Targeting the Adenosinergic Axis in Chronic Lymphocytic Leukemia: A Way to Disrupt the Tumor Niche?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
ATP/ADO Balance: Shifting from Immune-Activating to Immune-Suppressive Signals
2. The Purinergic/Adenosinergic Systems in the Tumor Microenvironment
3. Expression of the Adenosinergic Axis Machinery on CLL Cells
3.1. CD39 and CD73
3.2. CD26 and Nucleoside Transporters
3.3. P2 and P1 Receptors
4. ADO Signaling in CLL Cells
4.1. ADO Signaling Modulates CLL Cells Homing
4.2. ADO Signaling Rescues CLL Cells from Spontaneous- or Drug-Induced Apoptosis
5. ADO in the CLL Niche Is Part of a Network of Micro-Environmental Signals
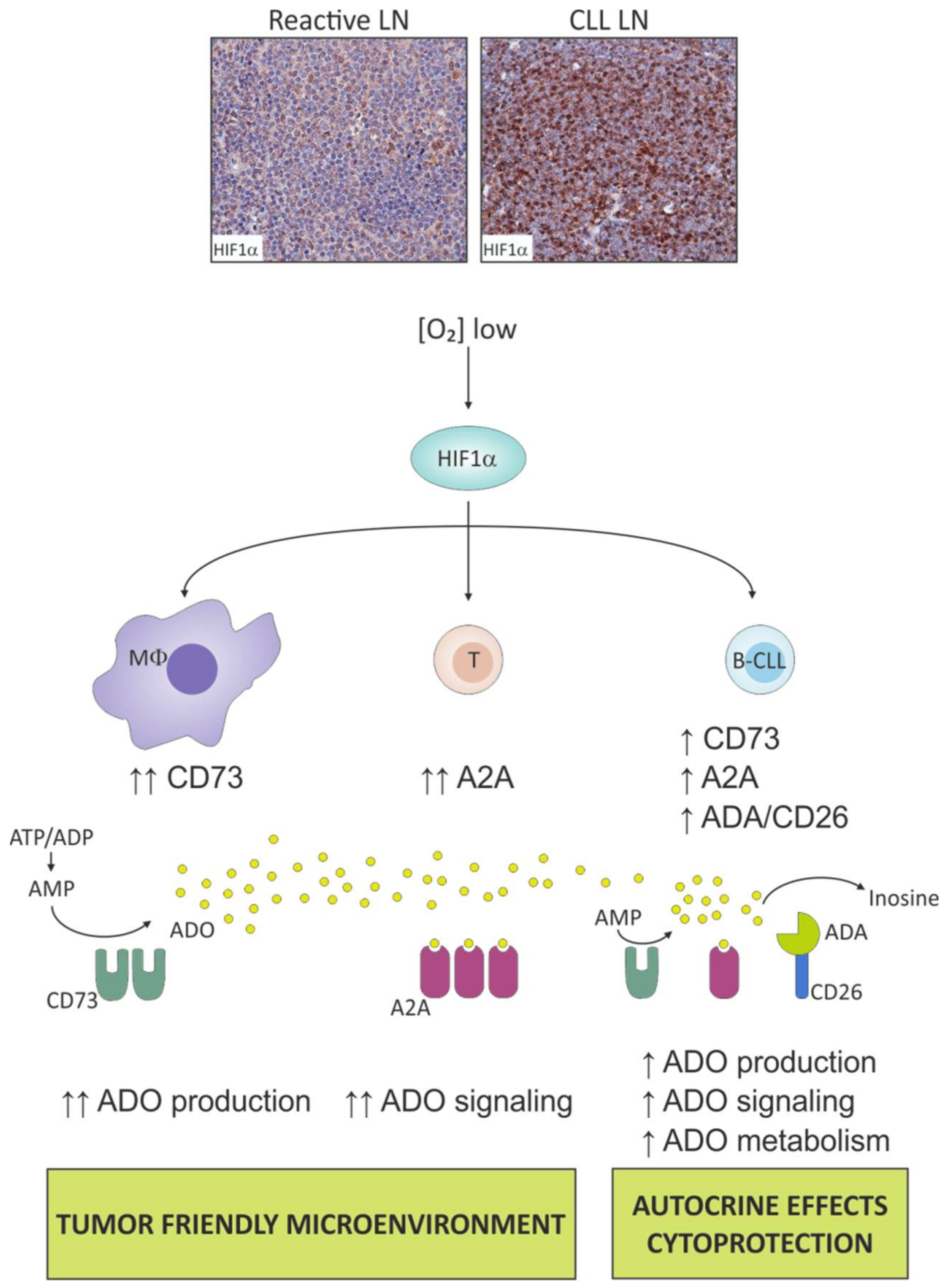
5.1. Hypoxia Boosts the Adenosinergic Axis in CLL Cells
5.2. Hypoxia Fosters the Adenosinergic Axis in “Not So Innocent” Bystander Cells of the Leukemic Niche
5.3. Hypoxia Contributes to Metabolic Skewing of Cells in the Leukemic Niche
5.4. Targeting the Adenosinergic Pathway
5.5. Future Perspectives
6. Concluding Remarks
Acknowledgments
Conflicts of Interest
References
- Trautmann, A. Extracellular ATP in the immune system: More than just a “danger signal”. Sci. Signal. 2009, 2, pe6. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Horenstein, A.L.; Vaisitti, T.; Brusa, D.; Rossi, D.; Laurenti, L.; D’Arena, G.; Coscia, M.; Tripodo, C.; Inghirami, G.; et al. CD73-generated extracellular adenosine in chronic lymphocytic leukemia creates local conditions counteracting drug-induced cell death. Blood 2011, 118, 6141–6152. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Vaisitti, T.; Audrito, V.; Bologna, C.; Buonincontri, R.; Chen, S.S.; Arruga, F.; Brusa, D.; Coscia, M.; Jaksic, O.; et al. Adenosine signaling mediates hypoxic responses in the chronic lymphocytic leukemia microenvironment. Blood Adv. 2016, 1, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Calissano, C.; Damle, R.N.; Hayes, G.; Murphy, E.J.; Hellerstein, M.K.; Moreno, C.; Sison, C.; Kaufman, M.S.; Kolitz, J.E.; Allen, S.L.; et al. In vivo intraclonal and interclonal kinetic heterogeneity in b-cell chronic lymphocytic leukemia. Blood 2009, 114, 4832–4842. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Gribben, J.G. The microenvironment in chronic lymphocytic leukemia (CLL) and other B cell malignancies: Insight into disease biology and new targeted therapies. Semin. Cancer Biol. 2014, 24, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Vaisitti, T.; Zucchetto, A.; Gattei, V.; Malavasi, F. CD38 as a molecular compass guiding topographical decisions of chronic lymphocytic leukemia cells. Semin. Cancer Biol. 2010, 20, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Nabhan, C.; Raca, G.; Wang, Y.L. Predicting prognosis in chronic lymphocytic leukemia in the contemporary era. JAMA Oncol. 2015, 1, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Chiaretti, S.; Marinelli, M.; Del Giudice, I.; Bonina, S.; Piciocchi, A.; Messina, M.; Vignetti, M.; Rossi, D.; Di Maio, V.; Mauro, F.R.; et al. NOTCH1, SF3B1, BIRC3 and TP53 mutations in patients with chronic lymphocytic leukemia undergoing first-line treatment: Correlation with biological parameters and response to treatment. Leuk. Lymphoma 2014, 55, 2785–2792. [Google Scholar] [CrossRef] [PubMed]
- Hallek, M. Chronic lymphocytic leukemia: 2017 update on diagnosis, risk stratification, and treatment. Am. J. Hematol. 2017, 92, 946–965. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.A.; Shanafelt, T.D. Prognostic factors and risk stratification in chronic lymphocytic leukemia. Semin. Oncol. 2016, 43, 233–240. [Google Scholar] [CrossRef] [PubMed]
- Gaidano, G.; Rossi, D. The mutational landscape of chronic lymphocytic leukemia and its impact on prognosis and treatment. Hematol. Am. Soc. Hematol. Educ. Program 2017, 2017, 329–337. [Google Scholar]
- Arruga, F.; Deaglio, S. Mechanisms of resistance to targeted therapies in chronic lymphocytic leukemia. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2017. [Google Scholar]
- Tripathi, R.; Lee-Verges, E.; Higashi, M.; Gimenez, N.; Rosich, L.; Lopez-Guerra, M.; Colomer, D. New drug discovery approaches targeting recurrent mutations in chronic lymphocytic leukemia. Expert Opin. Drug Discov. 2017, 12, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G. Physiology and pathophysiology of purinergic neurotransmission. Physiol. Rev. 2007, 87, 659–797. [Google Scholar] [CrossRef] [PubMed]
- Yegutkin, G.G. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Elliott, M.R.; Chekeni, F.B.; Trampont, P.C.; Lazarowski, E.R.; Kadl, A.; Walk, S.F.; Park, D.; Woodson, R.I.; Ostankovich, M.; Sharma, P.; et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009, 461, 282–286. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Varani, K.; Merighi, S.; Fogli, E.; Sacchetto, V.; Benini, A.; Leung, E.; Mac-Lennan, S.; Borea, P.A. Adenosine and lymphocyte regulation. Purinergic Signal. 2007, 3, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Stagg, J.; Darcy, P.K.; Smyth, M.J. CD73: A potent suppressor of antitumor immune responses. Trends Immunol. 2012, 33, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Robson, S.C.; Sevigny, J.; Zimmermann, H. The E-NTPDase family of ectonucleotidases: Structure function relationships and pathophysiological significance. Purinergic Signal. 2006, 2, 409–430. [Google Scholar] [CrossRef] [PubMed]
- Regateiro, F.S.; Cobbold, S.P.; Waldmann, H. CD73 and adenosine generation in the creation of regulatory microenvironments. Clin. Exp. Immunol. 2013, 171, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Merighi, S.; Sacchetto, V.; Simioni, C.; Borea, P.A. Adenosine receptors and cancer. Biochim. Biophys. Acta 2011, 1808, 1400–1412. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Gorelik, E.; Prasad, S.J.; Ronchese, F.; Lukashev, D.; Wong, M.K.; Huang, X.; Caldwell, S.; Liu, K.; Smith, P.; et al. A2A adenosine receptor protects tumors from antitumor T cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13132–13137. [Google Scholar] [CrossRef] [PubMed]
- Ohta, A.; Sitkovsky, M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature 2001, 414, 916–920. [Google Scholar] [CrossRef] [PubMed]
- Butler, M.; Sanmugalingam, D.; Burton, V.J.; Wilson, T.; Pearson, R.; Watson, R.P.; Smith, P.; Parkinson, S.J. Impairment of adenosine A3 receptor activity disrupts neutrophil migratory capacity and impacts innate immune function in vivo. Eur. J. Immunol. 2012, 42, 3358–3368. [Google Scholar] [CrossRef] [PubMed]
- Hoskin, D.W.; Mader, J.S.; Furlong, S.J.; Conrad, D.M.; Blay, J. Inhibition of T cell and natural killer cell function by adenosine and its contribution to immune evasion by tumor cells (review). Int. J. Oncol. 2008, 32, 527–535. [Google Scholar] [CrossRef] [PubMed]
- De Lera Ruiz, M.; Lim, Y.H.; Zheng, J. Adenosine A2A receptor as a drug discovery target. J. Med. Chem. 2014, 57, 3623–3650. [Google Scholar] [CrossRef] [PubMed]
- Abbracchio, M.P.; Burnstock, G.; Boeynaems, J.M.; Barnard, E.A.; Boyer, J.L.; Kennedy, C.; Knight, G.E.; Fumagalli, M.; Gachet, C.; Jacobson, K.A.; et al. International union of pharmacology LVIII: Update on the P2Y G protein-coupled nucleotide receptors: From molecular mechanisms and pathophysiology to therapy. Pharmacol. Rev. 2006, 58, 281–341. [Google Scholar] [CrossRef] [PubMed]
- Surprenant, A.; North, R.A. Signaling at purinergic P2X receptors. Annu. Rev. Physiol. 2009, 71, 333–359. [Google Scholar] [CrossRef] [PubMed]
- Coddou, C.; Yan, Z.; Obsil, T.; Huidobro-Toro, J.P.; Stojilkovic, S.S. Activation and regulation of purinergic P2X receptor channels. Pharmacol. Rev. 2011, 63, 641–683. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Knight, G.E. The potential of P2X7 receptors as a therapeutic target, including inflammation and tumour progression. Purinergic Signal. 2017, 14, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Burnstock, G.; Di Virgilio, F. Purinergic signalling and cancer. Purinergic Signal. 2013, 9, 491–540. [Google Scholar] [CrossRef] [PubMed]
- Junger, W.G. Immune cell regulation by autocrine purinergic signalling. Nat. Rev. Immunol. 2011, 11, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Vijayan, D.; Young, A.; Teng, M.W.L.; Smyth, M.J. Targeting immunosuppressive adenosine in cancer. Nat. Rev. Cancer 2017, 17, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Di Virgilio, F.; Adinolfi, E. Extracellular purines, purinergic receptors and tumor growth. Oncogene 2017, 36, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Kumar, V. Adenosine as an endogenous immunoregulator in cancer pathogenesis: Where to go? Purinergic Signal. 2013, 9, 145–165. [Google Scholar] [CrossRef] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef] [PubMed]
- Cairns, R.A.; Harris, I.S.; Mak, T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. [Google Scholar] [CrossRef] [PubMed]
- Young, A.; Mittal, D.; Stagg, J.; Smyth, M.J. Targeting cancer-derived adenosine: New therapeutic approaches. Cancer Discov. 2014, 4, 879–888. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Blandizzi, C.; Pacher, P.; Hasko, G. Immunity, inflammation and cancer: A leading role for adenosine. Nat. Rev. Cancer 2013, 13, 842–857. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Hasko, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Linden, J. Adenosine metabolism and cancer. Focus on “adenosine downregulates DPPIV on HT-29 colon cancer cells by stimulating protein tyrosine phosphatases and reducing ERK1/2 activity via a novel pathway”. Am. J. Physiol. Cell Physiol. 2006, 291, C405–C406. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Smyth, M.J. Extracellular adenosine triphosphate and adenosine in cancer. Oncogene 2010, 29, 5346–5358. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.; Bar-Yehuda, S.; Synowitz, M.; Powell, J.D.; Klotz, K.N.; Gessi, S.; Borea, P.A. Adenosine receptors and cancer. In Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; pp. 399–441. [Google Scholar]
- Wang, L.; Fan, J.; Thompson, L.F.; Zhang, Y.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 has distinct roles in nonhematopoietic and hematopoietic cells to promote tumor growth in mice. J. Clin. Investig. 2011, 121, 2371–2382. [Google Scholar] [CrossRef] [PubMed]
- Mikhailov, A.; Sokolovskaya, A.; Yegutkin, G.G.; Amdahl, H.; West, A.; Yagita, H.; Lahesmaa, R.; Thompson, L.F.; Jalkanen, S.; Blokhin, D.; et al. CD73 participates in cellular multiresistance program and protects against trail-induced apoptosis. J. Immunol. 2008, 181, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Mittal, D.; Sinha, D.; Barkauskas, D.; Young, A.; Kalimutho, M.; Stannard, K.; Caramia, F.; Haibe-Kains, B.; Stagg, J.; Khanna, K.K.; et al. Adenosine 2B receptor expression on cancer cells promotes metastasis. Cancer Res. 2016, 76, 4372–4382. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Longhi, M.S.; Robson, S.C.; Stagg, J. The ectonucleotidases CD39 and CD73: Novel checkpoint inhibitor targets. Immunol. Rev. 2017, 276, 121–144. [Google Scholar] [CrossRef] [PubMed]
- Caligaris-Cappio, F.; Ghia, P. Novel insights in chronic lymphocytic leukemia: Are we getting closer to understanding the pathogenesis of the disease? J. Clin. Oncol. 2008, 26, 4497–4503. [Google Scholar] [CrossRef] [PubMed]
- Soma, L.A.; Craig, F.E.; Swerdlow, S.H. The proliferation center microenvironment and prognostic markers in chronic lymphocytic leukemia/small lymphocytic lymphoma. Hum. Pathol. 2006, 37, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Seiffert, M.; Schulz, A.; Ohl, S.; Dohner, H.; Stilgenbauer, S.; Lichter, P. Soluble CD14 is a novel monocyte-derived survival factor for chronic lymphocytic leukemia cells, which is induced by CLL cells in vitro and present at abnormally high levels in vivo. Blood 2010, 116, 4223–4230. [Google Scholar] [CrossRef] [PubMed]
- Bennett, F.; Rawstron, A.; Plummer, M.; de Tute, R.; Moreton, P.; Jack, A.; Hillmen, P. B-cell chronic lymphocytic leukaemia cells show specific changes in membrane protein expression during different stages of cell cycle. Br. J. Haematol. 2007, 139, 600–604. [Google Scholar] [CrossRef] [PubMed]
- Damle, R.N.; Ghiotto, F.; Valetto, A.; Albesiano, E.; Fais, F.; Yan, X.J.; Sison, C.P.; Allen, S.L.; Kolitz, J.; Schulman, P.; et al. B-cell chronic lymphocytic leukemia cells express a surface membrane phenotype of activated, antigen-experienced B lymphocytes. Blood 2002, 99, 4087–4093. [Google Scholar] [CrossRef] [PubMed]
- Abousamra, N.K.; Salah El-Din, M.; Hamza Elzahaf, E.; Esmael, M.E. Ectonucleoside triphosphate diphosphohydrolase-1 (E-NTPDase1/CD39) as a new prognostic marker in chronic lymphocytic leukemia. Leuk. Lymphoma 2015, 56, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Olson, K.E.; Broekman, M.J.; Islam, N.; Ballard, H.S.; Furman, R.R.; Olson, A.E.; Marcus, A.J. CD39 activity correlates with stage and inhibits platelet reactivity in chronic lymphocytic leukemia. J. Transl. Med. 2007, 5, 23. [Google Scholar] [CrossRef] [PubMed]
- Pulte, D.; Furman, R.R.; Broekman, M.J.; Drosopoulos, J.H.; Ballard, H.S.; Olson, K.E.; Kizer, J.R.; Marcus, A.J. CD39 expression on T lymphocytes correlates with severity of disease in patients with chronic lymphocytic leukemia. Clin. Lymphoma Myeloma Leuk. 2011, 11, 367–372. [Google Scholar] [CrossRef] [PubMed]
- Perry, C.; Hazan-Halevy, I.; Kay, S.; Cipok, M.; Grisaru, D.; Deutsch, V.; Polliack, A.; Naparstek, E.; Herishanu, Y. Increased CD39 expression on CD4+ T lymphocytes has clinical and prognostic significance in chronic lymphocytic leukemia. Ann. Hematol. 2012, 91, 1271–1279. [Google Scholar] [CrossRef] [PubMed]
- Jak, M.; Mous, R.; Remmerswaal, E.B.; Spijker, R.; Jaspers, A.; Yague, A.; Eldering, E.; Van Lier, R.A.; Van Oers, M.H. Enhanced formation and survival of CD4+ CD25hi Foxp3+ T-cells in chronic lymphocytic leukemia. Leuk. Lymphoma 2009, 50, 788–801. [Google Scholar] [CrossRef] [PubMed]
- Mackey, J.R.; Galmarini, C.M.; Graham, K.A.; Joy, A.A.; Delmer, A.; Dabbagh, L.; Glubrecht, D.; Jewell, L.D.; Lai, R.; Lang, T.; et al. Quantitative analysis of nucleoside transporter and metabolism gene expression in chronic lymphocytic leukemia (CLL): Identification of fludarabine-sensitive and -insensitive populations. Blood 2005, 105, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Deaglio, S. HPLC-based assay to monitor extracellular nucleotide/nucleoside metabolism in human chronic lymphocytic leukemia cells. J. Vis. Exp. 2016. [Google Scholar] [CrossRef] [PubMed]
- Pastor-Anglada, M.; Molina-Arcas, M.; Casado, F.J.; Bellosillo, B.; Colomer, D.; Gil, J. Nucleoside transporters in chronic lymphocytic leukaemia. Leukemia 2004, 18, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Cortes, A.; Gracia, E.; Moreno, E.; Mallol, J.; Lluis, C.; Canela, E.I.; Casado, V. Moonlighting adenosine deaminase: A target protein for drug development. Med. Res. Rev. 2015, 35, 85–125. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Umeki, K.; Yamamoto, I.; Sakamoto, F.; Noguchi, S.; Ohtaki, S. CD26 (dipeptidyl peptidase IV/DPP IV) as a novel molecular marker for differentiated thyroid carcinoma. Int. J. Cancer 1995, 64, 326–331. [Google Scholar] [CrossRef] [PubMed]
- Stecca, B.A.; Nardo, B.; Chieco, P.; Mazziotti, A.; Bolondi, L.; Cavallari, A. Aberrant dipeptidyl peptidase IV (DPP IV/CD26) expression in human hepatocellular carcinoma. J. Hepatol. 1997, 27, 337–345. [Google Scholar] [CrossRef]
- Cheung, A.H.; Iyer, D.N.; Lam, C.S.; Ng, L.; Wong, S.K.M.; Lee, H.S.; Wan, T.; Man, J.; Chow, A.K.M.; Poon, R.T.; et al. Emergence of CD26+ cancer stem cells with metastatic properties in colorectal carcinogenesis. Int. J. Mol. Sci. 2017, 18, 1106. [Google Scholar] [CrossRef] [PubMed]
- Angevin, E.; Isambert, N.; Trillet-Lenoir, V.; You, B.; Alexandre, J.; Zalcman, G.; Vielh, P.; Farace, F.; Valleix, F.; Podoll, T.; et al. First-in-human phase 1 of YS110, a monoclonal antibody directed against CD26 in advanced CD26-expressing cancers. Br. J. Cancer 2017, 116, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Matuszak, M.; Lewandowski, K.; Czyz, A.; Kiernicka-Parulska, J.; Przybylowicz-Chalecka, A.; Jarmuz-Szymczak, M.; Lewandowska, M.; Komarnicki, M. The prognostic significance of surface dipeptidylpeptidase IV (CD26) expression in B-cell chronic lymphocytic leukemia. Leuk. Res. 2016, 47, 166–171. [Google Scholar] [CrossRef] [PubMed]
- Ibrahem, L.; Elderiny, W.E.; Elhelw, L.; Ismail, M. CD49d and CD26 are independent prognostic markers for disease progression in patients with chronic lymphocytic leukemia. Blood Cells Mol. Dis. 2015, 55, 154–160. [Google Scholar] [CrossRef] [PubMed]
- Cro, L.; Morabito, F.; Zucal, N.; Fabris, S.; Lionetti, M.; Cutrona, G.; Rossi, F.; Gentile, M.; Ferrario, A.; Ferrarini, M.; et al. CD26 expression in mature B-cell neoplasia: Its possible role as a new prognostic marker in B-CLL. Hematol. Oncol. 2009, 27, 140–147. [Google Scholar] [CrossRef] [PubMed]
- Durinx, C.; Lambeir, A.M.; Bosmans, E.; Falmagne, J.B.; Berghmans, R.; Haemers, A.; Scharpe, S.; De Meester, I. Molecular characterization of dipeptidyl peptidase activity in serum: Soluble CD26/dipeptidyl peptidase IV is responsible for the release of X-Pro dipeptides. Eur. J. Biochem. 2000, 267, 5608–5613. [Google Scholar] [CrossRef] [PubMed]
- Molica, S.; Digiesi, G.; Mirabelli, R.; Cutrona, G.; Antenucci, A.; Molica, M.; Giannarelli, D.; Sperduti, I.; Morabito, F.; Neri, A.; et al. Serum level of CD26 predicts time to first treatment in early B-chronic lymphocytic leukemia. Eur. J. Haematol. 2009, 83, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Bellosillo, B.; Casado, F.J.; Montserrat, E.; Gil, J.; Colomer, D.; Pastor-Anglada, M. Fludarabine uptake mechanisms in B-cell chronic lymphocytic leukemia. Blood 2003, 101, 2328–2334. [Google Scholar] [CrossRef] [PubMed]
- Molina-Arcas, M.; Marce, S.; Villamor, N.; Huber-Ruano, I.; Casado, F.J.; Bellosillo, B.; Montserrat, E.; Gil, J.; Colomer, D.; Pastor-Anglada, M. Equilibrative nucleoside transporter-2 (hENT2) protein expression correlates with ex vivo sensitivity to fludarabine in chronic lymphocytic leukemia (CLL) cells. Leukemia 2005, 19, 64–68. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Capece, M.; Amoroso, F.; De Marchi, E.; Franceschini, A. Emerging roles of P2X receptors in cancer. Curr. Med. Chem. 2015, 22, 878–890. [Google Scholar] [CrossRef] [PubMed]
- Roger, S.; Jelassi, B.; Couillin, I.; Pelegrin, P.; Besson, P.; Jiang, L.H. Understanding the roles of the P2X7 receptor in solid tumour progression and therapeutic perspectives. Biochim. Biophys. Acta 2015, 1848, 2584–2602. [Google Scholar] [CrossRef] [PubMed]
- Markwardt, F.; Lohn, M.; Bohm, T.; Klapperstuck, M. Purinoceptor-operated cationic channels in human B lymphocytes. J. Physiol. 1997, 498 Pt 1, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.; Munerati, M.; Melchiorri, L.; Hanau, S.; di Virgilio, F.; Baricordi, O.R. Responses to extracellular ATP of lymphoblastoid cell lines from duchenne muscular dystrophy patients. Am. J. Physiol. 1994, 267, C886–C892. [Google Scholar] [CrossRef] [PubMed]
- Ghalali, A.; Wiklund, F.; Zheng, H.; Stenius, U.; Hogberg, J. Atorvastatin prevents ATP-driven invasiveness via P2X7 and EHBP1 signaling in PTEN-expressing prostate cancer cells. Carcinogenesis 2014, 35, 1547–1555. [Google Scholar] [CrossRef] [PubMed]
- Chong, J.H.; Zheng, G.G.; Zhu, X.F.; Guo, Y.; Wang, L.; Ma, C.H.; Liu, S.Y.; Xu, L.L.; Lin, Y.M.; Wu, K.F. Abnormal expression of P2X family receptors in chinese pediatric acute leukemias. Biochem. Biophys. Res. Commun. 2010, 391, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Cabrini, G.; Falzoni, S.; Forchap, S.L.; Pellegatti, P.; Balboni, A.; Agostini, P.; Cuneo, A.; Castoldi, G.; Baricordi, O.R.; Di Virgilio, F. A His-155 to Tyr polymorphism confers gain-of-function to the human P2X7 receptor of human leukemic lymphocytes. J. Immunol. 2005, 175, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Adinolfi, E.; Melchiorri, L.; Falzoni, S.; Chiozzi, P.; Morelli, A.; Tieghi, A.; Cuneo, A.; Castoldi, G.; Di Virgilio, F.; Baricordi, O.R. P2X7 receptor expression in evolutive and indolent forms of chronic B lymphocytic leukemia. Blood 2002, 99, 706–708. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.S.; Dao-Ung, L.P.; Gu, B.J.; Sluyter, R.; Shemon, A.N.; Li, C.; Taper, J.; Gallo, J.; Manoharan, A. A loss-of-function polymorphic mutation in the cytolytic P2X7 receptor gene and chronic lymphocytic leukaemia: A molecular study. Lancet 2002, 359, 1114–1119. [Google Scholar] [CrossRef]
- Baricordi, O.R.; Melchiorri, L.; Adinolfi, E.; Falzoni, S.; Chiozzi, P.; Buell, G.; Di Virgilio, F. Increased proliferation rate of lymphoid cells transfected with the P2X(7) ATP receptor. J. Biol. Chem. 1999, 274, 33206–33208. [Google Scholar] [CrossRef] [PubMed]
- Wiley, J.S.; Dubyak, G.R. Extracellular adenosine triphosphate increases cation permeability of chronic lymphocytic leukemic lymphocytes. Blood 1989, 73, 1316–1323. [Google Scholar] [PubMed]
- Gu, B.J.; Zhang, W.; Worthington, R.A.; Sluyter, R.; Dao-Ung, P.; Petrou, S.; Barden, J.A.; Wiley, J.S. A Glu-496 to Ala polymorphism leads to loss of function of the human P2X7 receptor. J. Biol. Chem. 2001, 276, 11135–11142. [Google Scholar] [CrossRef] [PubMed]
- Thunberg, U.; Tobin, G.; Johnson, A.; Soderberg, O.; Padyukov, L.; Hultdin, M.; Klareskog, L.; Enblad, G.; Sundstrom, C.; Roos, G.; et al. Polymorphism in the P2X7 receptor gene and survival in chronic lymphocytic leukaemia. Lancet 2002, 360, 1935–1939. [Google Scholar] [CrossRef]
- Starczynski, J.; Pepper, C.; Pratt, G.; Hooper, L.; Thomas, A.; Hoy, T.; Milligan, D.; Bentley, P.; Fegan, C. The P2X7 receptor gene polymorphism 1513 A→C has no effect on clinical prognostic markers, in vitro sensitivity to fludarabine, bcl-2 family protein expression or survival in b-cell chronic lymphocytic leukaemia. Br. J. Haematol. 2003, 123, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.Y.; Ibbotson, R.E.; Orchard, J.A.; Gardiner, A.C.; Seear, R.V.; Chase, A.J.; Oscier, D.G.; Cross, N.C. P2X7 polymorphism and chronic lymphocytic leukaemia: Lack of correlation with incidence, survival and abnormalities of chromosome 12. Leukemia 2003, 17, 2097–2100. [Google Scholar] [CrossRef] [PubMed]
- Nuckel, H.; Frey, U.H.; Durig, J.; Duhrsen, U.; Siffert, W. 1513A/C polymorphism in the P2X7 receptor gene in chronic lymphocytic leukemia: Absence of correlation with clinical outcome. Eur. J. Haematol. 2004, 72, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Hasko, G.; Linden, J.; Cronstein, B.; Pacher, P. Adenosine receptors: Therapeutic aspects for inflammatory and immune diseases. Nat. Rev. Drug Discov. 2008, 7, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Ernst, P.B.; Garrison, J.C.; Thompson, L.F. Much ado about adenosine: Adenosine synthesis and function in regulatory T cell biology. J. Immunol. 2010, 185, 1993–1998. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chu, X.; Deng, F.; Tong, L.; Tong, G.; Yi, Y.; Liu, J.; Tang, J.; Tang, Y.; Xia, Y.; et al. The adenosine A2B receptor promotes tumor progression of bladder urothelial carcinoma by enhancing MAPK signaling pathway. Oncotarget 2017, 8, 48755–48768. [Google Scholar] [CrossRef] [PubMed]
- Joos, G.; Jakim, J.; Kiss, B.; Szamosi, R.; Papp, T.; Felszeghy, S.; Saghy, T.; Nagy, G.; Szondy, Z. Involvement of adenosine A3 receptors in the chemotactic navigation of macrophages towards apoptotic cells. Immunol. Lett. 2017, 183, 62–72. [Google Scholar] [CrossRef] [PubMed]
- By, Y.; Durand-Gorde, J.M.; Condo, J.; Lejeune, P.J.; Fenouillet, E.; Guieu, R.; Ruf, J. Monoclonal antibody-assisted stimulation of adenosine A2A receptors induces simultaneous downregulation of CXCR4 and CCR5 on CD4+ T-cells. Hum. Immunol. 2010, 71, 1073–1076. [Google Scholar] [CrossRef] [PubMed]
- Zhang, N.; Yang, D.; Dong, H.; Chen, Q.; Dimitrova, D.I.; Rogers, T.J.; Sitkovsky, M.; Oppenheim, J.J. Adenosine A2A receptors induce heterologous desensitization of chemokine receptors. Blood 2006, 108, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Burger, J.A.; Kipps, T.J. Cxcr4: A key receptor in the crosstalk between tumor cells and their microenvironment. Blood 2006, 107, 1761–1767. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Hoffmann, C.; Cattabeni, F.; Abbracchio, M.P. Adenosine-induced cell death: Evidence for receptor-mediated signalling. Apoptosis 1999, 4, 197–211. [Google Scholar] [CrossRef] [PubMed]
- Himer, L.; Csoka, B.; Selmeczy, Z.; Koscso, B.; Pocza, T.; Pacher, P.; Nemeth, Z.H.; Deitch, E.A.; Vizi, E.S.; Cronstein, B.N.; et al. Adenosine A2A receptor activation protects CD4+ T lymphocytes against activation-induced cell death. FASEB J. 2010, 24, 2631–2640. [Google Scholar] [CrossRef] [PubMed]
- Kalouche, G.; Boucher, C.; Coste, A.; Debussche, L.; Orsini, C.; Baudouin, C.; Debeir, T.; Vige, X.; Rostene, W. Prostaglandin EP2 receptor signaling protects human trabecular meshwork cells from apoptosis induced by ER stress through down-regulation of p53. Biochim. Biophys. Acta 2016, 1863, 2322–2332. [Google Scholar] [CrossRef] [PubMed]
- Naderi, A.; Chia, K.M.; Liu, J. Synergy between inhibitors of androgen receptor and mek has therapeutic implications in estrogen receptor-negative breast cancer. Breast Cancer Res. 2011, 13, R36. [Google Scholar] [CrossRef] [PubMed]
- Shachar, I.; Cohen, S.; Marom, A.; Becker-Herman, S. Regulation of CLL survival by hypoxia-inducible factor and its target genes. FEBS Lett. 2012, 586, 2906–2910. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Valsecchi, R.; Coltella, N.; Belloni, D.; Ponente, M.; Ten Hacken, E.; Scielzo, C.; Scarfo, L.; Bertilaccio, M.T.; Brambilla, P.; Lenti, E.; et al. HIF-1α regulates the interaction of chronic lymphocytic leukemia cells with the tumor microenvironment. Blood 2016, 127, 1987–1997. [Google Scholar] [CrossRef] [PubMed]
- Kontos, C.K.; Papageorgiou, S.G.; Diamantopoulos, M.A.; Scorilas, A.; Bazani, E.; Vasilatou, D.; Gkontopoulos, K.; Glezou, E.; Stavroulaki, G.; Dimitriadis, G.; et al. mRNA overexpression of the hypoxia inducible factor 1 alpha subunit gene (HIF1A): An independent predictor of poor overall survival in chronic lymphocytic leukemia. Leuk. Res. 2017, 53, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, W.; Wang, Y.; Zou, T.; Zhang, B.; Xu, Y.; Pang, T.; Hu, Q.; Chen, M.; Wang, L.; et al. Hypoxia-induced miR-214 expression promotes tumour cell proliferation and migration by enhancing the Warburg effect in gastric carcinoma cells. Cancer Lett. 2018, 414, 44–56. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.F.; Poonit, N.; Zhang, Y.C.; Ye, C.Y.; Cai, H.L.; Yu, C.Y.; Zhou, Y.H.; Wu, B.B.; Cai, J.; Cai, X.H. Activating adenosine A1 receptor accelerates PC12 cell injury via ADORA1/PKC/KATP pathway after intermittent hypoxia exposure. Mol. Cell. Biochem. 2018, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Multhoff, G. Accomplices of the hypoxic tumor microenvironment compromising antitumor immunity: Adenosine, lactate, acidosis, vascular endothelial growth factor, potassium ions, and phosphatidylserine. Front. Immunol. 2017, 8, 1887. [Google Scholar] [CrossRef] [PubMed]
- Bowser, J.L.; Phan, L.H.; Eltzschig, H.K. The hypoxia-adenosine link during intestinal inflammation. J. Immunol. 2018, 200, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Kjaergaard, J.; Lukashev, D.; Ohta, A. Hypoxia-adenosinergic immunosuppression: Tumor protection by T regulatory cells and cancerous tissue hypoxia. Clin. Cancer Res. 2008, 14, 5947–5952. [Google Scholar] [CrossRef] [PubMed]
- Poth, J.M.; Brodsky, K.; Ehrentraut, H.; Grenz, A.; Eltzschig, H.K. Transcriptional control of adenosine signaling by hypoxia-inducible transcription factors during ischemic or inflammatory disease. J. Mol. Med. 2013, 91, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Synnestvedt, K.; Furuta, G.T.; Comerford, K.M.; Louis, N.; Karhausen, J.; Eltzschig, H.K.; Hansen, K.R.; Thompson, L.F.; Colgan, S.P. Ecto-5′-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J. Clin. Investig. 2002, 110, 993–1002. [Google Scholar] [CrossRef] [PubMed]
- Boissard, F.; Fournie, J.J.; Laurent, C.; Poupot, M.; Ysebaert, L. Nurse like cells: Chronic lymphocytic leukemia associated macrophages. Leuk. Lymphoma 2015, 56, 1570–1572. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Romano, A.; De Simone, S.; Procaccini, C.; La Rocca, C.; et al. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J. Clin. Investig. 2013, 123, 3664–3671. [Google Scholar] [CrossRef] [PubMed]
- Koczula, K.M.; Ludwig, C.; Hayden, R.; Cronin, L.; Pratt, G.; Parry, H.; Tennant, D.; Drayson, M.; Bunce, C.M.; Khanim, F.L.; et al. Metabolic plasticity in CLL: Adaptation to the hypoxic niche. Leukemia 2016, 30, 65–73. [Google Scholar] [CrossRef] [PubMed]
- DiLillo, D.J.; Weinberg, J.B.; Yoshizaki, A.; Horikawa, M.; Bryant, J.M.; Iwata, Y.; Matsushita, T.; Matta, K.M.; Chen, Y.; Venturi, G.M.; et al. Chronic lymphocytic leukemia and regulatory B cells share IL-10 competence and immunosuppressive function. Leukemia 2013, 27, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Beavis, P.A.; Divisekera, U.; Liu, M.C.; Moller, A.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice are resistant to carcinogenesis. Cancer Res. 2012, 72, 2190–2196. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Divisekera, U.; Duret, H.; Sparwasser, T.; Teng, M.W.; Darcy, P.K.; Smyth, M.J. CD73-deficient mice have increased antitumor immunity and are resistant to experimental metastasis. Cancer Res. 2011, 71, 2892–2900. [Google Scholar] [CrossRef] [PubMed]
- Stagg, J.; Divisekera, U.; McLaughlin, N.; Sharkey, J.; Pommey, S.; Denoyer, D.; Dwyer, K.M.; Smyth, M.J. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 1547–1552. [Google Scholar] [CrossRef] [PubMed]
- Hilchey, S.P.; Kobie, J.J.; Cochran, M.R.; Secor-Socha, S.; Wang, J.C.; Hyrien, O.; Burack, W.R.; Mosmann, T.R.; Quataert, S.A.; Bernstein, S.H. Human follicular lymphoma CD39+-infiltrating T cells contribute to adenosine-mediated T cell hyporesponsiveness. J. Immunol. 2009, 183, 6157–6166. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Martins, I.; Sukkurwala, A.Q.; Adjemian, S.; Ma, Y.; Pellegatti, P.; Shen, S.; Kepp, O.; Scoazec, M.; Mignot, G.; et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011, 334, 1573–1577. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Wu, Y.; Gao, W.; Enjyoji, K.; Csizmadia, E.; Muller, C.E.; Murakami, T.; Robson, S.C. CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 2010, 139, 1030–1040. [Google Scholar] [CrossRef] [PubMed]
- Bastid, J.; Cottalorda-Regairaz, A.; Alberici, G.; Bonnefoy, N.; Eliaou, J.F.; Bensussan, A. ENTPD1/CD39 is a promising therapeutic target in oncology. Oncogene 2013, 32, 1743–1751. [Google Scholar] [CrossRef] [PubMed]
- Hayes, G.M.; Cairns, B.; Levashova, Z.; Chinn, L.; Perez, M.; Theunissen, J.W.; Liao-Chan, S.; Bermudez, A.; Flory, M.R.; Schweighofer, K.J.; et al. CD39 is a promising therapeutic antibody target for the treatment of soft tissue sarcoma. Am. J. Transl. Res. 2015, 7, 1181–1188. [Google Scholar] [PubMed]
- Hausler, S.F.; Del Barrio, I.M.; Diessner, J.; Stein, R.G.; Strohschein, J.; Honig, A.; Dietl, J.; Wischhusen, J. Anti-CD39 and anti-CD73 antibodies A1 and 7G2 improve targeted therapy in ovarian cancer by blocking adenosine-dependent immune evasion. Am. J. Transl. Res. 2014, 6, 129–139. [Google Scholar] [PubMed]
- Loi, S.; Pommey, S.; Haibe-Kains, B.; Beavis, P.A.; Darcy, P.K.; Smyth, M.J.; Stagg, J. CD73 promotes anthracycline resistance and poor prognosis in triple negative breast cancer. Proc. Natl. Acad. Sci. USA 2013, 110, 11091–11096. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Turcotte, M.; Spring, K.; Pommey, S.; Royal, I.; Stagg, J. Anti-CD73 therapy impairs tumor angiogenesis. Int. J. Cancer 2014, 134, 1466–1473. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Pommey, S.; Smyth, M.J.; Stagg, J. Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin. Cancer Res. 2013, 19, 5626–5635. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, L.; Yegutkin, G.G.; Pacher, P.; Blandizzi, C.; Hasko, G. Anti-CD73 in cancer immunotherapy: Awakening new opportunities. Trends Cancer 2016, 2, 95–109. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.; Fan, J.; Wang, L.; Thompson, L.F.; Liu, A.; Daniel, B.J.; Shin, T.; Curiel, T.J.; Zhang, B. CD73 on tumor cells impairs antitumor T-cell responses: A novel mechanism of tumor-induced immune suppression. Cancer Res. 2010, 70, 2245–2255. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Zhi, X.; Zhou, P.; Chen, S.; Zhao, F.; Shao, Z.; Ou, Z.; Yin, L. Effects of ecto-5′-nucleotidase on human breast cancer cell growth in vitro and in vivo. Oncol. Rep. 2007, 17, 1341–1346. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hay, C.M.; Sult, E.; Huang, Q.; Mulgrew, K.; Fuhrmann, S.R.; McGlinchey, K.A.; Hammond, S.A.; Rothstein, R.; Rios-Doria, J.; Poon, E.; et al. Targeting CD73 in the tumor microenvironment with MEDI9447. Oncoimmunology 2016, 5, e1208875. [Google Scholar] [CrossRef] [PubMed]
- Barnhart, B.C.; Sega, E.; Yamniuk, A.; Hatcher, S.; Lei, M.; Ghermazien, H.; Lewin, A.; Wang, X.T.; Huang, H.; Zhang, P.; et al. Abstract 1476: A therapeutic antibody that inhibits CD73 activity by dual mechanisms. Cancer Res. 2016, 76. [Google Scholar] [CrossRef]
- Beavis, P.A.; Milenkovski, N.; Henderson, M.A.; John, L.B.; Allard, B.; Loi, S.; Kershaw, M.H.; Stagg, J.; Darcy, P.K. Adenosine receptor 2A blockade increases the efficacy of anti-PD-1 through enhanced antitumor T-cell responses. Cancer Immunol. Res. 2015, 3, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Beavis, P.A.; Divisekera, U.; Paget, C.; Chow, M.T.; John, L.B.; Devaud, C.; Dwyer, K.; Stagg, J.; Smyth, M.J.; Darcy, P.K. Blockade of A2A receptors potently suppresses the metastasis of CD73+ tumors. Proc. Natl. Acad. Sci. USA 2013, 110, 14711–14716. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Alme, A.; Senaldi, L.; Zarek, P.E.; Horton, M.; Powell, J.D. Enhancement of tumor immunotherapy by deletion of the A2A adenosine receptor. Cancer Immunol. Immunother. 2012, 61, 917–926. [Google Scholar] [CrossRef] [PubMed]
- Cekic, C.; Sag, D.; Li, Y.; Theodorescu, D.; Strieter, R.M.; Linden, J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J. Immunol. 2012, 188, 198–205. [Google Scholar] [CrossRef] [PubMed]
- Iannone, R.; Miele, L.; Maiolino, P.; Pinto, A.; Morello, S. Blockade of A2B adenosine receptor reduces tumor growth and immune suppression mediated by myeloid-derived suppressor cells in a mouse model of melanoma. Neoplasia 2013, 15, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Ryzhov, S.; Novitskiy, S.V.; Zaynagetdinov, R.; Goldstein, A.E.; Carbone, D.P.; Biaggioni, I.; Dikov, M.M.; Feoktistov, I. Host A2B adenosine receptors promote carcinoma growth. Neoplasia 2008, 10, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.J.; Tan, J.B.; Becker, A.; Yi, F.; Leleti, M.L.; Rosen, B.; Sharif, E.; Debien, L.; Young, S.; Lim, W.H.; et al. Abstract 4572: Characterization of the potent and selective A2AR antagonist AB928 for the treatment of cancer. Cancer Res. 2017, 77. [Google Scholar] [CrossRef]
- Sitkovsky, M.; Ohta, A. Targeting the hypoxia-adenosinergic signaling pathway to improve the adoptive immunotherapy of cancer. J. Mol. Med. 2013, 91, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Allard, B.; Beavis, P.A.; Darcy, P.K.; Stagg, J. Immunosuppressive activities of adenosine in cancer. Curr. Opin. Pharmacol. 2016, 29, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Allard, D.; Allard, B.; Gaudreau, P.O.; Chrobak, P.; Stagg, J. CD73-adenosine: A next-generation target in immuno-oncology. Immunotherapy 2016, 8, 145–163. [Google Scholar] [CrossRef] [PubMed]
- Sitkovsky, M.V.; Hatfield, S.; Abbott, R.; Belikoff, B.; Lukashev, D.; Ohta, A. Hostile, hypoxia-A2-adenosinergic tumor biology as the next barrier to overcome for tumor immunologists. Cancer Immunol. Res. 2014, 2, 598–605. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Kepp, O.; Zitvogel, L. Immunogenic cell death in cancer therapy. Annu. Rev. Immunol. 2013, 31, 51–72. [Google Scholar] [CrossRef] [PubMed]
- Verbrugge, I.; Hagekyriakou, J.; Sharp, L.L.; Galli, M.; West, A.; McLaughlin, N.M.; Duret, H.; Yagita, H.; Johnstone, R.W.; Smyth, M.J.; et al. Radiotherapy increases the permissiveness of established mammary tumors to rejection by immunomodulatory antibodies. Cancer Res. 2012, 72, 3163–3174. [Google Scholar] [CrossRef] [PubMed]
- Balakrishnan, K.; Stellrecht, C.M.; Genini, D.; Ayres, M.; Wierda, W.G.; Keating, M.J.; Leoni, L.M.; Gandhi, V. Cell death of bioenergetically compromised and transcriptionally challenged CLL lymphocytes by chlorinated ATP. Blood 2005, 105, 4455–4462. [Google Scholar] [CrossRef] [PubMed]
- Stellrecht, C.M.; Chen, L.S.; Ayres, M.L.; Dennison, J.B.; Shentu, S.; Chen, Y.; Keating, M.J.; Wierda, W.G.; Gandhi, V. Chlorinated adenosine analogue induces ampk and autophagy in chronic lymphocytic leukaemia cells during therapy. Br. J. Haematol. 2017, 179, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Bichi, R.; Shinton, S.A.; Martin, E.S.; Koval, A.; Calin, G.A.; Cesari, R.; Russo, G.; Hardy, R.R.; Croce, C.M. Human chronic lymphocytic leukemia modeled in mouse by targeted tcl1 expression. Proc. Natl. Acad. Sci. USA 2002, 99, 6955–6960. [Google Scholar] [CrossRef] [PubMed]
- Vaisitti, T.; Serra, S.; Bologna, C.; Vitale, N.; Gobessi, S.; Efremov, D.G.; Deaglio, S. Abstract 1721: Hyper-activation of the adenosinergic axis in the Eμ-TCL1 chronic lymphocytic leukemia mouse model offers therapeutic opportunities. Blood 2017, 130, 1721. [Google Scholar]
- McClanahan, F.; Riches, J.C.; Miller, S.; Day, W.P.; Kotsiou, E.; Neuberg, D.; Croce, C.M.; Capasso, M.; Gribben, J.G. Mechanisms of PD-L1/PD-1-mediated CD8 T-cell dysfunction in the context of aging-related immune defects in the emicro-TCL1 CLL mouse model. Blood 2015, 126, 212–221. [Google Scholar] [CrossRef] [PubMed]
- Gorgun, G.; Ramsay, A.G.; Holderried, T.A.; Zahrieh, D.; Le Dieu, R.; Liu, F.; Quackenbush, J.; Croce, C.M.; Gribben, J.G. Eμ-TCL1 mice represent a model for immunotherapeutic reversal of chronic lymphocytic leukemia-induced T-cell dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 6250–6255. [Google Scholar] [CrossRef] [PubMed]
- Hanna, B.S.; McClanahan, F.; Yazdanparast, H.; Zaborsky, N.; Kalter, V.; Rossner, P.M.; Benner, A.; Durr, C.; Egle, A.; Gribben, J.G.; et al. Depletion of CLL-associated patrolling monocytes and macrophages controls disease development and repairs immune dysfunction in vivo. Leukemia 2016, 30, 570–579. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vaisitti, T.; Arruga, F.; Deaglio, S. Targeting the Adenosinergic Axis in Chronic Lymphocytic Leukemia: A Way to Disrupt the Tumor Niche? Int. J. Mol. Sci. 2018, 19, 1167. https://doi.org/10.3390/ijms19041167
Vaisitti T, Arruga F, Deaglio S. Targeting the Adenosinergic Axis in Chronic Lymphocytic Leukemia: A Way to Disrupt the Tumor Niche? International Journal of Molecular Sciences. 2018; 19(4):1167. https://doi.org/10.3390/ijms19041167
Chicago/Turabian StyleVaisitti, Tiziana, Francesca Arruga, and Silvia Deaglio. 2018. "Targeting the Adenosinergic Axis in Chronic Lymphocytic Leukemia: A Way to Disrupt the Tumor Niche?" International Journal of Molecular Sciences 19, no. 4: 1167. https://doi.org/10.3390/ijms19041167
APA StyleVaisitti, T., Arruga, F., & Deaglio, S. (2018). Targeting the Adenosinergic Axis in Chronic Lymphocytic Leukemia: A Way to Disrupt the Tumor Niche? International Journal of Molecular Sciences, 19(4), 1167. https://doi.org/10.3390/ijms19041167