of Brca1 and Brca2 in Prostate Cancer. Front. Biosci. 2013, 18, 1445–1459. [Google Scholar]
- Thompson, D.; Easton, D.F.; Breast Cancer Linkage, C. Cancer Incidence in Brca1 Mutation Carriers. J. Natl. Cancer Inst. 2002, 94, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Linkage Consortium. Cancer Risks in Brca2 Mutation Carriers. J. Natl. Cancer Inst. 1999, 91, 1310–1316. [Google Scholar]
- Douglas, J.A.; Levin, A.M.; Zuhlke, K.A.; Ray, A.M.; Johnson, G.R.; Lange, E.M.; Wood, D.P.; Cooney, K.A. Common Variation in the Brca1 Gene and Prostate Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1510–1516. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Struewing, J.P.; Hartge, P.; Wacholder, S.; Baker, S.M.; Berlin, M.; McAdams, M.; Timmerman, M.M.; Brody, L.C.; Tucker, M.A. The Risk of Cancer Associated with Specific Mutations of Brca1 and Brca2 among Ashkenazi Jews. N. Engl. J. Med. 1997, 336, 1401–1408. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, T.; Kauff, N.D.; Mitra, N.; Nafa, K.; Huang, H.; Palmer, C.; Gulati, T.; Wadsworth, E.; Donat, S.; Robson, M.E.; et al. Brca Mutations and Risk of Prostate Cancer in Ashkenazi Jews. Clin. Cancer Res. 2004, 10, 2918–2921. [Google Scholar] [CrossRef] [PubMed]
- Agalliu, I.; Gern, R.; Leanza, S.; Burk, R.D. Associations of High-Grade Prostate Cancer with Brca1 and Brca2 Founder Mutations. Clin. Cancer Res. 2009, 15, 1112–1120. [Google Scholar] [CrossRef] [PubMed]
- John, E.M.; Miron, A.; Gong, G.; Phipps, A.I.; Felberg, A.; Li, F.P.; West, D.W.; Whittemore, A.S. Prevalence of Pathogenic Brca1 Mutation Carriers in 5 Us Racial/Ethnic Groups. JAMA 2007, 298, 2869–2876. [Google Scholar] [CrossRef] [PubMed]
- Sigurdsson, S.; Thorlacius, S.; Tomasson, J.; Tryggvadottir, L.; Benediktsdottir, K.; Eyfjord, J.E.; Jonsson, E. Brca2 Mutation in Icelandic Prostate Cancer Patients. J. Mol. Med. 1997, 75, 758–761. [Google Scholar] [CrossRef] [PubMed]
- Gayther, S.A.; de Foy, K.A.; Harrington, P.; Pharoah, P.; Dunsmuir, W.D.; Edwards, S.M.; Gillett, C.; Ardern-Jones, A.; Dearnaley, D.P.; Easton, D.F.; et al. The Frequency of Germ-Line Mutations in the Breast Cancer Predisposition Genes Brca1 and Brca2 in Familial Prostate Cancer. The Cancer Research Campaign/British Prostate Group United Kingdom Familial Prostate Cancer Study Collaborators. Cancer Res. 2000, 60, 4513–4518. [Google Scholar] [PubMed]
- Gronberg, H.; Ahman, A.K.; Emanuelsson, M.; Bergh, A.; Damber, J.E.; Borg, A. Brca2 Mutation in a Family with Hereditary Prostate Cancer. Genes Chromosomes Cancer 2001, 30, 299–301. [Google Scholar] [CrossRef]
- Bancroft, E.K.; Page, E.C.; Castro, E.; Lilja, H.; Vickers, A.; Sjoberg, D.; Assel, M.; Foster, C.S.; Mitchell, G.; Drew, K.; et al. Targeted Prostate Cancer Screening in Brca1 and Brca2 Mutation Carriers: Results from the Initial Screening Round of the Impact Study. Eur. Urol. 2014, 66, 489–499. [Google Scholar] [CrossRef] [PubMed]
- Akbari, M.R.; Wallis, C.J.; Toi, A.; Trachtenberg, J.; Sun, P.; Narod, S.A.; Nam, R.K. The Impact of a Brca2 Mutation on Mortality from Screen-Detected Prostate Cancer. Br. J. Cancer 2014, 111, 1238–1240. [Google Scholar] [CrossRef] [PubMed]
- Leao, R.R.N.; Price, A.J.; James Hamilton, R. Germline Brca Mutation in Male Carriers-Ripe for Precision Oncology? Prostate Cancer Prostatic Dis. 2017. [Google Scholar] [CrossRef]
- Grindedal, E.M.; Moller, P.; Eeles, R.; Stormorken, A.T.; Bowitz-Lothe, I.M.; Landro, S.M.; Clark, N.; Kvale, R.; Shanley, S.; Maehle, L. Germ-Line Mutations in Mismatch Repair Genes Associated with Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2009, 18, 2460–2467. [Google Scholar] [CrossRef] [PubMed]
- Win, A.K.; Lindor, N.M.; Young, J.P.; Macrae, F.A.; Young, G.P.; Williamson, E.; Parry, S.; Goldblatt, J.; Lipton, L.; Winship, I.; et al. Risks of Primary Extracolonic Cancers Following Colorectal Cancer in Lynch Syndrome. J. Natl. Cancer Inst. 2012, 104, 1363–1372. [Google Scholar] [CrossRef] [PubMed]
- Bauer, C.M.; Ray, A.M.; Halstead-Nussloch, B.A.; Dekker, R.G.; Raymond, V.M.; Gruber, S.B.; Cooney, K.A. Hereditary Prostate Cancer as a Feature of Lynch Syndrome. Fam. Cancer 2011, 10, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Langeberg, W.J.; Kwon, E.M.; Koopmeiners, J.S.; Ostrander, E.A.; Stanford, J.L. Population-Based Study of the Association of Variants in Mismatch Repair Genes with Prostate Cancer Risk and Outcomes. Cancer Epidemiol. Biomark. Prev. 2010, 19, 258–264. [Google Scholar] [CrossRef] [PubMed]
- Dong, X.; Wang, L.; Taniguchi, K.; Wang, X.; Cunningham, J.M.; McDonnell, S.K.; Qian, C.; Marks, A.F.; Slager, S.L.; Peterson, B.J.; et al. Mutations in Chek2 Associated with Prostate Cancer Risk. Am. J. Hum. Genet. 2003, 72, 270–280. [Google Scholar] [CrossRef] [PubMed]
- Seppala, E.H.; Ikonen, T.; Mononen, N.; Autio, V.; Rokman, A.; Matikainen, M.P.; Tammela, T.L.; Schleutker, J. Chek2 Variants Associate with Hereditary Prostate Cancer. Br. J. Cancer 2003, 89, 1966–1970. [Google Scholar] [CrossRef] [PubMed]
- Angele, S.; Falconer, A.; Edwards, S.M.; Dork, T.; Bremer, M.; Moullan, N.; Chapot, B.; Muir, K.; Houlston, R.; Norman, A.R.; et al. Atm Polymorphisms as Risk Factors for Prostate Cancer Development. Br. J. Cancer 2004, 91, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Blackford, A.N.; Jackson, S.P. Atm, Atr, and DNA-Pk: The Trinity at the Heart of the DNA Damage Response. Mol. Cell 2017, 66, 801–817. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative Clinical Genomics of Advanced Prostate Cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Holcomb, I.N.; Young, J.M.; Coleman, I.M.; Salari, K.; Grove, D.I.; Hsu, L.; True, L.D.; Roudier, M.P.; Morrissey, C.M.; Higano, C.S.; et al. Comparative Analyses of Chromosome Alterations in Soft-Tissue Metastases within and across Patients with Castration-Resistant Prostate Cancer. Cancer Res. 2009, 69, 7793–7802. [Google Scholar] [CrossRef] [PubMed]
- Robbins, C.M.; Tembe, W.A.; Baker, A.; Sinari, S.; Moses, T.Y.; Beckstrom-Sternberg, S.; Beckstrom-Sternberg, J.; Barrett, M.; Long, J.; Chinnaiyan, A.; et al. Copy Number and Targeted Mutational Analysis Reveals Novel Somatic Events in Metastatic Prostate Tumors. Genome Res. 2011, 21, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kan, Z.; Jaiswal, B.S.; Stinson, J.; Janakiraman, V.; Bhatt, D.; Stern, H.M.; Yue, P.; Haverty, P.M.; Bourgon, R.; Zheng, J.; et al. Diverse Somatic Mutation Patterns and Pathway Alterations in Human Cancers. Nature 2010, 466, 869–873. [Google Scholar] [CrossRef] [PubMed]
- Dong, J.T. Chromosomal Deletions and Tumor Suppressor Genes in Prostate Cancer. Cancer Metastasis Rev. 2001, 20, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Nupponen, N.N.; Visakorpi, T. Molecular Cytogenetics of Prostate Cancer. Microsc. Res. Tech. 2000, 51, 456–463. [Google Scholar] [CrossRef]
- Ren, G.; Liu, X.; Mao, X.; Zhang, Y.; Stankiewicz, E.; Hylands, L.; Song, R.; Berney, D.M.; Clark, J.; Cooper, C.; et al. Identification of Frequent Braf Copy Number Gain and Alterations of Raf Genes in Chinese Prostate Cancer. Genes Chromosomes Cancer 2012, 51, 1014–1023. [Google Scholar] [CrossRef] [PubMed]
- Petrovics, G.; Liu, A.; Shaheduzzaman, S.; Furusato, B.; Sun, C.; Chen, Y.; Nau, M.; Ravindranath, L.; Chen, Y.; Dobi, A.; et al. Frequent Overexpression of Ets-Related Gene-1 (Erg1) in Prostate Cancer Transcriptome. Oncogene 2005, 24, 3847–3852. [Google Scholar] [CrossRef] [PubMed]
- Tomlins, S.A.; Rhodes, D.R.; Perner, S.; Dhanasekaran, S.M.; Mehra, R.; Sun, X.W.; Varambally, S.; Cao, X.; Tchinda, J.; Kuefer, R.; et al. Recurrent Fusion of Tmprss2 and Ets Transcription Factor Genes in Prostate Cancer. Science 2005, 310, 644–648. [Google Scholar] [CrossRef] [PubMed]
- International Cancer Genome Consortium; Hudson, T.J.; Anderson, W.; Artez, A.; Barker, A.D.; Bell, C.; Bernabe, R.R.; Bhan, M.K.; Calvo, F.; Eerola, I.; et al. International Network of Cancer Genome Projects. Nature 2010, 464, 993–998. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (Tcga): An Immeasurable Source of Knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Berger, M.F.; Lawrence, M.S.; Demichelis, F.; Drier, Y.; Cibulskis, K.; Sivachenko, A.Y.; Sboner, A.; Esgueva, R.; Pflueger, D.; Sougnez, C.; et al. The Genomic Complexity of Primary Human Prostate Cancer. Nature 2011, 470, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome Sequencing Identifies Recurrent SPOP, FOXA1 and MED12 Mutations in Prostate Cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef] [PubMed]
- Baca, S.C.; Prandi, D.; Lawrence, M.S.; Mosquera, J.M.; Romanel, A.; Drier, Y.; Park, K.; Kitabayashi, N.; MacDonald, T.Y.; Ghandi, M.; et al. Punctuated Evolution of Prostate Cancer Genomes. Cell 2013, 153, 666–677. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Research Network. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar]
- Fraser, M.; Sabelnykova, V.Y.; Yamaguchi, T.N.; Heisler, L.E.; Livingstone, J.; Huang, V.; Shiah, Y.J.; Yousif, F.; Lin, X.; Masella, A.P.; et al. Genomic Hallmarks of Localized, Non-Indolent Prostate Cancer. Nature 2017, 541, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of Mutational Processes in Human Cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Korbel, J.O.; Campbell, P.J. Criteria for Inference of Chromothripsis in Cancer Genomes. Cell 2013, 152, 1226–1236. [Google Scholar] [CrossRef] [PubMed]
- Khani, F.; Mosquera, J.M.; Park, K.; Blattner, M.; O’Reilly, C.; MacDonald, T.Y.; Chen, Z.; Srivastava, A.; Tewari, A.K.; Barbieri, C.E.; et al. Evidence for Molecular Differences in Prostate Cancer between African American and Caucasian Men. Clin. Cancer Res. 2014, 20, 4925–4934. [Google Scholar] [CrossRef] [PubMed]
- Magi-Galluzzi, C.; Tsusuki, T.; Elson, P.; Simmerman, K.; LaFargue, C.; Esgueva, R.; Klein, E.; Rubin, M.A.; Zhou, M. Tmprss2-Erg Gene Fusion Prevalence and Class Are Significantly Different in Prostate Cancer of Caucasian, African-American and Japanese Patients. Prostate 2011, 71, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Rosen, P.; Pfister, D.; Young, D.; Petrovics, G.; Chen, Y.; Cullen, J.; Bohm, D.; Perner, S.; Dobi, A.; McLeod, D.G.; et al. Differences in Frequency of Erg Oncoprotein Expression between Index Tumors of Caucasian and African American Patients with Prostate Cancer. Urology 2012, 80, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Petrovics, G.; Li, H.; Stumpel, T.; Tan, S.H.; Young, D.; Katta, S.; Li, Q.; Ying, K.; Klocke, B.; Ravindranath, L.; et al. A Novel Genomic Alteration of Lsamp Associates with Aggressive Prostate Cancer in African American Men. EBioMedicine 2015, 2, 1957–1964. [Google Scholar] [CrossRef] [PubMed]
- Barøy, T.; Kresse, S.H.; Skarn, M.; Stabell, M.; Castro, R.; Lauvrak, S.; Llombart-Bosch, A.; Myklebost, O.; Meza-Zepeda, L.A. Reexpression of Lsamp Inhibits Tumor Growth in a Preclinical Osteosarcoma Model. Mol. Cancer 2014, 13, 93. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Lui, W.O.; Vos, M.D.; Clark, G.J.; Takahashi, M.; Schoumans, J.; Khoo, S.K.; Petillo, D.; Lavery, T.; Sugimura, J.; et al. The T(1;3) Breakpoint-Spanning Genes Lsamp and Nore1 Are Involved in Clear Cell Renal Cell Carcinomas. Cancer Cell 2003, 4, 405–413. [Google Scholar] [CrossRef]
- Ren, S.; Wei, G.H.; Liu, D.; Wang, L.; Hou, Y.; Zhu, S.; Peng, L.; Zhang, Q.; Cheng, Y.; Su, H.; et al. Whole-Genome and Transcriptome Sequencing of Prostate Cancer Identify New Genetic Alterations Driving Disease Progression. Eur. Urol. 2017, 73, 322–339. [Google Scholar] [CrossRef] [PubMed]
- Blattner, M.; Lee, D.J.; O’Reilly, C.; Park, K.; MacDonald, T.Y.; Khani, F.; Turner, K.R.; Chiu, Y.L.; Wild, P.J.; Dolgalev, I.; et al. SPOP Mutations in Prostate Cancer across Demographically Diverse Patient Cohorts. Neoplasia 2014, 16, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Grasso, C.S.; Wu, Y.M.; Robinson, D.R.; Cao, X.; Dhanasekaran, S.M.; Khan, A.P.; Quist, M.J.; Jing, X.; Lonigro, R.J.; Brenner, J.C.; et al. The Mutational Landscape of Lethal Castration-Resistant Prostate Cancer. Nature 2012, 487, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Lindberg, J.; Sui, G.; Luo, J.; Egevad, L.; Li, T.; Xie, C.; Wan, M.; Kim, S.T.; Wang, Z.; et al. Identification of Novel Chd1-Associated Collaborative Alterations of Genomic Structure and Functional Assessment of Chd1 in Prostate Cancer. Oncogene 2012, 31, 3939–3948. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, L.U.; Rider, L.; Nieto, C.; Romero, L.; Karimpour-Fard, A.; Loda, M.; Lucia, M.S.; Wu, M.; Shi, L.; Cimic, A.; et al. Coordinate Loss of Map3k7 and Chd1 Promotes Aggressive Prostate Cancer. Cancer Res. 2015, 75, 1021–1034. [Google Scholar] [CrossRef] [PubMed]
- Kari, V.; Mansour, W.Y.; Raul, S.K.; Baumgart, S.J.; Mund, A.; Grade, M.; Sirma, H.; Simon, R.; Will, H.; Dobbelstein, M.; et al. Loss of Chd1 Causes DNA Repair Defects and Enhances Prostate Cancer Therapeutic Responsiveness. EMBO Rep. 2016, 17, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, T.R.; Boysen, G.; Wang, M.Y.; Xu, Q.Z.; Guo, W.; Koh, F.M.; Wang, C.; Zhang, L.Z.; Wang, Y.; Gil, V.; et al. Chd1 Loss Sensitizes Prostate Cancer to DNA Damaging Therapy by Promoting Error-Prone Double-Strand Break Repair. Ann. Oncol. 2017, 28, 1495–1507. [Google Scholar] [CrossRef] [PubMed]
- Lindquist, K.J.; Paris, P.L.; Hoffmann, T.J.; Cardin, N.J.; Kazma, R.; Mefford, J.A.; Simko, J.P.; Ngo, V.; Chen, Y.; Levin, A.M.; et al. Mutational Landscape of Aggressive Prostate Tumors in African American Men. Cancer Res. 2016, 76, 1860–1868. [Google Scholar] [CrossRef] [PubMed]
- Schulz, W.A.; Elo, J.P.; Florl, A.R.; Pennanen, S.; Santourlidis, S.; Engers, R.; Buchardt, M.; Seifert, H.H.; Visakorpi, T. Genomewide DNA Hypomethylation Is Associated with Alterations on Chromosome 8 in Prostate Carcinoma. Genes Chromosomes Cancer 2002, 35, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.W.; Mosquera, J.M.; Garofalo, A.; Oh, C.; Baco, M.; Amin-Mansour, A.; Rabasha, B.; Bahl, S.; Mullane, S.A.; Robinson, B.D.; et al. Exome Sequencing of African-American Prostate Cancer Reveals Loss-of-Function Erf Mutations. Cancer Discov. 2017, 7, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; White, T.A.; MacKenzie, A.P.; Clegg, N.; Lee, C.; Dumpit, R.F.; Coleman, I.; Ng, S.B.; Salipante, S.J.; Rieder, M.J.; et al. Exome Sequencing Identifies a Spectrum of Mutation Frequencies in Advanced and Lethal Prostate Cancers. Proc. Natl. Acad. Sci. USA 2011, 108, 17087–17092. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, C.C.; Morrissey, C.; Kumar, A.; Zhang, X.; Smith, C.; Coleman, I.; Salipante, S.J.; Milbank, J.; Yu, M.; Grady, W.M.; et al. Complex Msh2 and Msh6 Mutations in Hypermutated Microsatellite Unstable Advanced Prostate Cancer. Nat. Commun. 2014, 5, 4988. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Coleman, I.; Morrissey, C.; Zhang, X.; True, L.D.; Gulati, R.; Etzioni, R.; Bolouri, H.; Montgomery, B.; White, T.; et al. Substantial Interindividual and Limited Intraindividual Genomic Diversity among Tumors from Men with Metastatic Prostate Cancer. Nat. Med. 2016, 22, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging Mechanisms of Resistance to Androgen Receptor Inhibitors in Prostate Cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Boudadi, K.; Antonarakis, E.S. Resistance to Novel Antiandrogen Therapies in Metastatic Castration-Resistant Prostate Cancer. Clin. Med. Insights Oncol. 2016, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Antonarakis, E.S.; Morris, M.J.; Attard, G. Emerging Molecular Biomarkers in Advanced Prostate Cancer: Translation to the Clinic. Am. Soc. Clin. Oncol. Educ. Book 2016, 35, 131–141. [Google Scholar] [CrossRef] [PubMed]
- Abida, W.; Armenia, J.; Gopalan, A.; Brennan, R.; Walsh, M.; Barron, D.; Danila, D.; Rathkopf, D.; Morris, M.; Slovin, S.; et al. Prospective Genomic Profiling of Prostate Cancer across Disease States Reveals Germline and Somatic Alterations That May Affect Clinical Decision Making. JCO Precis. Oncol. 2017, 2017. [Google Scholar] [CrossRef] [PubMed]
- Romanel, A.; Garritano, S.; Stringa, B.; Blattner, M.; Dalfovo, D.; Chakravarty, D.; Soong, D.; Cotter, K.A.; Petris, G.; Dhingra, P.; et al. Inherited Determinants of Early Recurrent Somatic Mutations in Prostate Cancer. Nat. Commun. 2017, 8, 48. [Google Scholar] [CrossRef] [PubMed]
- Wei, L.; Wang, J.; Lampert, E.; Schlanger, S.; DePriest, A.D.; Hu, Q.; Gomez, E.C.; Murakam, M.; Glenn, S.T.; Conroy, J.; et al. Intratumoral and Intertumoral Genomic Heterogeneity of Multifocal Localized Prostate Cancer Impacts Molecular Classifications and Genomic Prognosticators. Eur. Urol. 2017, 71, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Banks, P.; Xu, W.; Murphy, D.; James, P.; Sandhu, S. Relevance of DNA Damage Repair in the Management of Prostate Cancer. Curr. Probl. Cancer 2017, 41, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Boysen, G.; Barbieri, C.E.; Bryant, H.E.; Castro, E.; Nelson, P.S.; Olmos, D.; Pritchard, C.C.; Rubin, M.A.; de Bono, J.S. DNA Repair in Prostate Cancer: Biology and Clinical Implications. Eur. Urol. 2017, 71, 417–425. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Yap, T.A.; Boss, D.S.; Carden, C.P.; Mergui-Roelvink, M.; Gourley, C.; De Greve, J.; Lubinski, J.; Shanley, S.; Messiou, C.; et al. Poly(Adp)-Ribose Polymerase Inhibition: Frequent Durable Responses in Brca Carrier Ovarian Cancer Correlating with Platinum-Free Interval. J. Clin. Oncol. 2010, 28, 2512–2519. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef] [PubMed]
- Beltran, H.; Eng, K.; Mosquera, J.M.; Sigaras, A.; Romanel, A.; Rennert, H.; Kossai, M.; Pauli, C.; Faltas, B.; Fontugne, J.; et al. Whole-Exome Sequencing of Metastatic Cancer and Biomarkers of Treatment Response. JAMA Oncol. 2015, 1, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.H.; Pritchard, C.C.; Boyd, T.; Nelson, P.S.; Montgomery, B. Biallelic Inactivation of Brca2 in Platinum-Sensitive Metastatic Castration-Resistant Prostate Cancer. Eur. Urol. 2016, 69, 992–995. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; Goldberg, M.S.; Konstantinopoulos, P.A.; D’Andrea, A.D. DNA Damage and Repair Biomarkers of Immunotherapy Response. Cancer Discov. 2017, 7, 675–693. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch Repair Deficiency Predicts Response of Solid Tumors to Pd-1 Blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.W.; Swales, K.; Decordova, S.; DeYoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A First-Time-in-Human Study of Gsk2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the Pi3k Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Redig, A.J.; Janne, P.A. Basket Trials and the Evolution of Clinical Trial Design in an Era of Genomic Medicine. J. Clin. Oncol. 2015, 33, 975–977. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.T.; Chau, C.H.; Price, D.K.; Figg, W.D. Precision Oncology Medicine: The Clinical Relevance of Patient-Specific Biomarkers Used to Optimize Cancer Treatment. J. Clin. Pharmacol. 2016, 56, 1484–1499. [Google Scholar] [CrossRef] [PubMed]
- Mathieson, I.; Reich, D. Differences in the Rare Variant Spectrum among Human Populations. PLoS Genet. 2017, 13, e1006581. [Google Scholar] [CrossRef] [PubMed]
- Lawrence, M.S.; Stojanov, P.; Mermel, C.H.; Robinson, J.T.; Garraway, L.A.; Golub, T.R.; Meyerson, M.; Gabriel, S.B.; Lander, E.S.; Getz, G. Discovery and Saturation Analysis of Cancer Genes across 21 Tumour Types. Nature 2014, 505, 495–501. [Google Scholar] [CrossRef] [PubMed]
- Haga, S.B. Impact of Limited Population Diversity of Genome-Wide Association Studies. Genet. Med. 2010, 12, 81–84. [Google Scholar] [CrossRef] [PubMed]
- National Institutes of Health. Nih Policy and Guidelines on the Inclusion of Women and Minorities as Subjects in Clinical Research—Amended 28 November 2017. Available online: https://grants.nih.gov/grants/funding/women_min/guidelines.htm (accessed on 19 March 2018).
- Knerr, S.; Wayman, D.; Bonham, V.L. Inclusion of Racial and Ethnic Minorities in Genetic Research: Advance the Spirit by Changing the Rules? J. Law Med. Ethics 2011, 39, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Patten, E. Who Is Multiracial? Depends on How You Ask; Pew Research Center: Washington, DC, USA, 2015. [Google Scholar]
- Bamshad, M.; Wooding, S.P. Signatures of Natural Selection in the Human Genome. Nat. Rev. Genet. 2003, 4, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Baharian, S.; Barakatt, M.; Gignoux, C.R.; Shringarpure, S.; Errington, J.; Blot, W.J.; Bustamante, C.D.; Kenny, E.E.; Williams, S.M.; Aldrich, M.C.; et al. The Great Migration and African-American Genomic Diversity. PLoS Genet. 2016, 12, e1006059. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.D.; Hirschman, C. The Changing Racial and Ethnic Composition of the Us Population: Emerging American Identities. Popul. Dev. Rev. 2009, 35, 1–51. [Google Scholar] [CrossRef] [PubMed]
- Elliott, C.; Brodwin, P. Identity and Genetic Ancestry Tracing. BMJ 2002, 325, 1469–1471. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bamshad, M.J.; Wooding, S.; Watkins, W.S.; Ostler, C.T.; Batzer, M.A.; Jorde, L.B. Human Population Genetic Structure and Inference of Group Membership. Am. J. Hum. Genet. 2003, 72, 578–589. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal Components Analysis Corrects for Stratification in Genome-Wide Association Studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of Population Structure Using Multilocus Genotype Data. Genetics 2000, 155, 945–959. [Google Scholar] [PubMed]
- Raj, A.; Stephens, M.; Pritchard, J.K. Faststructure: Variational Inference of Population Structure in Large Snp Data Sets. Genetics 2014, 197, 573–589. [Google Scholar] [CrossRef] [PubMed]
- Alexander, D.H.; Lange, K. Enhancements to the Admixture Algorithm for Individual Ancestry Estimation. BMC Bioinf. 2011, 12, 246. [Google Scholar] [CrossRef] [PubMed]
- Byun, J.; Han, Y.; Gorlov, I.P.; Busam, J.A.; Seldin, M.F.; Amos, C.I. Ancestry Inference Using Principal Component Analysis and Spatial Analysis: A Distance-Based Analysis to Account for Population Substructure. BMC Genom. 2017, 18, 789. [Google Scholar] [CrossRef] [PubMed]
- Polite, B.N.; Adams-Campbell, L.L.; Brawley, O.W.; Bickell, N.; Carethers, J.M.; Flowers, C.R.; Foti, M.; Gomez, S.L.; Griggs, J.J.; Lathan, C.S.; et al. Charting the Future of Cancer Health Disparities Research: A Position Statement from the American Association for Cancer Research, the American Cancer Society, the American Society of Clinical Oncology, and the National Cancer Institute. J. Clin. Oncol. 2017, 35, 3075–3082. [Google Scholar] [CrossRef] [PubMed]
- Race Ethnicity Genetics Working Group. The Use of Racial, Ethnic, and Ancestral Categories in Human Genetics Research. Am. J. Hum. Genet. 2005, 77, 519–532. [Google Scholar]




© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tan, S.-H.; Petrovics, G.; Srivastava, S. Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. Int. J. Mol. Sci. 2018, 19, 1255. https://doi.org/10.3390/ijms19041255
Tan S-H, Petrovics G, Srivastava S. Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. International Journal of Molecular Sciences. 2018; 19(4):1255. https://doi.org/10.3390/ijms19041255
Chicago/Turabian StyleTan, Shyh-Han, Gyorgy Petrovics, and Shiv Srivastava. 2018. "Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities" International Journal of Molecular Sciences 19, no. 4: 1255. https://doi.org/10.3390/ijms19041255
APA StyleTan, S.-H., Petrovics, G., & Srivastava, S. (2018). Prostate Cancer Genomics: Recent Advances and the Prevailing Underrepresentation from Racial and Ethnic Minorities. International Journal of Molecular Sciences, 19(4), 1255. https://doi.org/10.3390/ijms19041255