Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes

 , ,
, ,  , and
, and 
Abstract
1. Deoxyribonucleic Acid as Hereditary Material
2. Cell Growth
3. Cell Cycle Control and Checkpoints
4. Disruption of Genome Integrity
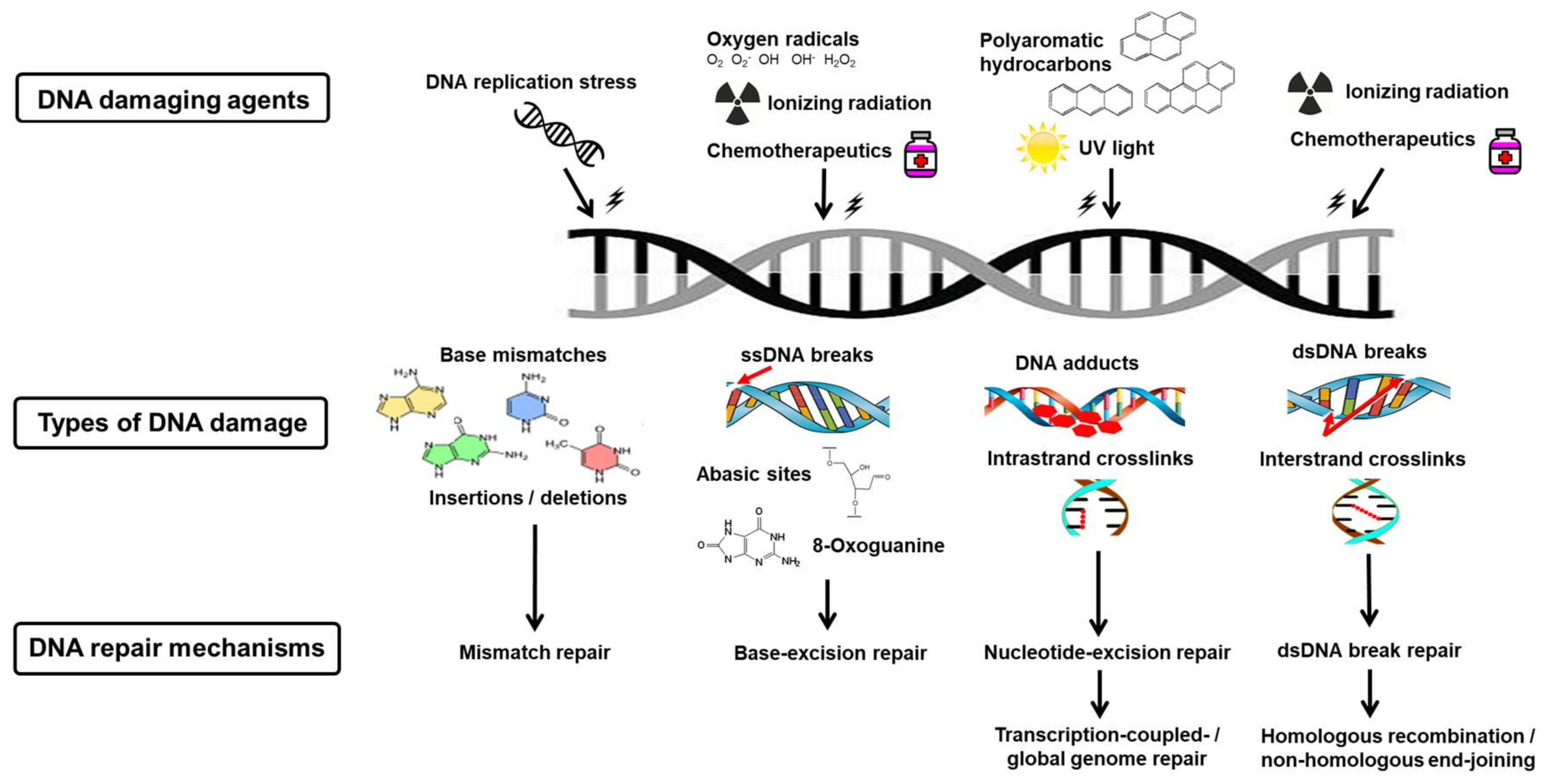
5. Endogenous Deoxyribonucleic Acid Damage
6. Exogenous Deoxyribonucleic Acid Damage
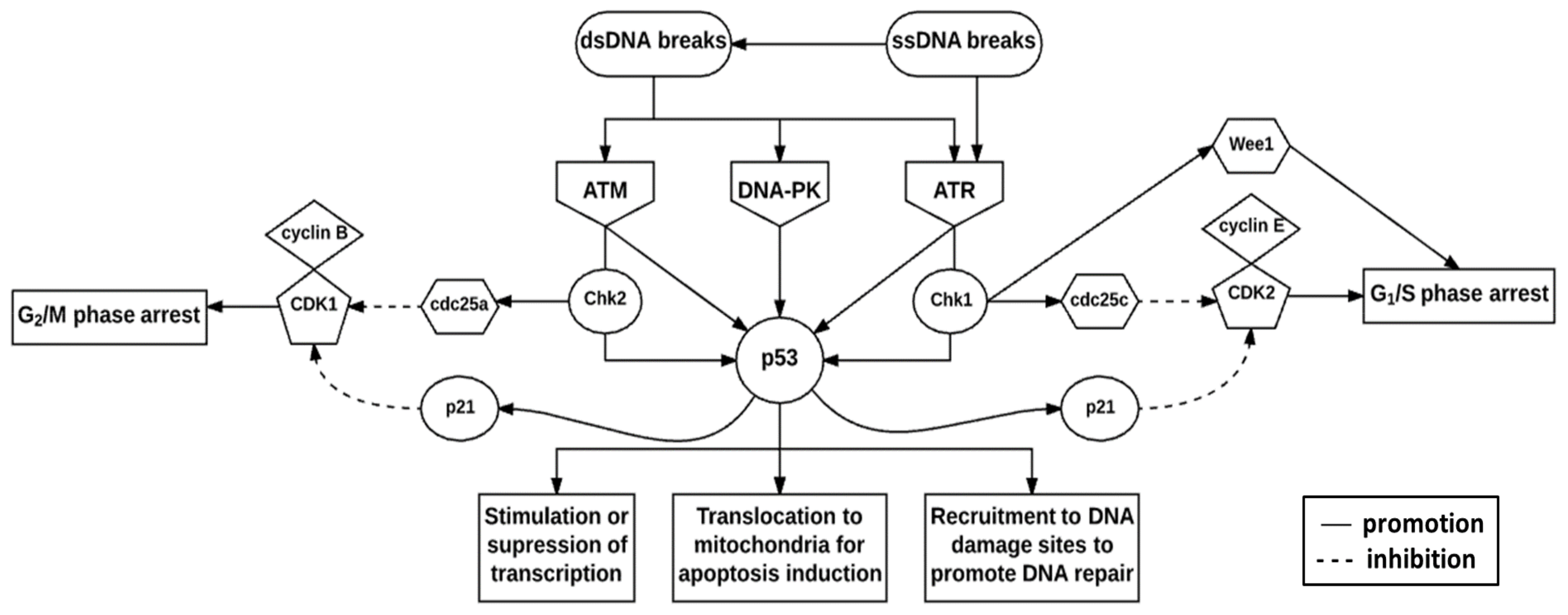
7. Deoxyribonucleic Acid Damage Response Pathway
8. Preservation of Genome Integrity
9. Mismatch Repair
10. Base-Excision Repair
11. Nucleotide-Excision Repair
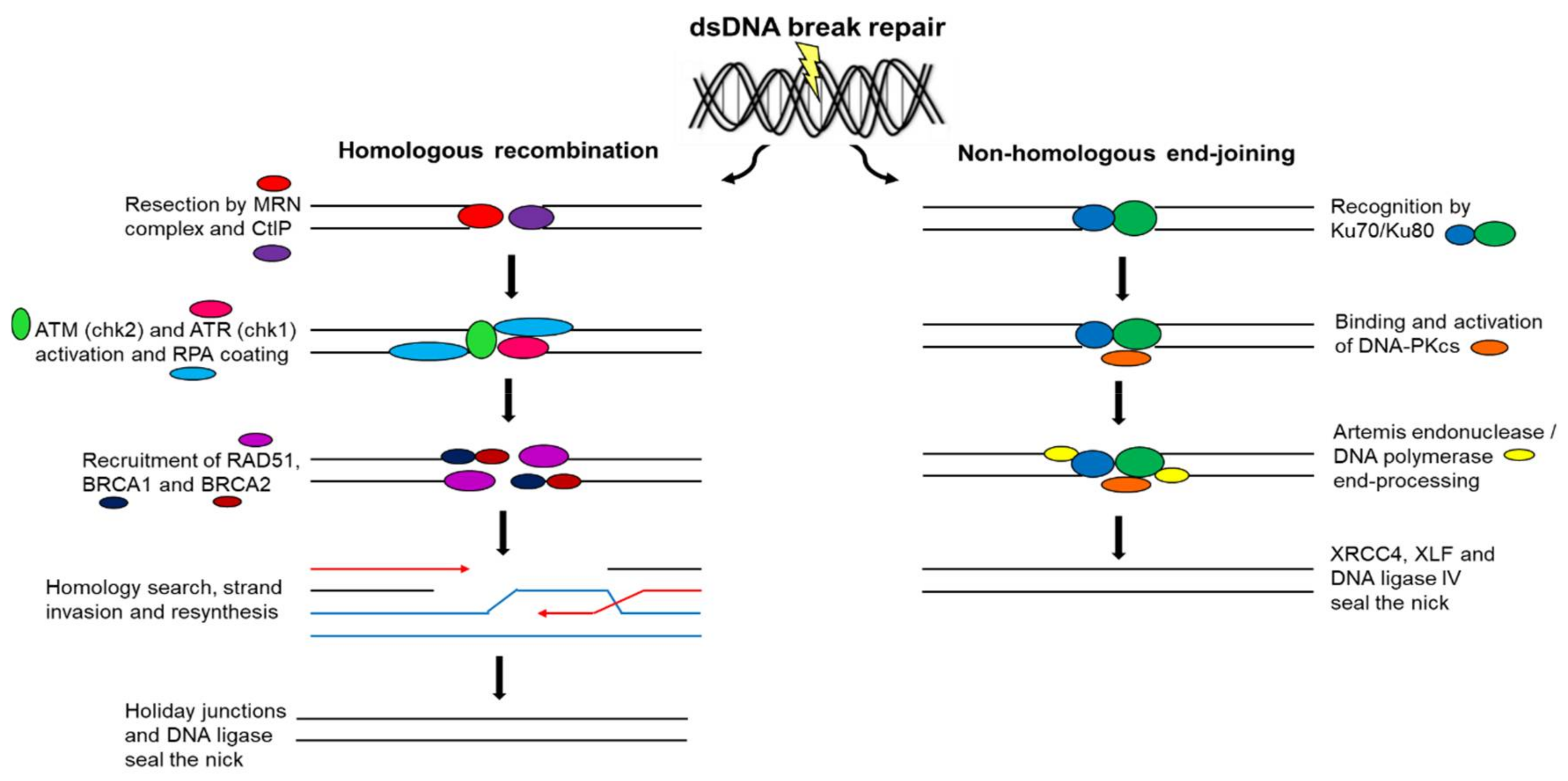
12. Double-Strand Deoxyribonucleic Acid Break Repair
13. Pathophysiology of Deoxyribonucleic Acid Repair Failure
14. Deoxyribonucleic Acid Repair Pathways as Therapeutic Targets and Future Directions
15. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| alt-NHEJ | alternative non-homologous end-joining |
| AOA1 | ataxia with oculomotor apraxia 1 |
| Arg | arginine |
| AsiDNA | a signal interfering DNA |
| ATM | ataxia telangiectasia mutated |
| ATR | ataxia telangiectasia and rad3-related |
| AUNIP | aurora kinase A and ninein interacting protein |
| BRCA1 | breast cancer gene 1 |
| BRCA2 | breast cancer gene 2 |
| CAKs | CDK activating kinases |
| Cas | CRISPR-associated nuclease |
| cdc25 | cell division cycle 25 |
| CDKs | cyclin-dependent kinases |
| CDKIs | CDK inhibitors |
| Chk1 | checkpoint kinase 1 |
| Chk2 | checkpoint kinase 2 |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| CtIP | C-terminal-binding protein interacting protein |
| Dbait | DNA strand break bait |
| DDR | DNA damage response |
| DNA | deoxyribonucleic acid |
| DNA-PK | DNA-dependent protein kinase |
| DNA-PKCS | DNA-PK catalytic subunit |
| dsDNA | double-strand DNA |
| ERCC1 | excision-repair cross-complementation group 1 |
| Exo1 | exonuclease 1 |
| FDA | Food and Drug Administration |
| FEN1 | flap endonuclease 1 |
| G | glycine |
| G0 | resting |
| G1 | growth 1/gap 1 |
| G2 | pre-mitotic/gap 2 |
| gRNA | guide RNA |
| H | histidine |
| H2O2 | hydrogen peroxide |
| HR | homologous recombination |
| INK | CDK4 inhibitor |
| iPSCs | induced pluripotent stem cells |
| KIP | CDK inhibitor |
| LDL | low density lipoprotein |
| Lys | lysine |
| M | mitotic |
| mHtt | mutant huntingtin gene |
| MLH1 | MutL homolog 1 |
| MMEJ | microhomology-mediated end-joining |
| MRE11 | dsDNA break repair nuclease MRE11 |
| MRN | MRE11-RAD50-NBS1 |
| MSH1 | MutS homolog 1 |
| MSH2 | MutS homolog 2 |
| MSH3 | MutS homolog 3 |
| MSH6 | MutS homolog 6 |
| mTOR | mammalian target of rapamycin |
| MutH | Mutator H |
| MutL | Mutator L |
| MutS | Mutator S |
| MutSα | MSH2-MSH6 heteroduplex |
| MutSβ | MSH2-MSH3 heteroduplex |
| Myt1 | myelin transcription factor 1 |
| N | asparagine |
| NAD+ | nicotinamide adenine dinucleotide |
| NBS1 | nibrin |
| NHEJ | non-homologous end-joining |
| O2 | oxygen |
| O2− | superoxide |
| PAM | protospacer adjacent motif |
| PARP1 | poly (ADP-ribose) polymerase 1 |
| PAXX | paralog of XRCC4 and XLF |
| PCNA | proliferating cell nuclear antigen |
| PI3K | phosphatidylinositol 3 kinase |
| PIKKs | phosphoinositide 3 kinase-related kinases |
| RAD50 | dsDNA break repair protein RAD50 |
| RNA | ribonucleic acid |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| RPA | replication protein A |
| S | DNA synthesis |
| SCAN1 | spinocerebellar ataxia with axonal neuropathy |
| Ser | serine |
| SSA | single-strand annealing |
| SSAP | single-strand annealing protein |
| ssDNA | single-strand DNA |
| TFIIH | transcription factor II human |
| Tim | Timeless |
| Tipin | Tim-interacting protein |
| USP4 | ubiquitin-specific protease 4 |
| XLF | XRCC4-like factor |
| XPA | xeroderma pigmentosum group A |
| XPB | xeroderma pigmentosum group B |
| XPC-RAD23B | xeroderma pigmentosum group C-RAD23 homolog B |
| XPD | xeroderma pigmentosum group D |
| XPF | xeroderma pigmentosum group F |
| XPG | xeroderma pigmentosum group G |
| XRCC1 | X-ray repair cross-complementation protein 1 |
| XRCC4 | X-ray repair cross-complementation protein 4 |
| Zn | zinc |
References
- Watson, J.D.; Crick, F.H. Genetical implications of the structure of deoxyribonucleic acid. Nature 1953, 171, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Clancy, S. DNA damage and repair: Mechanisms for maintaining DNA integrity. Nat. Educ. 2008, 1, 103. [Google Scholar]
- Wang, J.; Lindahl, T. Maintenance of genome stability. Genom. Proteom. Bioinform. 2016, 14, 119–121. [Google Scholar] [CrossRef] [PubMed]
- Kim, N.; Abdulovic, A.L.; Gealy, R.; Lippert, M.J.; Jinks-Robertson, S. Transcription-associated mutagenesis in yeast is directly proportional to the level of gene expression and influenced by the direction of DNA replication. DNA Repair 2007, 6, 1285–1296. [Google Scholar] [CrossRef] [PubMed]
- Khanna, K.K.; Shiloh, Y. The DNA Damage Response: Implications on Cancer Formation and Treatment; Springer: Dordrecht, The Netherlands, 2009. [Google Scholar]
- Ekim, B.; Magnuson, B.; Acosta-Jaquez, H.A.; Keller, J.A.; Feener, E.P.; Fingar, D.C. mTOR kinase domain phosphorylation promotes mTORC1 signaling, Cell Growth, and Cell Cycle Progression. Mol. Cell. Biol. 2011, 31, 2787–2801. [Google Scholar] [CrossRef] [PubMed]
- Ly, T.; Ahmad, Y.; Shlien, A.; Soroka, D.; Mills, A.; Emanuele, M.J.; Stratton, M.R.; Lamond, A.I. A proteomic chronology of gene expression through the cell cycle in human myeloid leukemia cells. eLife 2014, 3, e01630. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, E.; Gadaleta, M.C. Cell Cycle Control: Mechanisms and Protocols; Springer: New York, NY, USA, 2014. [Google Scholar]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Gabrielli, B.; Brooks, K.; Pavey, S. Defective cell cycle checkpoints as targets for anti-cancer therapies. Front. Pharmacol. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Barnum, K.; O’Connell, M. Cell cycle regulation by checkpoints. In Cell Cycle Control. Methods in Molecular Biology; Noguchi, E., Gadaleta, M.C., Eds.; Springer: New York, NY, USA, 2014; pp. 29–40. [Google Scholar]
- Robert, J. Cell Cycle Control. Textbook of Cell Signalling in Cancer; Springer: Cham, Switzerland, 2015; pp. 203–219. [Google Scholar]
- Morgan, D.O. The Cell Cycle: Principles of Control; New Science Press: London, UK, 2007. [Google Scholar]
- Levy-Cohen, G.; Blank, M. Functional analysis of protein ubiquitination. Anal. Biochem. 2015, 484, 37–39. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.; Gomes, A. PEST sequences in calmodulin-binding proteins. In Signal Transduction Mechanisms. Developments in Molecular and Cellular Biochemistry; Barnes, J., Coore, H., Mohammed, A., Sharma, R., Eds.; Springer: New York, NY, USA, 1995; pp. 17–27. [Google Scholar]
- Weis, M.C.; Avva, J.; Jacobberger, J.W.; Sreenath, S.N. A data-driven, mathematical model of mammalian cell cycle regulation. PLoS ONE 2014, 9, e97130. [Google Scholar] [CrossRef] [PubMed]
- Goodman, S.R. Medical Cell Biology, 3rd ed.; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Zachos, G.; Black, E.J.; Walker, M.; Scott, M.T.; Vagnarelli, P.; Earnshaw, W.C.; Gillespie, D.A. Chk1 is required for spindle checkpoint function. Dev. Cell 2007, 12, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Siede, W.; Doetsch, P.W. DNA Damage Recognition; CRC Press: Boca Raton, FL, USA, 2005. [Google Scholar]
- Geacintov, N.E.; Broyde, S. The Chemical Biology of DNA Damage; Wiley: Hoboken, NJ, USA, 2011. [Google Scholar]
- Speidel, D. The role of DNA damage responses in p53 biology. Arch. Toxicol. 2015, 89, 501–517. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Tropp, B.E. Molecular Biology: Genes to Proteins, 3rd ed.; Jones and Bartlett Publishers: Burlington, MA, USA, 2008. [Google Scholar]
- Henriksson, S.; Groth, P.; Gustafsson, N.; Helleday, T. Distinct mechanistic responses to replication fork stalling induced by either nucleotide or protein deprivation. Cell Cycle 2017. [Google Scholar] [CrossRef] [PubMed]
- Iyer, D.R.; Rhind, N. Replication fork slowing and stalling are distinct, checkpoint-independent consequences of replicating damaged DNA. PLoS Genet. 2017, 13, e1006958. [Google Scholar] [CrossRef] [PubMed]
- Ünsal-Kaçmaz, K.; Chastain, P.D.; Qu, P.-P.; Minoo, P.; Cordeiro-Stone, M.; Sancar, A.; Kaufmann, W.K. The human Tim/Tipin complex coordinates an intra-S checkpoint response to UV that slows replication fork displacement. Mol. Cell. Biol. 2007, 27, 3131–3142. [Google Scholar] [CrossRef] [PubMed]
- Gagou, M.E.; Zuazua-Villar, P.; Meuth, M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol. Biol. Cell 2010, 21, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Karl, S.; Pritschow, Y.; Volcic, M.; Häcker, S.; Baumann, B.; Wiesmüller, L.; Debatin, K.M.; Fulda, S. Identification of a novel pro-apopotic function of NF-κB in the DNA damage response. J. Cell. Mol. Med. 2009, 13, 4239–4256. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Musich, P.R.; Zou, Y. Differential DNA damage responses in p53 proficient and deficient cells: Cisplatin-induced nuclear import of XPA is independent of ATR checkpoint in p53-deficient lung cancer cells. Int. J. Biochem. Mol. Biol. 2011, 2, 138–145. [Google Scholar] [PubMed]
- Pabla, N.; Huang, S.; Mi, Q.-S.; Daniel, R.; Dong, Z. ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis. J. Biol. Chem. 2008, 283, 6572–6583. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. DNA damage checkpoints: From initiation to recovery or adaptation. Curr. Opin. Cell Biol. 2007, 19, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Rodier, F.; Campisi, J.; Bhaumik, D. Two faces of p53: Aging and tumor suppression. Nucleic Acids Res. 2007, 35, 7475–7484. [Google Scholar] [CrossRef] [PubMed]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Coates, J.; Jackson, S.P. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005, 434, 605. [Google Scholar] [CrossRef] [PubMed]
- Uziel, T.; Lerenthal, Y.; Moyal, L.; Andegeko, Y.; Mittelman, L.; Shiloh, Y. Requirement of the MRN complex for ATM activation by DNA damage. EMBO J. 2003, 22, 5612–5621. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, S.V.; Graham, M.E.; Jakob, B.; Tobias, F.; Kijas, A.W.; Tanuji, M.; Chen, P.; Robinson, P.J.; Taucher-Scholz, G.; Suzuki, K.; et al. Autophosphorylation and ATM activation: Additional sites add to the complexity. J. Biol. Chem. 2011, 286, 9107–9119. [Google Scholar] [CrossRef] [PubMed]
- Riballo, E.; Kühne, M.; Rief, N.; Doherty, A.; Smith, G.C.; Recio, M.J.; Reis, C.; Dahm, K.; Fricke, A.; Krempler, A.; et al. A pathway of double-strand break rejoining dependent upon ATM, Artemis, and proteins locating to gamma-H2AX foci. Mol. Cell 2004, 16, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Beucher, A.; Birraux, J.; Tchouandong, L.; Barton, O.; Shibata, A.; Conrad, S.; Goodarzi, A.A.; Krempler, A.; Jeggo, P.A.; Löbrich, M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J. 2009, 28, 3413–3427. [Google Scholar] [CrossRef] [PubMed]
- Maréchal, A.; Zou, L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb. Perspect. Biol. 2013, 5, a012716. [Google Scholar] [CrossRef] [PubMed]
- Bartek, J.; Lukas, J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer Cell 2003, 3, 421–429. [Google Scholar] [CrossRef]
- Busino, L.; Donzelli, M.; Chiesa, M.; Guardavaccaro, D.; Ganoth, D.; Dorrello, N.V.; Hershko, A.; Pagano, M.; Draetta, G.F. Degradation of Cdc25A by β-TrCP during S phase and in response to DNA damage. Nature 2003, 426, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Mjelle, R.; Hegre, S.A.; Aas, P.A.; Slupphaug, G.; Drabløs, F.; Sætrom, P.; Krokan, H.E. Cell cycle regulation of human DNA repair and chromatin remodeling genes. DNA Repair 2015, 30, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Torgovnick, A.; Schumacher, B. DNA repair mechanisms in cancer development and therapy. Front. Genet. 2015, 6, 157. [Google Scholar] [CrossRef] [PubMed]
- Modrich, P. Mechanisms and biological effects of mismatch repair. Annu. Rev. Genet. 1991, 25, 229–253. [Google Scholar] [CrossRef] [PubMed]
- Tropp, B.E. Molecular Biology: Genes to Proteins, 4th ed.; Jones & Bartlett Learning: Burlington, MA, USA, 2012. [Google Scholar]
- Spies, M.; Fishel, R. Mismatch Repair during homologous and homeologous recombination. Cold Spring Harb. Perspect. Biol. 2015, 7, a022657. [Google Scholar] [CrossRef] [PubMed]
- Constantin, N.; Dzantiev, L.; Kadyrov, F.A.; Modrich, P. Human mismatch repair: Reconstitution of a nick-directed bidirectional reaction. J. Biol. Chem. 2005, 280, 39752–39761. [Google Scholar] [CrossRef] [PubMed]
- Wei, K.; Clark, A.B.; Wong, E.; Kane, M.F.; Mazur, D.J.; Parris, T.; Kolas, N.K.; Russel, R.; Hou, H.; Kneitz, B.; et al. Inactivation of exonuclease 1 in mice results in DNA mismatch repair defects, increased cancer susceptibility, and male and female sterility. Genes Dev. 2003, 17, 603–614. [Google Scholar] [CrossRef] [PubMed]
- Dherin, C.; Gueneau, E.; Francin, M.; Nunez, M.; Miron, S.; Liberti, S.E.; Rasmussen, L.J.; Zinn-Justin, S.; Gilquin, B.; Charbonnier, J.B.; et al. Characterization of a highly conserved binding site of Mlh1 required for exonuclease I-dependent mismatch repair. Mol. Cell. Biol. 2009, 29, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Ganten, D.; Ruckpaul, K. Encyclopedic Reference of Genomics and Proteomics in Molecular Medicine; Springer: Berlin, Germany, 2006. [Google Scholar]
- Wood, R.D.; Mitchell, M.; Sgouros, J.; Lindahl, T. Human DNA repair genes. Science 2001, 291, 1284–1289. [Google Scholar] [CrossRef] [PubMed]
- Bowers, J.; Tran, P.T.; Joshi, A.; Liskay, R.M.; Alani, E. MSH-MLH complexes formed at a DNA mismatch are disrupted by the PCNA sliding clamp. J. Mol. Biol. 2001, 306, 957–968. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Predeus, A.V.; Kovacs, N.; Feig, M. Differential mismatch recognition specificities of eukaryotic MutS homologs, MutSα and MutSβ. Biophys. J. 2014, 106, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709. [Google Scholar] [CrossRef] [PubMed]
- Odell, I.D.; Barbour, J.-E.; Murphy, D.L.; Della-Maria, J.A.; Sweasy, J.B.; Tomkinson, A.E.; Wallace, S.S.; Pederson, D.S. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell. Biol. 2011, 31, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Ying, S.; Chen, Z.; Medhurst, A.L.; Neal, J.A.; Bao, Z.; Mortusewicz, O.; McGouran, J.; Song, X.; Shen, H.; Hamdy, F.C.; et al. DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016, 76, 1078–1088. [Google Scholar] [CrossRef] [PubMed]
- Krokan, H.E.; Bjørås, M. Base excision repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef] [PubMed]
- Aguiar, P.H.N.; Furtado, C.; Repolês, B.M.; Ribeiro, G.A.; Mendes, I.C.; Peloso, E.F.; Gadelha, F.R.; Macedo, A.M.; Franco, G.R.; Pena, S.D.; et al. Oxidative stress and DNA lesions: The role of 8-oxoguanine lesions in trypanosoma cruzi cell viability. PLoS Negl. Trop. Dis. 2013, 7, e2279. [Google Scholar] [CrossRef] [PubMed]
- Dianova, I.I.; Sleeth, K.M.; Allinson, S.L.; Parsons, J.L.; Breslin, C.; Caldecott, K.W.; Dianov, G.L. XRCC1–DNA polymerase β interaction is required for efficient base excision repair. Nucleic Acids Res. 2004, 32, 2550–2555. [Google Scholar] [CrossRef] [PubMed]
- Asagoshi, K.; Tano, K.; Chastain, P.D.; Adachi, N.; Sonoda, E.; Kikuchi, K.; Koyama, H.; Nagata, K.; Kaufman, D.G.; Takeda, S.; et al. FEN1 functions in long patch base excision repair under conditions of oxidative stress in vertebrate cells. Mol. Cancer Res. 2010, 8, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Sancar, A. Nucleotide excision repair: From, E. coli to man. Biochimie 1999, 81, 15–25. [Google Scholar] [CrossRef]
- Ranes, M.; Boeing, S.; Wang, Y.; Wienholz, F.; Menoni, H.; Walker, J.; Encheva, V.; Chakravarty, P.; Mari, P.O.; Stewart, A.; et al. A ubiquitylation site in Cockayne syndrome B required for repair of oxidative DNA damage, but not for transcription-coupled nucleotide excision repair. Nucleic Acids Res. 2016, 44, 5246–5255. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.; Li, H.; Wang, S.; Jiang, X.; Zhang, S.; Zhang, R.; Fu, P.P.; Sun, X. Ultrasensitive UPLC-MS-MS method for the quantitation of etheno-DNA adducts in human urine. Int. J. Environ. Res. Public Health 2014, 11, 10902. [Google Scholar] [CrossRef] [PubMed]
- Chaim, I.A.; Gardner, A.; Wu, J.; Iyama, T.; Wilson, D.M.; Samson, L.D. A novel role for transcription-coupled nucleotide excision repair for the in vivo repair of 3,N4-ethenocytosine. Nucleic Acids Res. 2017, 45, 3242–3252. [Google Scholar] [PubMed]
- Bee, L.; Marini, S.; Pontarin, G.; Ferraro, P.; Costa, R.; Albrecht, U.; Celotti, L. Nucleotide excision repair efficiency in quiescent human fibroblasts is modulated by circadian clock. Nucleic Acids Res. 2015, 43, 2126–2137. [Google Scholar] [CrossRef] [PubMed]
- Hoogstraten, D.; Bergink, S.; Ng, J.M.Y.; Verbiest, V.H.M.; Luijsterburg, M.S.; Geverts, B.; Raams, A.; Dinant, C.; Hoeijmakers, J.H.; Vermeulen, W.; et al. Versatile DNA damage detection by the global genome nucleotide excision repair protein XPC. J. Cell Sci. 2008, 121, 2850–2859. [Google Scholar] [CrossRef] [PubMed]
- Sugasawa, K.; Okamoto, T.; Shimizu, Y.; Masutani, C.; Iwai, S.; Hanaoka, F. A multistep damage recognition mechanism for global genomic nucleotide excision repair. Genes Dev. 2001, 15, 507–521. [Google Scholar] [CrossRef] [PubMed]
- Lans, H.; Marteijn, J.A.; Schumacher, B.; Hoeijmakers, J.H.J.; Jansen, G.; Vermeulen, W. Involvement of global genome repair, transcription coupled repair, and chromatin remodeling in UV DNA damage response changes during development. PLoS Genet. 2010, 6, e1000941. [Google Scholar] [CrossRef] [PubMed]
- Aparicio, T.; Baer, R.; Gautier, J. DNA double-strand break repair pathway choice and cancer. DNA Repair 2014, 19, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Weterings, E.; Gallegos, A.C.; Dominick, L.N.; Cooke, L.S.; Bartels, T.N.; Vagner, J.; Matsunaga, T.O.; Mahadevan, D. A novel small molecule inhibitor of the DNA repair protein Ku70/80. DNA Repair 2016, 43, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.L.; Barrasa, M.I.; Orr-Weaver, T.L. Replication fork progression during re-replication requires the DNA damage checkpoint and double-strand break repair. Curr. Biol. 2015, 25, 1654–1660. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.M.; Misri, S.; Meyer, B.; Raj, S.; Zobel, C.L.; Sleckman, B.P.; Hallahan, D.E.; Sharma, G.G. Unique epigenetic influence of H2AX phosphorylation and H3K56 acetylation on normal stem cell radioresponses. Mol. Biol. Cell 2016, 27, 1332–1345. [Google Scholar] [CrossRef] [PubMed]
- Burma, S.; Chen, B.P.; Murphy, M.; Kurimasa, A.; Chen, D.J. ATM phosphorylates histone H2AX in response to DNA double-strand breaks. J. Biol. Chem. 2001, 276, 42462–42467. [Google Scholar] [CrossRef] [PubMed]
- Redon, C.E.; Nakamura, A.J.; Zhang, Y.-W.; Ji, J.; Bonner, W.M.; Kinders, R.J.; Parchment, R.E.; Doroshow, J.H.; Pommier, Y. Histone γH2AX and poly(ADP-Rribose) as clinical pharmacodynamic biomarkers. Clin. Cancer Res. 2010, 16, 4532–4542. [Google Scholar] [CrossRef] [PubMed]
- Löbrich, M.; Shibata, A.; Beucher, A.; Fisher, A.; Ensminger, M.; Goodarzi, A.A.; Barton, O.; Jeggo, P.A. γH2AX foci analysis for monitoring DNA double-strand break repair: Strengths, limitations and optimization. Cell Cycle 2010, 9, 662–669. [Google Scholar] [CrossRef] [PubMed]
- Cleaver, J.E. γH2Ax: Biomarker of damage or functional participant in DNA repair “all that glitters is not gold!”. Photochem. Photobiol. 2011, 87, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Kass, E.M.; Helgadottir, H.R.; Chen, C.-C.; Barbera, M.; Wang, R.; Westermark, U.K.; Ludwig, T.; Moynahan, M.E.; Jasin, M. Double-strand break repair by homologous recombination in primary mouse somatic cells requires BRCA1 but not the ATM kinase. Proc. Natl. Acad. Sci. USA 2013, 110, 5564–5569. [Google Scholar] [CrossRef] [PubMed]
- Mao, Z.; Bozzella, M.; Seluanov, A.; Gorbunova, V. Comparison of nonhomologous end joining and homologous recombination in human cells. DNA Repair 2008, 7, 1765–1771. [Google Scholar] [CrossRef] [PubMed]
- Adachi, N.; Suzuki, H.; Iiizumi, S.; Koyama, H. Hypersensitivity of nonhomologous DNA end-joining mutants to VP-16 and ICRF-193: Implications for the repair of topoisomerase II-mediated DNA damage. J. Biol. Chem. 2003, 278, 35897–35902. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lu, H.; Tippin, B.; Goodman, M.F.; Shimazaki, N.; Koiwai, O.; Hsieh, C.L.; Schwarz, K.; Lieber, M.R. A biochemically defined system for mammalian nonhomologous DNA end joining. Mol. Cell 2004, 16, 701–713. [Google Scholar] [CrossRef] [PubMed]
- Schulte-Uentrop, L.; El-Awady, R.A.; Schliecker, L.; Willers, H.; Dahm-Daphi, J. Distinct roles of XRCC4 and Ku80 in non-homologous end-joining of endonuclease- and ionizing radiation-induced DNA double-strand breaks. Nucleic Acids Res. 2008, 36, 2561–2569. [Google Scholar] [CrossRef] [PubMed]
- Brandsma, I.; van Gent, D.C. Pathway choice in DNA double strand break repair: Observations of a balancing act. Genome Integr. 2012, 3, 9. [Google Scholar] [CrossRef] [PubMed]
- Ochi, T.; Blackford, A.N.; Coates, J.; Jhujh, S.; Mehmood, S.; Tamura, N.; Travers, J.; Wu, Q.; Draviam, V.M.; Robinson, C.V.; et al. PAXX, a paralog of XRCC4 and XLF, interacts with Ku to promote DNA double-strand break repair. Science 2015, 347, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Shao, Z.; Jiang, W.; Lee, B.J.; Zha, S. PAXX promotes Ku accumulation at DNA breaks and is essential for end-joining in XLF-deficient mice. Nat. Commun. 2017, 8, 13816. [Google Scholar] [CrossRef] [PubMed]
- Falck, J.; Forment, J.V.; Coates, J.; Mistrik, M.; Lukas, J.; Bartek, J.; Jackson, S.P. Cyclin-dependent kinase targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep. 2012, 13, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Limbo, O.; Chahwan, C.; Yamada, Y.; de Bruin, R.A.; Wittenberg, C.; Russell, P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol. Cell 2007, 28, 134–146. [Google Scholar] [CrossRef] [PubMed]
- Shibata, A.; Moiani, D.; Arvai, A.S.; Perry, J.; Harding, S.M.; Genois, M.M.; Maity, R.; van Rossum-Fikkert, S.; Kertokalio, A.; Romoli, F.; et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol. Cell 2014, 53, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Stark, J.M.; Pierce, A.J.; Oh, J.; Pastink, A.; Jasin, M. Genetic steps of mammalian homologous repair with distinct mutagenic consequences. Mol. Cell. Biol. 2004, 24, 9305–9316. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanism. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef] [PubMed]
- Ander, M.; Subramaniam, S.; Fahmy, K.; Stewart, A.F.; Schäffer, E. A single-strand annealing protein clamps DNA to detect and secure homology. PLoS Biol. 2015, 13, e1002213. [Google Scholar] [CrossRef] [PubMed]
- Morrical, S.W. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 2015, 7, a016444. [Google Scholar] [CrossRef] [PubMed]
- Nussenzweig, A.; Nussenzweig, M.C. A backup DNA repair pathway moves to the forefront. Cell 2007, 131, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Biehs, R.; Steinlage, M.; Barton, O.; Juhász, S.; Künzel, J.; Spies, J.; Shibata, A.; Jeggo, P.A.; Löbrich, M. DNA double-strand break resection occurs during non-homologous end joining in G1 but is distinct from resection during homologous recombination. Mol. Cell 2017, 65, 671–684. [Google Scholar] [CrossRef] [PubMed]
- Newman, E.A.; Lu, F.; Bashllari, D.; Wang, L.; Opipari, A.W.; Castle, V.P. Alternative NHEJ pathway components are therapeutic targets in high-risk neuroblastoma. Mol. Cancer Res. 2015, 13, 470–482. [Google Scholar] [CrossRef] [PubMed]
- Lou, J.; Chen, H.; Han, J.; He, H.; Huen, M.S.Y.; Feng, X.; Liu, T.; Huang, J. AUNIP/C1orf135 directs DNA double-strand breaks towards the homologous recombination repair pathway. Nat. Commun. 2017, 8, 895. [Google Scholar] [CrossRef] [PubMed]
- Lieu, A.S.; Chen, T.S.; Chou, C.H.; Wu, C.H.; Hsu, C.Y.; Huang, C.Y.; Chang, L.K.; Loh, J.K.; Chang, C.S.; Hsu, C.M.; et al. Functional characterization of AIBp, a novel Aurora-A binding protein in centrosome structure and spindle formation. Int. J. Oncol. 2010, 37, 429–436. [Google Scholar] [PubMed]
- Wijnhoven, P.; Konietzny, R.; Blackford, A.N.; Travers, J.; Kessler, B.M.; Nishi, R.; Jackson, S.P. USP4 auto-deubiquitylation promotes homologous recombination. Mol. Cell 2015, 60, 362–373. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhang, H.; Wang, X.; Tian, Q.; Hu, Z.; Peng, C.; Jiang, P.; Wang, T.; Guo, W.; Chen, Y.; et al. The deubiquitylating enzyme USP4 cooperates with CtIP in DNA double-strand break end resection. Cell Rep. 2015, 13, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, A.J.F.; Miller, J.H.; Suzuki, D.T.; Lewontin, R.C.; Gelbart, W.M. An Introduction to Genetic Analysis, 7th ed.; W.H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Maletzki, C.; Huehns, M.; Bauer, I.; Ripperger, T.; Mork, M.M.; Vilar, E.; Klöcking, S.; Zettl, H.; Prall, F.; Linnebacher, M. Frameshift mutational target gene analysis identifies similarities and differences in constitutional mismatch repair-deficiency and Lynch syndrome. Mol. Carcinog. 2017, 56, 1753–1764. [Google Scholar] [CrossRef] [PubMed]
- Bowden, N.A.; Beveridge, N.J.; Ashton, K.A.; Baines, K.J.; Scott, R.J. Understanding xeroderma pigmentosum complementation groups using gene expression profiling after UV-light exposure. Int. J. Mol. Sci. 2015, 16, 15985–15996. [Google Scholar] [CrossRef] [PubMed]
- Hirano, R.; Interthal, H.; Huang, C.; Nakamura, T.; Deguchi, K.; Choi, K.; Bhattacharjee, M.B.; Arimura, K.; Umehara, F.; Izumo, S.; et al. Spinocerebellar ataxia with axonal neuropathy: Consequence of a Tdp1 recessive neomorphic mutation? EMBO J. 2007, 26, 4732–4743. [Google Scholar] [CrossRef] [PubMed]
- Çağlayan, M.; Horton, J.K.; Prasad, R.; Wilson, S.H. Complementation of aprataxin deficiency by base excision repair enzymes. Nucleic Acids Res. 2015, 43, 2271–2281. [Google Scholar] [CrossRef] [PubMed]
- Akouchekian, M.; Hemati, S.; Jafari, D.; Jalilian, N.; Dehghan Manshadi, M. Does PTEN gene mutation play any role in Li-Fraumeni syndrome. Med. J. Islam. Repub. Iran 2016, 30, 378. [Google Scholar] [PubMed]
- Lawrence, K.S.; Chau, T.; Engebrecht, J. DNA damage response and spindle assembly checkpoint function throughout the cell cycle to ensure genomic integrity. PLoS Genet. 2015, 11, e1005150. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Steffen, J.D.; McCauley, M.M.; Pascal, J.M. Fluorescent sensors of PARP-1 structural dynamics and allosteric regulation in response to DNA damage. Nucleic Acids Res. 2016, 44, 9771–9783. [Google Scholar] [CrossRef] [PubMed]
- Langelier, M.F.; Ruhl, D.D.; Planck, J.L.; Kraus, W.L.; Pascal, J.M. The Zn3 domain of human poly(ADP-ribose) polymerase-1 (PARP-1) functions in both DNA-dependent poly(ADP-ribose) synthesis activity and chromatin compaction. J. Biol. Chem. 2010, 285, 18877–18887. [Google Scholar] [CrossRef] [PubMed]
- Heale, J.T.; Ball, A.R.; Schmiesing, J.A.; Kim, J.-S.; Kong, X.; Zhou, S.; Hudson, D.F.; Earnshaw, W.C.; Yokomori, K. Condensin I interacts with the PARP-1-XRCC1 complex and functions in DNA single-strand break repair. Mol. Cell 2006, 21, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Audeh, M.W.; Carmichael, J.; Penson, R.T.; Friedlander, M.; Powell, B.; Bell-McGuinn, K.M.; Scott, C.; Weitzel, J.N.; Oaknin, A.; Loman, N.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and recurrent ovarian cancer: A proof-of-concept trial. Lancet 2010, 376, 245–251. [Google Scholar] [CrossRef]
- Tutt, A.; Robson, M.; Garber, J.E.; Domchek, S.M.; Audeh, M.W.; Weitzel, J.N.; Friedlander, M.; Arun, B.; Loman, N.; Schmutzler, R.K.; et al. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: A proof-of-concept trial. Lancet 2010, 376, 235–244. [Google Scholar] [CrossRef]
- Wiener, D.; Gajardo-Meneses, P.; Ortega-Hernández, V.; Herrera-Cares, C.B.; Díaz, S.N.; Fernández, W.; Cornejo, V.; Gamboa, J.; Tapia, T.; Alvarez, C.; et al. BRCA1 and BARD1 colocalize mainly in the cytoplasm of breast cancer tumors, and their isoforms show differential expression. Breast Cancer Res. Treat. 2015, 153, 669–678. [Google Scholar] [CrossRef] [PubMed]
- Sartori, A.A.; Lukas, C.; Coates, J.; Mistrik, M.; Fu, S.; Bartek, J.; Baer, R.; Lukas, J.; Jackson, S.P. Human CtIP promotes DNA end resection. Nature 2007, 450, 509. [Google Scholar] [CrossRef] [PubMed]
- Farmer, H.; McCabe, N.; Lord, C.J.; Tutt, A.N.; Johnson, D.A.; Richardson, T.B.; Santarosa, M.; Dillon, K.J.; Hickson, I.; Knights, C.; et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005, 434, 917. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature 2005, 434, 913. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, R.; Bakkenist, C.J.; McKinnon, P.J.; Kastan, M.B. Phosphorylation of SMC1 is a critical downstream event in the ATM–NBS1–BRCA1 pathway. Genes Dev. 2004, 18, 1423–1438. [Google Scholar] [CrossRef] [PubMed]
- Williamson, C.T.; Kubota, E.; Hamill, J.D.; Klimowicz, A.; Ye, R.; Muzik, H.; Dean, M.; Tu, L.; Gilley, D.; Magliocco, A.M.; et al. Enhanced cytotoxicity of PARP inhibition in mantle cell lymphoma harbouring mutations in both ATM and p53. EMBO Mol. Med. 2012, 4, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Ronson, G.; Piberger, A.L.; Higgs, M.; Olsen, A.; Stewart, G.; McHugh, P.; Petermann, E.; Lakin, N. PARP1 and PARP2 stabilise replication forks at base excision repair intermediates through Fbh1-dependent Rad51 regulation. Nat. Commun. 2018, 9, 746. [Google Scholar] [CrossRef] [PubMed]
- Ray Chaudhuri, A.; Callen, E.; Ding, X.; Gogola, E.; Duarte, A.A.; Lee, J.E.; Wong, N.; Lafarga, V.; Calvo, J.A.; Panzarino, N.J.; et al. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature 2016, 535, 382. [Google Scholar] [CrossRef] [PubMed]
- Bryant, H.E.; Petermann, E.; Schultz, N.; Jemth, A.-S.; Loseva, O.; Issaeva, N.; Johansson, F.; Fernandez, S.; McGlynn, P.; Helleday, T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009, 28, 2601–2615. [Google Scholar] [CrossRef] [PubMed]
- Takagi, M.; Yoshida, M.; Nemoto, Y.; Tamaichi, H.; Tsuchida, R.; Seki, M.; Uryu, K.; Nishii, R.; Miyamoto, S.; Saito, M.; et al. Loss of DNA damage response in neuroblastoma and utility of a PARP inhibitor. J. Natl. Cancer Inst. 2017, 109. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.; Ison, G.; McKee, A.E.; Zhang, H.; Tang, S.; Gwise, T.; Sridhara, R.; Lee, E.; Tzou, A.; Philip, R.; et al. FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA-mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin. Cancer Res. 2015, 21, 4257–4261. [Google Scholar] [CrossRef] [PubMed]
- Pujade-Lauraine, E.; Ledermann, J.A.; Selle, F.; Gebski, V.; Penson, R.T.; Oza, A.M.; Korach, J.; Huzarski, T.; Poveda, A.; Pignata, S.; et al. Olaparib tablets as maintenance therapy in patients with platinum-sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT-Ov21): A double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Oncol. 2017, 18, 1274–1284. [Google Scholar] [CrossRef]
- Bundred, N.; Gardovskis, J.; Jaskiewicz, J.; Eglitis, J.; Paramonov, V.; McCormack, P.; Swaisland, H.; Cavallin, M.; Parry, T.; Carmichael, J.; et al. Evaluation of the pharmacodynamics and pharmacokinetics of the PARP inhibitor olaparib: A phase I multicentre trial in patients scheduled for elective breast cancer surgery. Investig. New Drugs 2013, 31, 949–958. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-Ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Biau, J.; Devun, F.; Jdey, W.; Kotula, E.; Quanz, M.; Chautard, E.; Sayarath, M.; Sun, J.S.; Verrelle, P.; Dutreix, M. A preclinical study combining the DNA repair inhibitor Dbait with radiotherapy for the treatment of melanoma. Neoplasia 2014, 16, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Yao, H.; Qiu, H.; Shao, Z.; Wang, G.; Wang, J.; Yao, Y.; Xin, Y.; Zhou, M.; Wang, A.Z.; Zhang, L. Nanoparticle formulation of small DNA molecules, Dbait, improves the sensitivity of hormone-independent prostate cancer to radiotherapy. Nanomed. Nanotechnol. Biol. Med. 2016, 12, 2261–2271. [Google Scholar] [CrossRef] [PubMed]
- Herath, N.I.; Devun, F.; Herbette, A.; Lienafa, M.C.; Sun, J.S.; Dutreix, M.; Denys, A. Potentiation of doxorubicin efficacy in hepatocellular carcinoma by the DNA repair inhibitor DT01 in preclinical models. Eur. Radiol. 2017, 27, 4435–4444. [Google Scholar] [CrossRef] [PubMed]
- Herath, N.I.; Devun, F.; Lienafa, M.C.; Herbette, A.; Denys, A.; Sun, J.S.; Dutreix, M. The DNA repair inhibitor DT01 as a novel therapeutic strategy for chemosensitization of colorectal liver metastasis. Mol. Cancer Ther. 2016, 15, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Thierry, S.; Jdey, W.; Alculumbre, S.; Soumelis, V.; Noguiez-Hellin, P.; Dutreix, M. The DNA repair inhibitor Dbait is specific for malignant hematologic cells in blood. Mol. Cancer Ther. 2017, 16, 2817–2827. [Google Scholar] [CrossRef] [PubMed]
- Jdey, W.; Thierry, S.; Russo, C.; Devun, F.; Al Abo, M.; Noguiez-Hellin, P.; Sun, J.S.; Barillot, E.; Zinovyev, A.; Kuperstein, I.; et al. Drug driven synthetic lethality: Bypassing tumor cell genetics with a combination of Dbait and PARP inhibitors. Clin. Cancer Res. 2017, 23, 1001–1011. [Google Scholar] [CrossRef] [PubMed]
- Gavande, N.S.; VanderVere-Carozza, P.S.; Hinshaw, H.D.; Jalal, S.I.; Sears, C.R.; Pawelczak, K.S.; Turchi, J.J. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol. Ther. 2016, 160, 65–83. [Google Scholar] [CrossRef] [PubMed]
- Batey, M.A.; Zhao, Y.; Kyle, S.; Richardson, C.; Slade, A.; Martin, N.M.B.; Lau, A.; Newell, D.R.; Curtin, N.J. Preclinical evaluation of a novel ATM inhibitor, KU59403, in vitro and in vivo in p53 functional and dysfunctional models of human cancer. Mol. Cancer Ther. 2013, 12, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Biddlestone-Thorpe, L.; Sajjad, M.; Rosenberg, E.; Beckta, J.M.; Valerie, N.C.K.; Tokarz, M.; Adams, B.R.; Wagner, A.F.; Khalil, A.; Gilfor, D.; et al. ATM kinase inhibition preferentially sensitizes p53-mutant glioma to ionizing radiation. Clin. Cancer Res. 2013, 19, 3189–3200. [Google Scholar] [CrossRef] [PubMed]
- Foote, K.M.; Blades, K.; Cronin, A.; Fillery, S.; Guichard, S.S.; Hassall, L.; Hickson, I.; Jacq, X.; Jewsbury, P.J.; McGuire, T.M.; et al. Discovery of 4-{4-[(3R)-3-Methylmorpholin-4-yl]-6-[1-(methylsulfonyl)cyclopropyl]pyrimidin-2-yl}-1H-indole (AZ20): A potent and selective inhibitor of ATR protein kinase with monotherapy in vivo antitumor activity. J. Med. Chem. 2013, 56, 2125–2138. [Google Scholar] [CrossRef] [PubMed]
- Madhusudan, S.; Smart, F.; Shrimpton, P.; Parsons, J.L.; Gardiner, L.; Houlbrook, S.; Talbot, D.C.; Hammonds, T.; Freemont, P.A.; Sternberg, M.J.; et al. Isolation of a small molecule inhibitor of DNA base excision repair. Nucleic Acids Res. 2005, 33, 4711–4724. [Google Scholar] [CrossRef] [PubMed]
- Andrews, B.J.; Turchi, J.J. Development of a high-throughput screen for inhibitors of replication protein A and its role in nucleotide excision repair. Mol. Cancer Ther. 2004, 3, 385–391. [Google Scholar] [PubMed]
- Boeckman, H.J.; Trego, K.S.; Turchi, J.J. Cisplatin sensitizes cancer cells to ionizing radiation via inhibition of nonhomologous end joining. Mol. Cancer Res. 2005, 3, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Munck, J.M.; Batey, M.A.; Zhao, Y.; Jenkins, H.; Richardson, C.J.; Cano, C.; Tavecchio, M.; Barbeau, J.; Bardos, J.; Cornell, L.; et al. Chemosensitization of cancer cells by KU-0060648, a dual inhibitor of DNA-PK and PI-3K. Mol. Cancer Ther. 2012, 11, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Alagpulinsa, D.A.; Ayyadevara, S.; Shmookler Reis, R.J. A small-molecule inhibitor of RAD51 reduces homologous recombination and sensitizes multiple myeloma cells to doxorubicin. Front. Oncol. 2014, 4, 289. [Google Scholar] [CrossRef] [PubMed]
- Makarova, K.S.; Wolf, Y.I.; Alkhnbashi, O.S.; Costa, F.; Shah, S.A.; Saunders, S.J.; Barrangou, R.; Brouns, S.J.; Charpentier, E.; Haft, D.H.; et al. An updated evolutionary classification of CRISPR–Cas systems. Nat. Rev. Microbiol. 2015, 13, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; East, A.; Cheng, A.; Lin, S.; Ma, E.; Doudna, J. RNA-programmed genome editing in human cells. eLife 2013, 2, e00471. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.A.; Erdmann, S.; Mojica, F.J.M.; Garrett, R.A. Protospacer recognition motifs: Mixed identities and functional diversity. RNA Biol. 2013, 10, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Sternberg, S.H.; Redding, S.; Jinek, M.; Greene, E.C.; Doudna, J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 2014, 507, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Anders, C.; Niewoehner, O.; Duerst, A.; Jinek, M. Structural basis of PAM-dependent target DNA recognition by the Cas9 endonuclease. Nature 2014, 513, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Bothmer, A.; Phadke, T.; Barrera, L.A.; Margulies, C.M.; Lee, C.S.; Buquicchio, F.; Moss, S.; Abdulkerim, H.S.; Selleck, W.; Jayaram, H.; et al. Characterization of the interplay between DNA repair and CRISPR/Cas9-induced DNA lesions at an endogenous locus. Nat. Commun. 2017, 8, 13905. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; González, F.; Huangfu, D. The iCRISPR platform for rapid genome editing in human Pluripotent Stem Cells. Methods Enzymol. 2014, 546, 215–250. [Google Scholar] [PubMed]
- Yang, S.; Chang, R.; Yang, H.; Zhao, T.; Hong, Y.; Kong, H.E.; Sun, X.; Qin, Z.; Jin, P.; Li, S.; et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J. Clin. Investig. 2017, 127, 2719–2724. [Google Scholar] [CrossRef] [PubMed]
- Carroll, D. Genome editing: Progress and challenges for medical applications. Genome Med. 2016, 8, 120. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Xu, Y.; Zhang, X.; Ding, C.; Huang, R.; Zhang, Z.; Lv, J.; Xie, X.; Chen, Y.; Li, Y.; et al. CRISPR/Cas9-mediated gene editing in human tripronuclear zygotes. Protein Cell 2015, 6, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.; Tong, Y.; Liu, X.Z.; Wang, T.T.; Cheng, L.; Wang, B.Y.; Lv, X.; Huang, Y.; Liu, D.P. Both TALENs and CRISPR/Cas9 directly target the HBB IVS2–654 (C > T) mutation in β-thalassemia-derived iPSCs. Sci. Rep. 2015, 5, 12065. [Google Scholar] [CrossRef] [PubMed]
- Ou, Z.; Niu, X.; He, W.; Chen, Y.; Song, B.; Xian, Y.; Fan, D.; Tang, D.; Sun, X. The combination of CRISPR/Cas9 and iPSC technologies in the gene therapy of human β-thalassemia in mice. Sci. Rep. 2016, 6, 32463. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| DNA Repair Mechanism | Associated Disease/Disorder | Mutation/Deficiency Responsible | Clinical Presentation |
|---|---|---|---|
| Mismatch repair | Lynch syndrome Constitutional mismatch repair deficiency syndrome | MSH and MLH mutations [103] | Colorectal cancer, endometrial cancer [103] |
| Nucleotide-excision repair | Xeroderma pigmentosum disorder | Mutations in xeroderma pigmentosum complexes [104] | Neurodegeneration, photosensitivity, skin cancer [104] |
| Base-excision repair | Spinocerebellar ataxia with axonal neuropathy (SCAN1) Ataxia with oculomotor apraxia 1 (AOA1) | Tyrosyl-DNA phosphodiesterase deficiency [105] Aprataxin deficiency [106] | Ataxia, neurodegeneration [105,106] |
| DNA damage response (DDR) | Li-Fraumeni syndrome | p53 mutation [107] | Soft tissue sarcomas, breast cancer, brain tumours [107] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Helena, J.M.; Joubert, A.M.; Grobbelaar, S.; Nolte, E.M.; Nel, M.; Pepper, M.S.; Coetzee, M.; Mercier, A.E. Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes. Int. J. Mol. Sci. 2018, 19, 1148. https://doi.org/10.3390/ijms19041148
Helena JM, Joubert AM, Grobbelaar S, Nolte EM, Nel M, Pepper MS, Coetzee M, Mercier AE. Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes. International Journal of Molecular Sciences. 2018; 19(4):1148. https://doi.org/10.3390/ijms19041148
Chicago/Turabian StyleHelena, Jolene Michelle, Anna Margaretha Joubert, Simone Grobbelaar, Elsie Magdalena Nolte, Marcel Nel, Michael Sean Pepper, Magdalena Coetzee, and Anne Elisabeth Mercier. 2018. "Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes" International Journal of Molecular Sciences 19, no. 4: 1148. https://doi.org/10.3390/ijms19041148
APA StyleHelena, J. M., Joubert, A. M., Grobbelaar, S., Nolte, E. M., Nel, M., Pepper, M. S., Coetzee, M., & Mercier, A. E. (2018). Deoxyribonucleic Acid Damage and Repair: Capitalizing on Our Understanding of the Mechanisms of Maintaining Genomic Integrity for Therapeutic Purposes. International Journal of Molecular Sciences, 19(4), 1148. https://doi.org/10.3390/ijms19041148