Effects of Mutations and Ligands on the Thermostability of the l-Arginine/Agmatine Antiporter AdiC and Deduced Insights into Ligand-Binding of Human l-Type Amino Acid Transporters

Abstract

{kind=link}
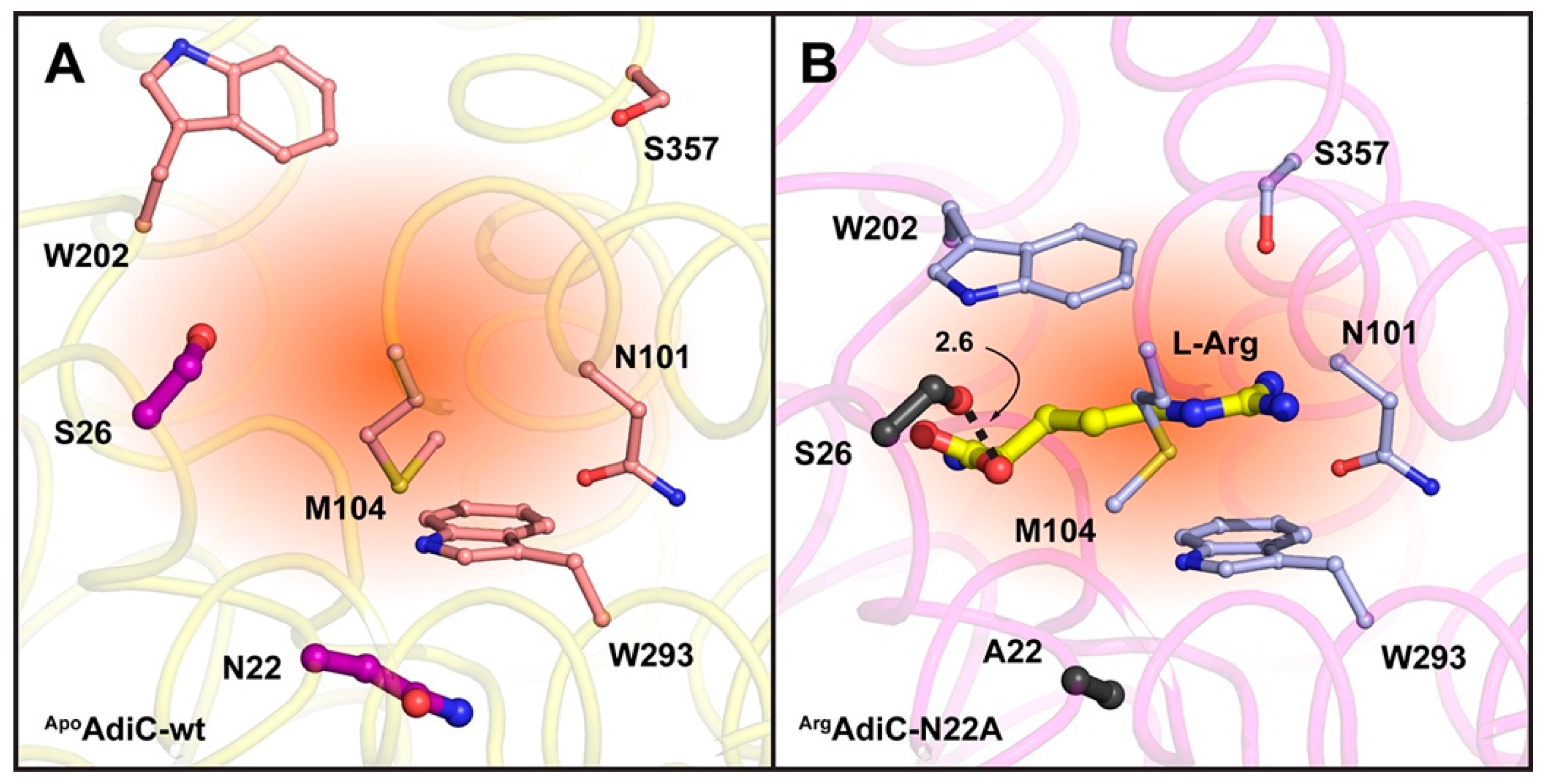{kind=link}
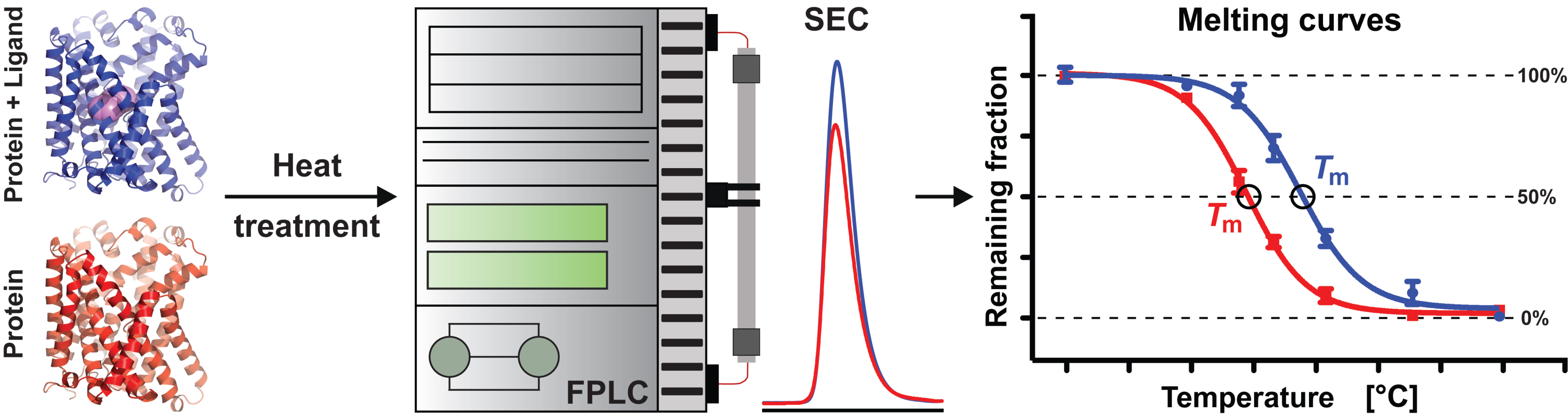{kind=link}
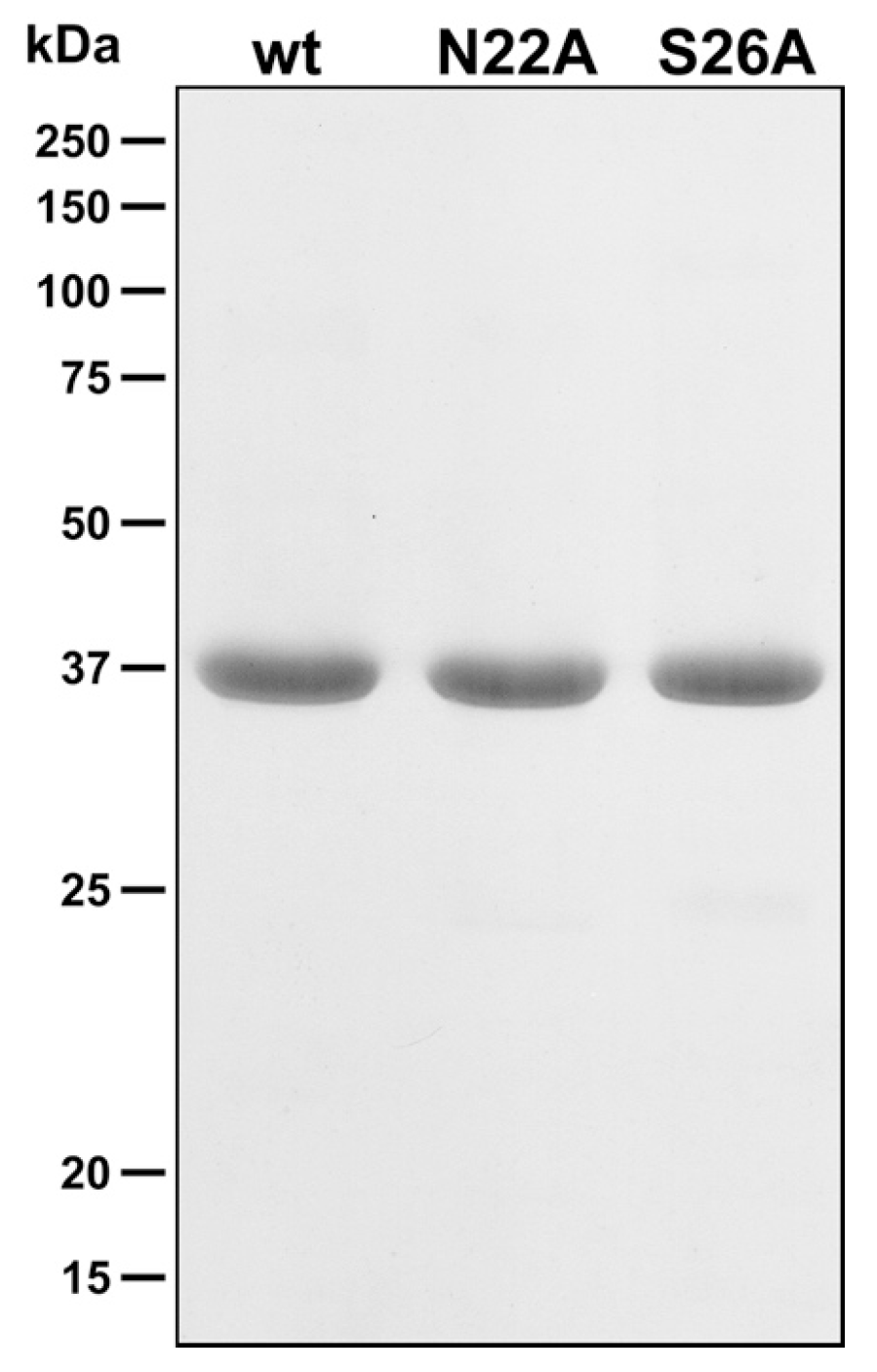{kind=link}
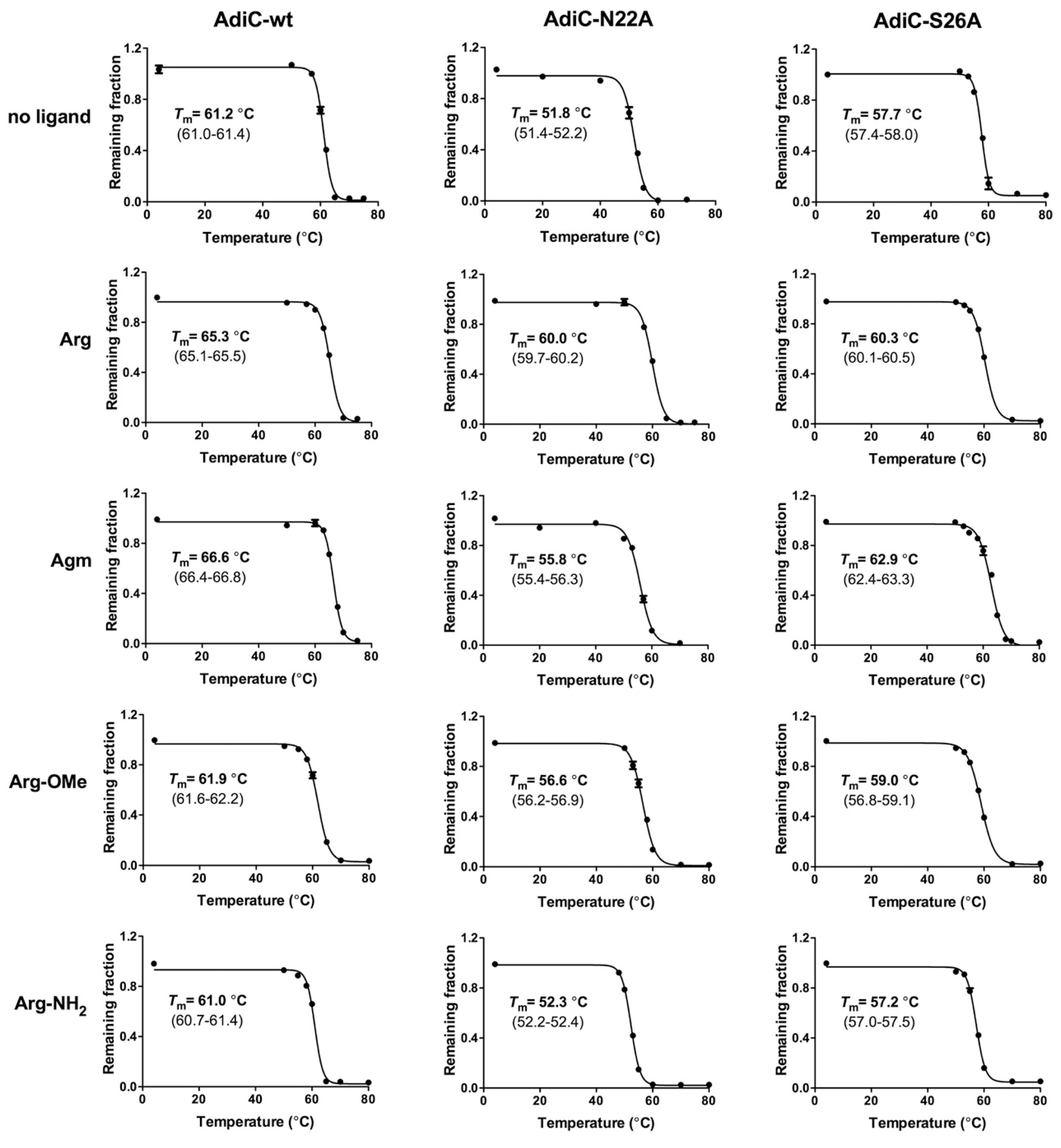{kind=link}
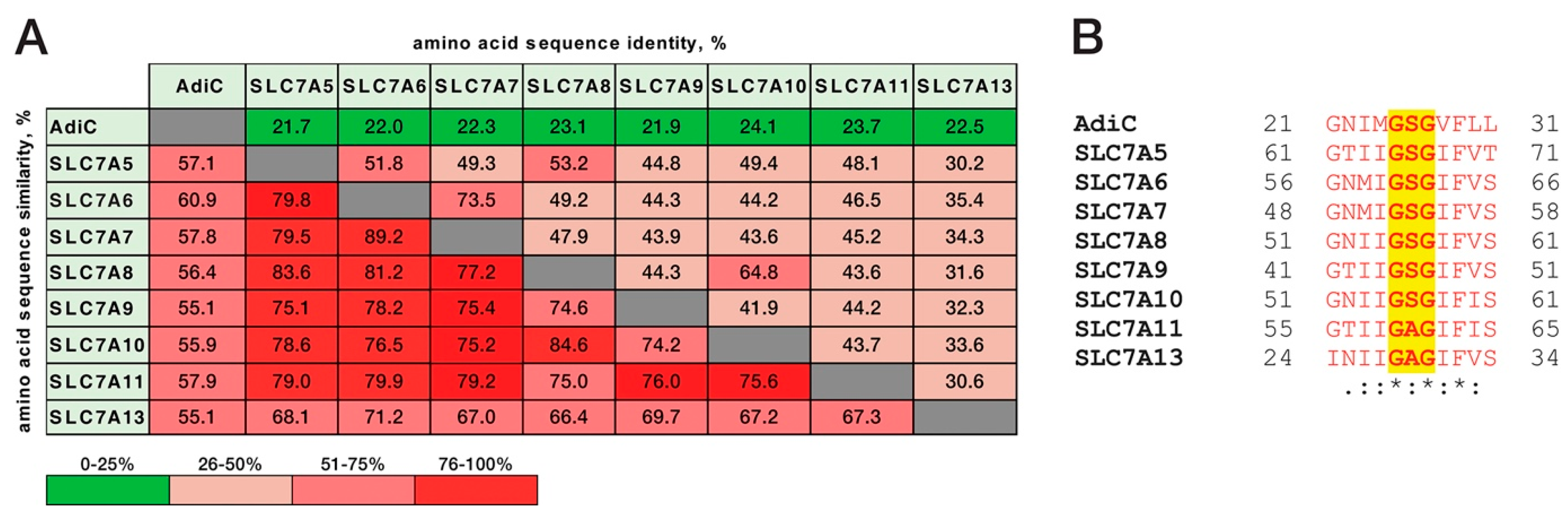{kind=link}
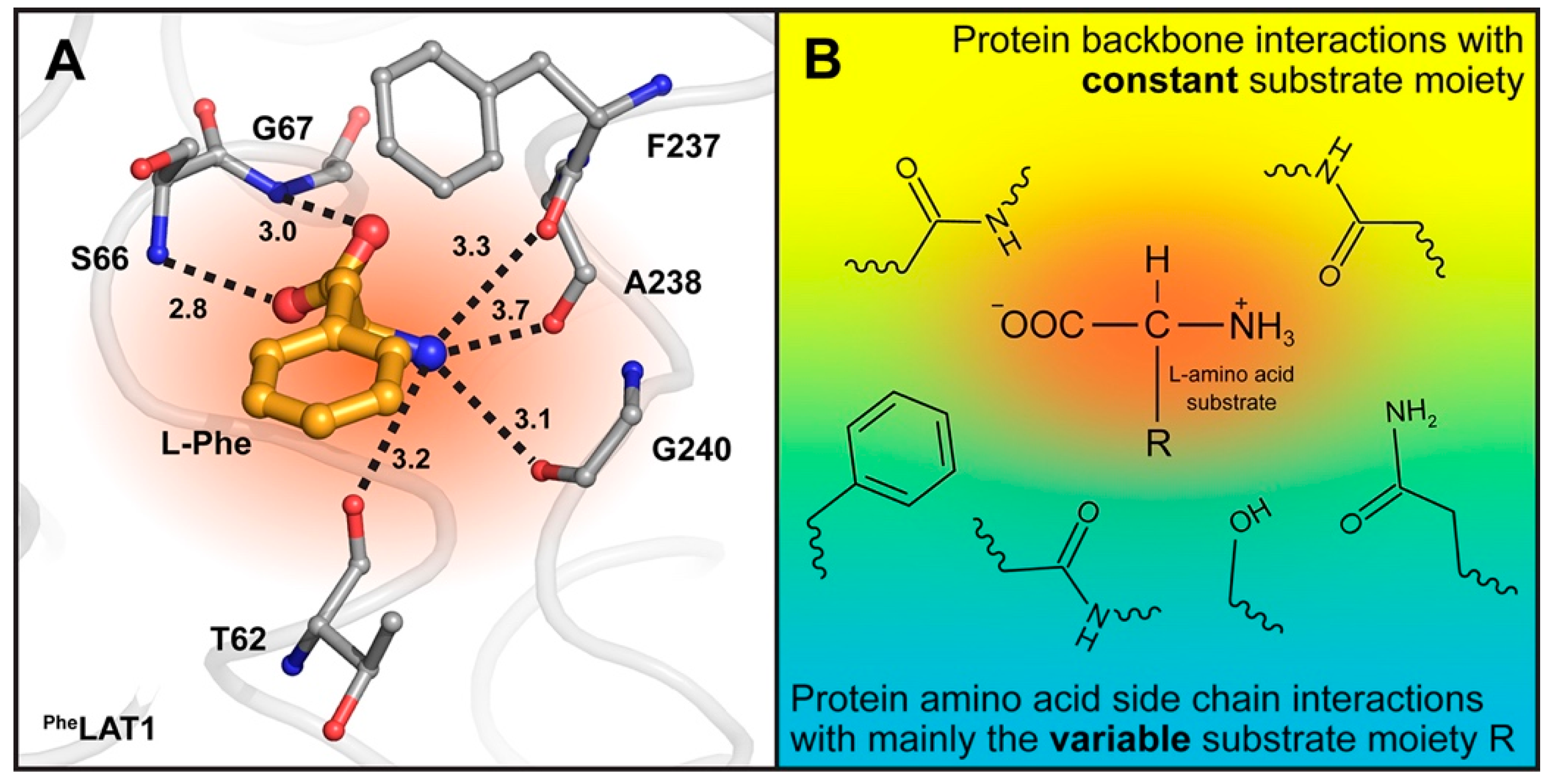{kind=link}
1. Introduction
2. Results and Discussion
3. Materials and Methods
3.1. Cloning and Overexpression of AdiC
3.2. Membrane Preparation and Purification of AdiC
3.3. Determination of AdiC Thermostability
3.4. Amino Acid Sequence Analysis
3.5. Homology Models and Ligand Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Foster, J.W. Escherichia coli acid resistance: Tales of an amateur acidophile. Nat. Rev. Microbiol. 2004, 2, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Casagrande, F.; Ratera, M.; Schenk, A.D.; Chami, M.; Valencia, E.; Lopez, J.M.; Torrents, D.; Engel, A.; Palacin, M.; Fotiadis, D. Projection structure of a member of the amino acid/polyamine/organocation transporter superfamily. J. Biol. Chem. 2008, 283, 33240–33248. [Google Scholar] [CrossRef] [PubMed]
- Fotiadis, D.; Kanai, Y.; Palacin, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Asp. Med. 2013, 34, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Lu, F.; Zhou, L.; Dang, S.; Sun, L.; Li, X.; Wang, J.; Shi, Y. Structure and mechanism of an amino acid antiporter. Science 2009, 324, 1565–1568. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Jayaram, H.; Shane, T.; Kolmakova-Partensky, L.; Wu, F.; Williams, C.; Xiong, Y.; Miller, C. Structure of a prokaryotic virtual proton pump at 3.2 Å resolution. Nature 2009, 460, 1040–1043. [Google Scholar] [CrossRef] [PubMed]
- Ilgü, H.; Jeckelmann, J.-M.; Gapsys, V.; Ucurum, Z.; de Groot, B.L.; Fotiadis, D. Insights into the molecular basis for substrate binding and specificity of the wild-type L-arginine/agmatine antiporter AdiC. Proc. Natl. Acad. Sci. USA 2016, 113, 10358–10363. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, L.; Ratera, M.; Paladino, A.; Bartoccioni, P.; Errasti-Murugarren, E.; Valencia, E.; Portella, G.; Bial, S.; Zorzano, A.; Fita, I.; et al. Molecular basis of substrate-induced permeation by an amino acid antiporter. Proc. Natl. Acad. Sci. USA 2011, 108, 3935–3940. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Zhou, L.; Jiao, X.; Lu, F.; Yan, C.; Zeng, X.; Wang, J.; Shi, Y. Mechanism of substrate recognition and transport by an amino acid antiporter. Nature 2010, 463, 828–832. [Google Scholar] [CrossRef] [PubMed]
- Geier, E.G.; Schlessinger, A.; Fan, H.; Gable, J.E.; Irwin, J.J.; Sali, A.; Giacomini, K.M. Structure-based ligand discovery for the large-neutral amino acid transporter 1, LAT-1. Proc. Natl. Acad. Sci. USA 2013, 110, 5480–5485. [Google Scholar] [CrossRef] [PubMed]
- Dickens, D.; Webb, S.D.; Antonyuk, S.; Giannoudis, A.; Owen, A.; Radisch, S.; Hasnain, S.S.; Pirmohamed, M. Transport of gabapentin by LAT1 (SLC7A5). Biochem. Pharmacol. 2013, 85, 1672–1683. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Scalise, M.; Galluccio, M.; Pochini, L.; Albanese, L.M.; Indiveri, C. LAT1 is the transport competent unit of the LAT1/CD98 heterodimeric amino acid transporter. Int. J. Biochem. Cell Biol. 2015, 67, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Augustyn, E.; Finke, K.; Zur, A.A.; Hansen, L.; Heeren, N.; Chien, H.C.; Lin, L.; Giacomini, K.M.; Colas, C.; Schlessinger, A.; et al. LAT-1 activity of meta-substituted phenylalanine and tyrosine analogs. Bioorg. Med. Chem. Lett. 2016, 26, 2616–2621. [Google Scholar] [CrossRef] [PubMed]
- Colas, C.; Ung, P.M.; Schlessinger, A. SLC transporters: Structure, function, and drug discovery. MedChemComm 2016, 7, 1069–1081. [Google Scholar] [CrossRef] [PubMed]
- Zur, A.A.; Chien, H.C.; Augustyn, E.; Flint, A.; Heeren, N.; Finke, K.; Hernandez, C.; Hansen, L.; Miller, S.; Lin, L.; et al. LAT1 activity of carboxylic acid bioisosteres: Evaluation of hydroxamic acids as substrates. Bioorg. Med. Chem. Lett. 2016, 26, 5000–5006. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Galluccio, M.; Scalise, M.; Parravicini, C.; Palazzolo, L.; Eberini, I.; Indiveri, C. Novel insights into the transport mechanism of the human amino acid transporter LAT1 (SLC7A5). Probing critical residues for substrate translocation. Biochim. Biophys. Acta 2017, 1861, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Napolitano, L.; Scalise, M.; Koyioni, M.; Koutentis, P.; Catto, M.; Eberini, I.; Parravicini, C.; Palazzolo, L.; Pisani, L.; Galluccio, M.; et al. Potent inhibitors of human LAT1 (SLC7A5) transporter based on dithiazole and dithiazine compounds for development of anticancer drugs. Biochem. Pharmacol. 2017, 143, 39–52. [Google Scholar] [CrossRef] [PubMed]
- Rosell, A.; Meury, M.; Alvarez-Marimon, E.; Costa, M.; Perez-Cano, L.; Zorzano, A.; Fernandez-Recio, J.; Palacin, M.; Fotiadis, D. Structural bases for the interaction and stabilization of the human amino acid transporter LAT2 with its ancillary protein 4F2hc. Proc. Natl. Acad. Sci. USA 2014, 111, 2966–2971. [Google Scholar] [CrossRef] [PubMed]
- Hinz, K.M.; Meyer, K.; Kinne, A.; Schulein, R.; Kohrle, J.; Krause, G. Structural insights into thyroid hormone transport mechanisms of the L-type amino acid transporter 2. Mol. Endocrinol. 2015, 29, 933–942. [Google Scholar] [CrossRef] [PubMed]
- Hinz, K.M.; Neef, D.; Rutz, C.; Furkert, J.; Kohrle, J.; Schulein, R.; Krause, G. Molecular features of the L-type amino acid transporter 2 determine different import and export profiles for thyroid hormones and amino acids. Mol. Cell. Endocrinol. 2017, 443, 163–174. [Google Scholar] [CrossRef] [PubMed]
- Krause, G.; Hinz, K.M. Thyroid hormone transport across L-type amino acid transporters: What can molecular modelling tell us? Mol. Cell. Endocrinol. 2017, 458, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Fang, Y.; Miller, C. Sided functions of an arginine-agmatine antiporter oriented in liposomes. Biochemistry 2012, 51, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.F.; Miller, C. Substrate selectivity in arginine-dependent acid resistance in enteric bacteria. Proc. Natl. Acad. Sci. USA 2013, 110, 5893–5897. [Google Scholar] [CrossRef] [PubMed]
- Magnani, F.; Serrano-Vega, M.J.; Shibata, Y.; Abdul-Hussein, S.; Lebon, G.; Miller-Gallacher, J.; Singhal, A.; Strege, A.; Thomas, J.A.; Tate, C.G. A mutagenesis and screening strategy to generate optimally thermostabilized membrane proteins for structural studies. Nat. Protoc. 2016, 11, 1554–1571. [Google Scholar] [CrossRef] [PubMed]
- Mancusso, R.; Karpowich, N.K.; Czyzewski, B.K.; Wang, D.N. Simple screening method for improving membrane protein thermostability. Methods 2011, 55, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Kolmakova-Partensky, L.; Miller, C. A bacterial arginine-agmatine exchange transporter involved in extreme acid resistance. J. Biol. Chem. 2007, 282, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, E.; Torrents, D.; Zorzano, A.; Palacin, M.; Chillaron, J. Identification and functional characterization of a novel low affinity aromatic-preferring amino acid transporter (arpAT). One of the few proteins silenced during primate evolution. J. Biol. Chem. 2005, 280, 19364–19372. [Google Scholar] [CrossRef] [PubMed]
- Nicklin, P.; Bergman, P.; Zhang, B.; Triantafellow, E.; Wang, H.; Nyfeler, B.; Yang, H.; Hild, M.; Kung, C.; Wilson, C.; et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 2009, 136, 521–534. [Google Scholar] [CrossRef] [PubMed]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–634. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, A.; Khuri, N.; Giacomini, K.M.; Sali, A. Molecular modeling and ligand docking for solute carrier (SLC) transporters. Curr. Top. Med. Chem. 2013, 13, 843–856. [Google Scholar] [CrossRef] [PubMed]
- Ilgü, H.; Jeckelmann, J.M.; Gachet, M.S.; Boggavarapu, R.; Ucurum, Z.; Gertsch, J.; Fotiadis, D. Variation of the detergent-binding capacity and phospholipid content of membrane proteins when purified in different detergents. Biophys. J. 2014, 106, 1660–1670. [Google Scholar] [CrossRef] [PubMed]
- McWilliam, H.; Li, W.; Uludag, M.; Squizzato, S.; Park, Y.M.; Buso, N.; Cowley, A.P.; Lopez, R. Analysis Tool Web Services from the EMBL-EBI. Nucleic Acids Res. 2013, 41, W597–W600. [Google Scholar] [CrossRef] [PubMed]
- Pei, J.; Kim, B.H.; Grishin, N.V. PROMALS3D: A tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 2008, 36, 2295–2300. [Google Scholar] [CrossRef] [PubMed]
- Eswar, N.; Webb, B.; Marti-Renom, M.A.; Madhusudhan, M.S.; Eramian, D.; Shen, M.Y.; Pieper, U.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2006. [Google Scholar]
- Krivov, G.G.; Shapovalov, M.V.; Dunbrack, R.L., Jr. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009, 77, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Meury, M.; Harder, D.; Ucurum, Z.; Boggavarapu, R.; Jeckelmann, J.-M.; Fotiadis, D. Structure determination of channel and transport proteins by high-resolution microscopy techniques. Biol. Chem. 2011, 392, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Harder, D.; Fotiadis, D. Measuring substrate binding and affinity of purified membrane transport proteins using the scintillation proximity assay. Nat. Protoc. 2012, 7, 1569–1578. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ilgü, H.; Jeckelmann, J.-M.; Colas, C.; Ucurum, Z.; Schlessinger, A.; Fotiadis, D. Effects of Mutations and Ligands on the Thermostability of the l-Arginine/Agmatine Antiporter AdiC and Deduced Insights into Ligand-Binding of Human l-Type Amino Acid Transporters. Int. J. Mol. Sci. 2018, 19, 918. https://doi.org/10.3390/ijms19030918
Ilgü H, Jeckelmann J-M, Colas C, Ucurum Z, Schlessinger A, Fotiadis D. Effects of Mutations and Ligands on the Thermostability of the l-Arginine/Agmatine Antiporter AdiC and Deduced Insights into Ligand-Binding of Human l-Type Amino Acid Transporters. International Journal of Molecular Sciences. 2018; 19(3):918. https://doi.org/10.3390/ijms19030918
Chicago/Turabian StyleIlgü, Hüseyin, Jean-Marc Jeckelmann, Claire Colas, Zöhre Ucurum, Avner Schlessinger, and Dimitrios Fotiadis. 2018. "Effects of Mutations and Ligands on the Thermostability of the l-Arginine/Agmatine Antiporter AdiC and Deduced Insights into Ligand-Binding of Human l-Type Amino Acid Transporters" International Journal of Molecular Sciences 19, no. 3: 918. https://doi.org/10.3390/ijms19030918
APA StyleIlgü, H., Jeckelmann, J.-M., Colas, C., Ucurum, Z., Schlessinger, A., & Fotiadis, D. (2018). Effects of Mutations and Ligands on the Thermostability of the l-Arginine/Agmatine Antiporter AdiC and Deduced Insights into Ligand-Binding of Human l-Type Amino Acid Transporters. International Journal of Molecular Sciences, 19(3), 918. https://doi.org/10.3390/ijms19030918