In-Silico Integration Approach to Identify a Key miRNA Regulating a Gene Network in Aggressive Prostate Cancer

 , ,
, ,  and
and
Abstract
1. Introduction
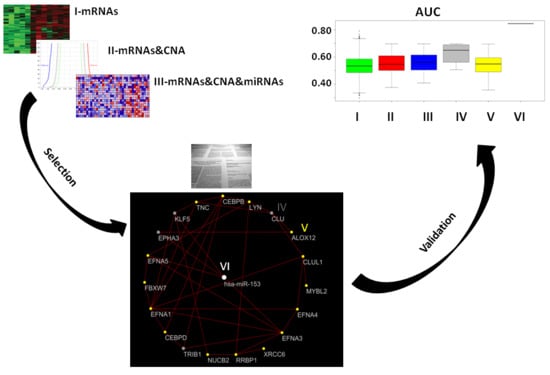
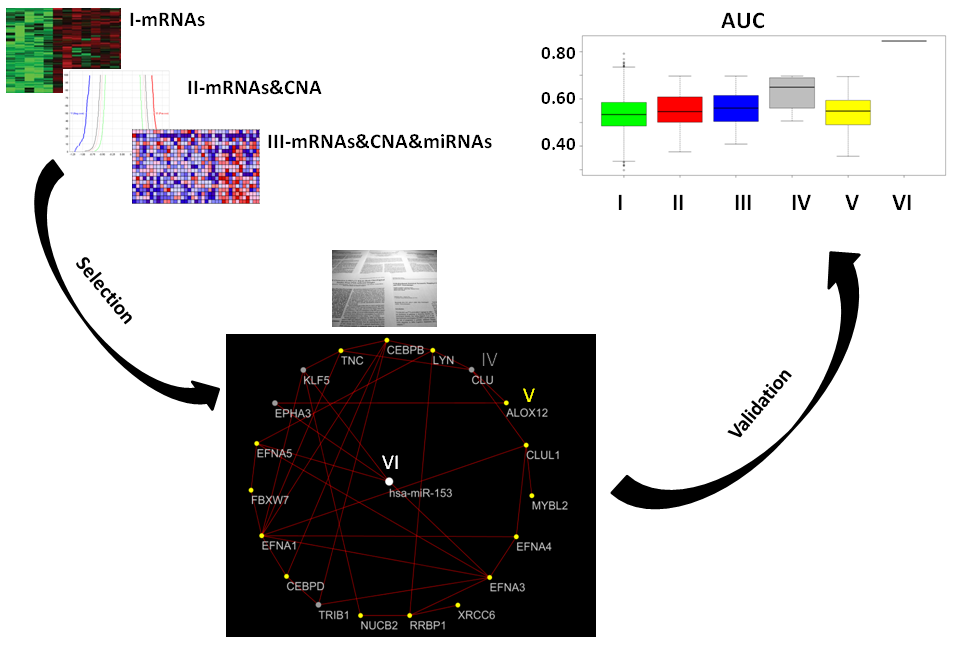
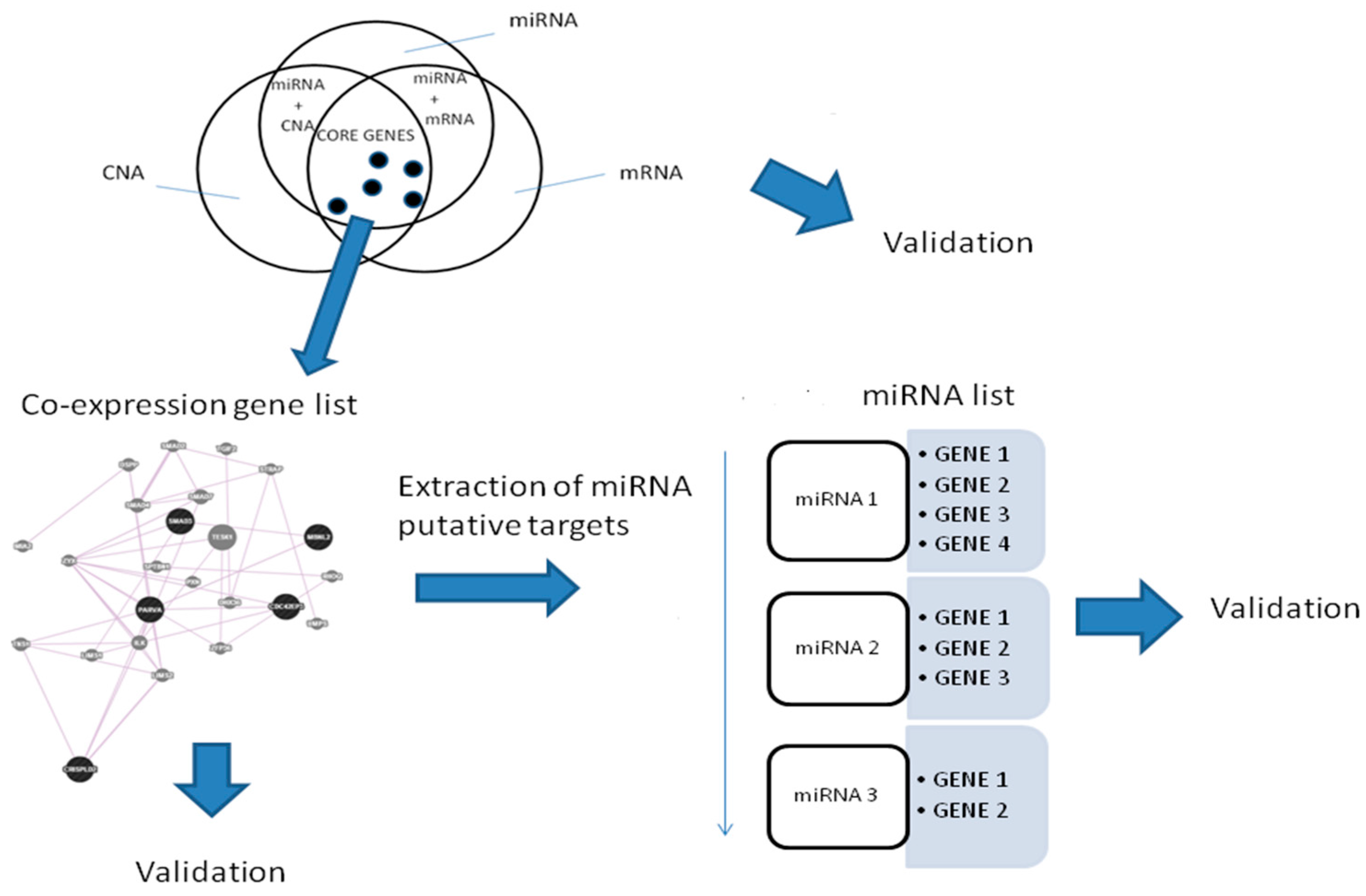
2. Results
2.1. Gene Expression, miRNA, and CNA Analyses
2.2. Combination of Gene Expression and CNA
2.3. Combination of Gene Expression or CNA and miRNAs
2.4. Combination of Gene Expression, CNA, and miRNA
2.5. Prostate Cancer Signatures
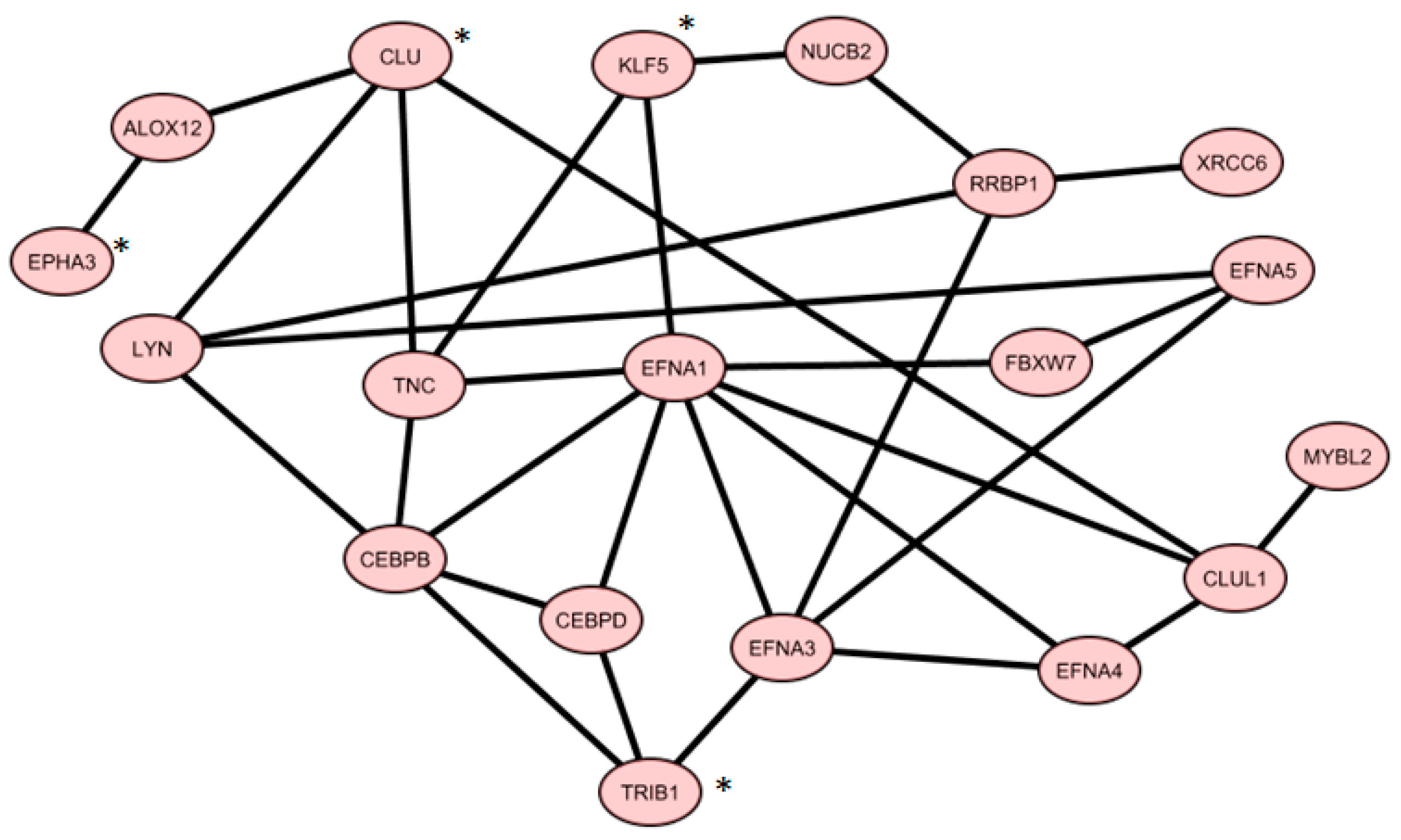
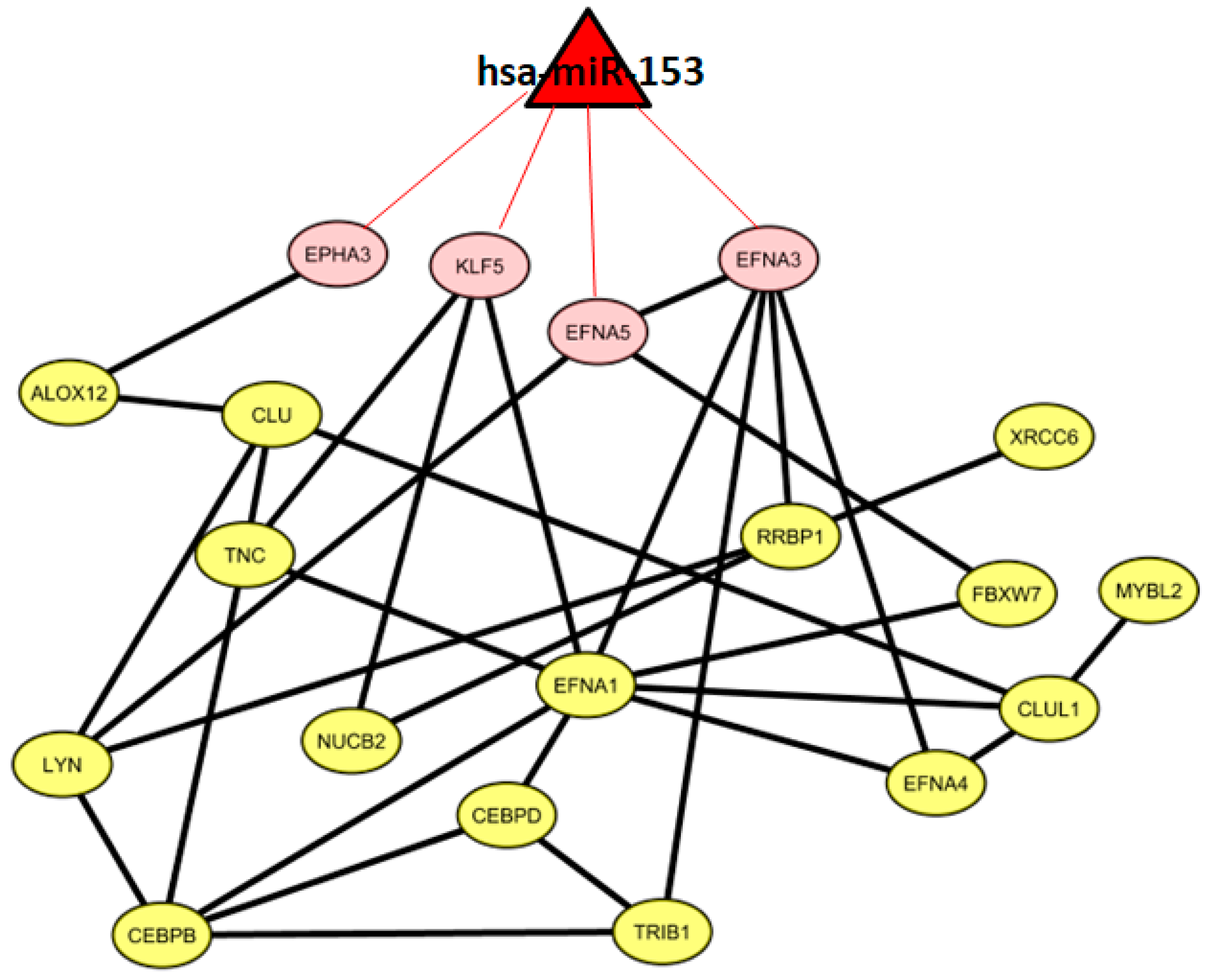
2.6. Co-Expressed Network
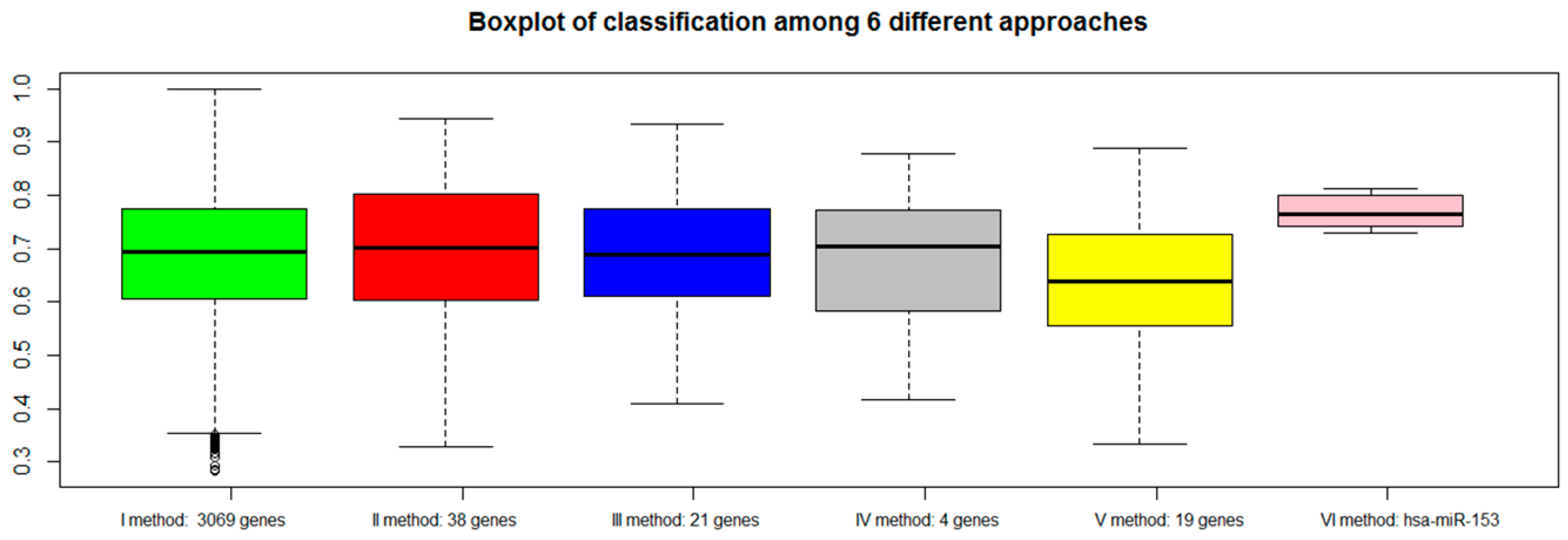
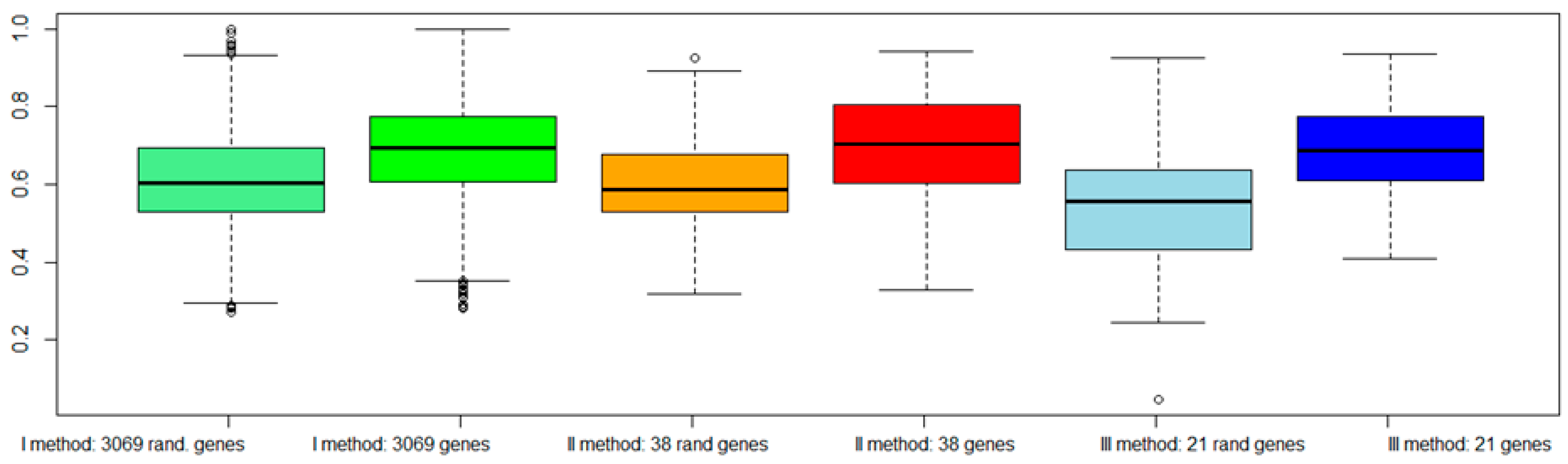
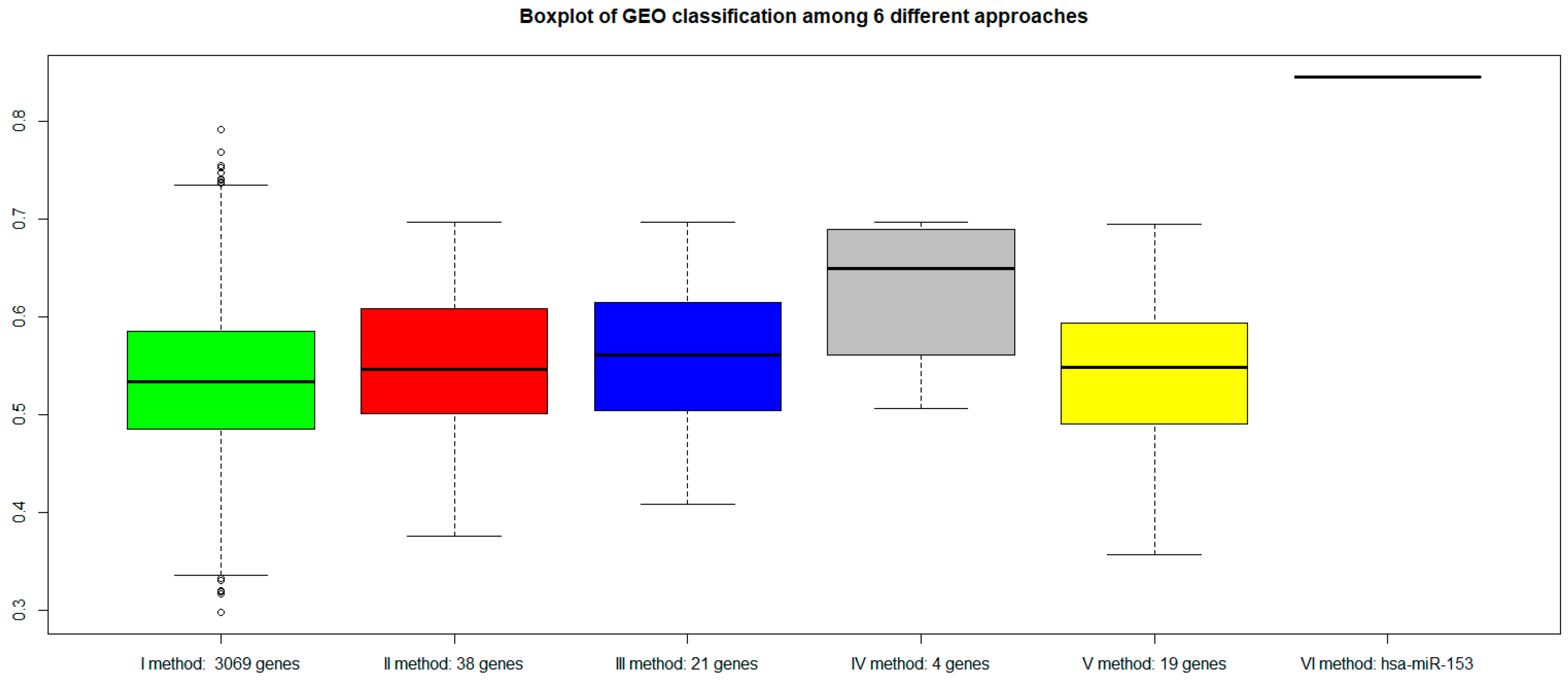
2.7. Classification of Normal and Aggressive Prostate Cancer Samples
3. Discussion
3.1. IV Four-Gene Signature
3.2. V 19-Gene Signature
- Phosphatidylinositol-3-kinases (PI3K/AKT) pathway, which include, for example, the ephrin family members (EFNA1, A3, A4, A5, EPHA3, …), the proto-oncogene LYN, and tenascin C (TNC);
- Cell cycle control and proliferation pathways, which include, for example, the Myb-related protein B (MYBL2) and the retinoic acid receptor beta (RARB);
- Protein Transcription and half-life pathways, which include, for instance, the enhancer binding proteins CEBPB and D, the ribosome binding protein 1 (RRBP1), the ubiquitin protein ligase FBXW7, and KLF5.
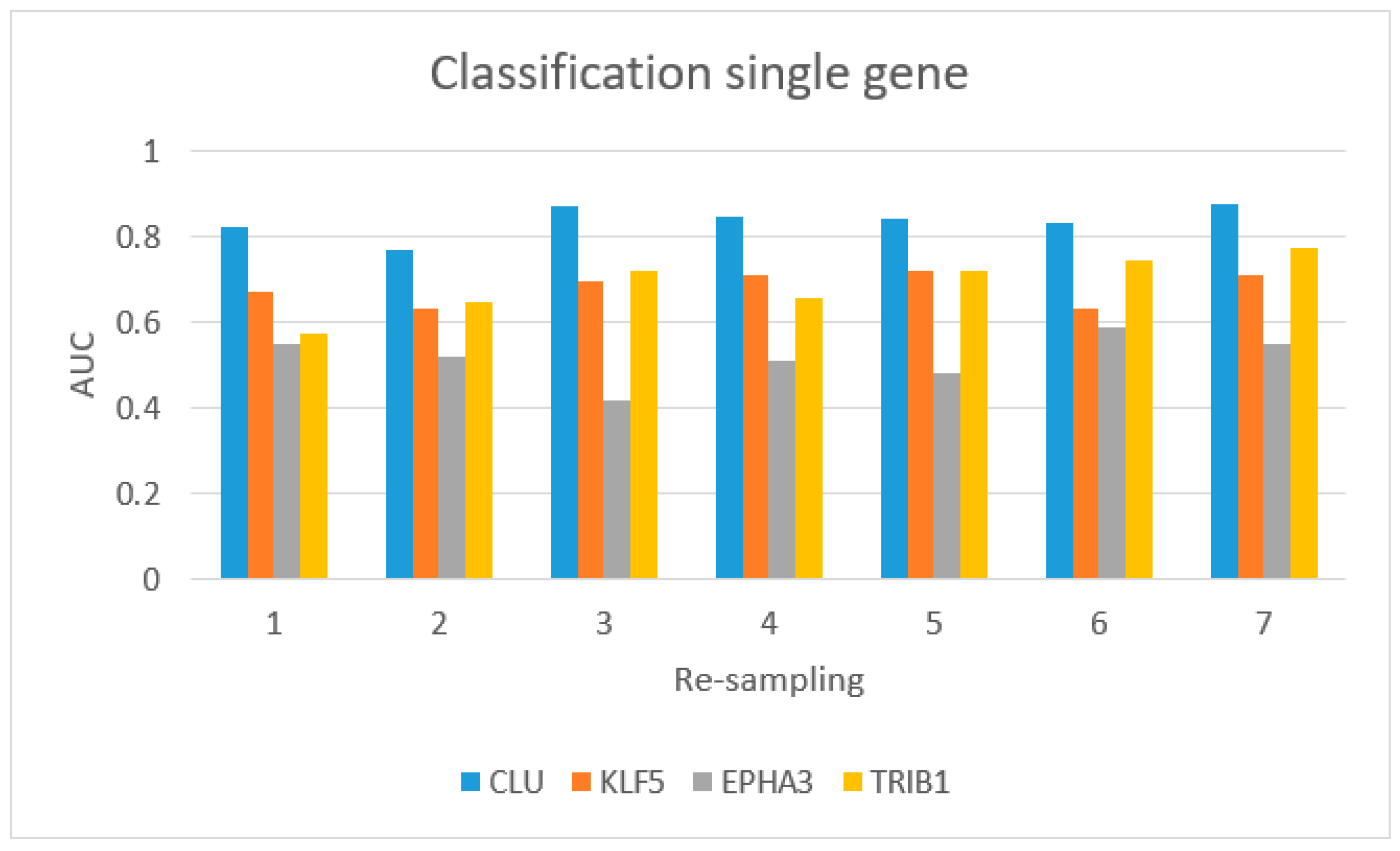
3.3. VI miRNA Signature: hsa-miR-153
4. Materials and Methods
4.1. Gene and miRNA Expression Analysis
4.2. Analysis of miRNA Targets
4.3. Copy Number Alterations Analysis
4.4. Combination of Gene Expression and Copy Number Alteration
4.5. Combination of Gene Expression, CNA, and miRNAs
4.6. Prostate Cancer Signatures
4.7. Co-Expressed Network
4.8. The Classifier
4.9. Evaluation of the Approaches
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| PC | Prostate Cancer |
| CNA | Copy Number Alteration |
| NS | Normal Sample |
| TCGA | The Cancer Genome Atlas |
References
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2012. CA Cancer J. Clin. 2012, 62, 10–29. [Google Scholar] [CrossRef] [PubMed]
- Antenor, J.A.; Roehl, K.A.; Eggener, S.E.; Kundu, S.D.; Han, M.; Catalona, W.J. Preoperative PSA and progression-free survival after radical prostatectomy for Stage T1c disease. Urology 2005, 66, 156–160. [Google Scholar] [CrossRef] [PubMed]
- McDonald, A.C.; Vira, M.A.; Vidal, A.C.; Gan, W.; Freedland, S.J.; Taioli, E. Association between systemic inflammatory markers and serum prostate-specific antigen in men without prostatic disease—The 2001–2008 National Health and Nutrition Examination Survey. Prostate 2014, 74, 561–567. [Google Scholar] [CrossRef] [PubMed]
- Chiam, K.; Ricciardelli, C.; Bianco-Miotto, T. Epigenetic biomarkers in prostate cancer: Current and future uses. Cancer Lett. 2014, 342, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Ciatto, S.; Zappa, M.; Bonardi, R.; Gervasi, G. Prostate cancer screening: The problem of overdiagnosis and lessons to be learned from breast cancer screening. Eur. J. Cancer 2000, 36, 1347–1350. [Google Scholar] [CrossRef]
- Dall’Era, M.A.; Cooperberg, M.R.; Chan, J.M.; Davies, B.J.; Albertsen, P.C.; Klotz, L.H.; Warlick, C.A.; Holmberg, L.; Bailey, D.E., Jr.; Wallace, M.E.; et al. Active surveillance for early-stage prostate cancer: Review of the current literature. Cancer 2008, 112, 1650–1659. [Google Scholar] [CrossRef] [PubMed]
- Bryant, R.J.; Pawlowski, T.; Catto, J.W.; Marsden, G.; Vessella, R.L.; Rhees, B.; Kuslich, C.; Visakorpi, T.; Hamdy, F.C. Changes in circulating microRNA levels associated with prostate cancer. Br. J. Cancer 2012, 106, 768–774. [Google Scholar] [CrossRef] [PubMed]
- Ploussard, G.; de la Taille, A. Urine biomarkers in prostate cancer. Nat. Rev. Urol. 2010, 7, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chun, F.K.; de la Taille, A.; van Poppel, H.; Marberger, M.; Stenzl, A.; Mulders, P.F.; Huland, H.; Abbou, C.C.; Stillebroer, A.B.; van Gils, M.P.; et al. Prostate cancer gene 3 (PCA3): Development and internal validation of a novel biopsy nomogram. Eur. Urol. 2009, 56, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Groskopf, J.; Aubin, S.M.; Deras, I.L.; Blase, A.; Bodrug, S.; Clark, C.; Brentano, S.; Mathis, J.; Pham, J.; Meyer, T.; et al. APTIMA PCA3 molecular urine test: Development of a method to aid in the diagnosis of prostate cancer. Clin. Chem. 2006, 52, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Laxman, B.; Morris, D.S.; Yu, J.; Siddiqui, J.; Cao, J.; Mehra, R.; Lonigro, R.J.; Tsodikov, A.; Wei, J.T.; Tomlins, S.A.; et al. A first-generation multiplex biomarker analysis of urine for the early detection of prostate cancer. Cancer Res. 2008, 68, 645–649. [Google Scholar] [CrossRef] [PubMed]
- Roobol, M.J.; Schröder, F.H.; van Leenders, G.L.; Hessels, D.; van den Bergh, R.C.; Wolters, T.; van Leeuwen, P.J. Performance of prostate cancer antigen 3 (PCA3) and prostate-specific antigen in Prescreened men: Reproducibility and detection characteristics for prostate cancer patients with high PCA3 scores (≥100). Eur. Urol. 2010, 58, 893–899. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; De Summa, S.; Petriella, D.; Tudoran, O.; Danza, K.; Tommasi, S. The value of new high-throughput technologies for diagnosis and prognosis in solid tumors. Cancer Biomark. 2014, 14, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Bertoli, G.; Cava, C.; Castiglioni, I. MicroRNAs as Biomarkers for Diagnosis, Prognosis and Theranostics in Prostate Cancer. Int. J. Mol. Sci. 2016, 17, 421. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Colaprico, A.; Bertoli, G.; Bontempi, G.; Mauri, G.; Castiglioni, I. How interacting pathways are regulated by miRNAs in breast cancer subtypes. BMC Bioinform. 2016, 17 (Suppl. S12), 348. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Castiglioni, I. Integrating genetics and epigenetics in breast cancer: Biological insights, experimental, computational methods and therapeutic potential. BMC Syst. Biol. 2015, 9, 62. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar] [CrossRef] [PubMed]
- Glinge, C.; Clauss, S.; Boddum, K.; Jabbari, R.; Jabbari, J.; Risgaard, B.; Tomsits, P.; Hildebrand, B.; Kääb, S.; Wakili, R.; et al. Stability of Circulating Blood-Based MicroRNAs—Pre-Analytic Methodological Considerations. PLoS ONE 2017, 12, e0167969. [Google Scholar] [CrossRef] [PubMed]
- Fesler, A.; Jiang, J.; Zhai, H.; Ju, J. Circulating microRNA testing for the early diagnosis and follow-up of colorectal cancer patients. Mol. Diagn. Ther. 2014, 18, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.B.; Li, T.B.; Zhang, Z.; Ren, K.D.; Zheng, Z.F.; Peng, J.; Luo, X.J. The Diagnostic Value of Circulating Brain-specific MicroRNAs for Ischemic Stroke. Intern. Med. 2016, 55, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yang, R.; Hu, B.L.; Lu, P.; Zhou, L.L.; He, Z.Y.; Wu, H.M.; Zhu, J.H. Reduced Circulating Levels of miR-433 and miR-133b Are Potential Biomarkers for Parkinson’s Disease. Front. Cell. Neurosci. 2017, 11, 170. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Quiroz, E.; Pacheco-Lugo, L.; Navarro-Quiroz, R.; Lorenzi, H.; España-Puccini, P.; Díaz-Olmos, Y.; Almendrales, L.; Olave, V.; Gonzalez-Torres, H.; Diaz-Perez, A.; et al. Profiling analysis of circulating microRNA in peripheral blood of patients with class IV lupus nephritis. PLoS ONE 2017, 12, e0187973. [Google Scholar] [CrossRef] [PubMed]
- True, L.; Coleman, I.; Hawley, S.; Huang, C.Y.; Gifford, D.; Coleman, R.; Beer, T.M.; Gelmann, E.; Datta, M.; Mostaghel, E.; et al. A molecular correlate to the Gleason grading system for prostate adenocarcinoma. Proc. Natl. Acad. Sci. USA 2006, 103, 10991–10996. [Google Scholar] [CrossRef] [PubMed]
- Cuzick, J.; Swanson, G.P.; Fisher, G.; Brothman, A.R.; Berney, D.M.; Reid, J.E.; Mesher, D.; Speights, V.O.; Stankiewicz, E.; Foster, C.S. Transatlantic Prostate Group. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: A retrospective study. Lancet Oncol. 2011, 12, 245–255. [Google Scholar] [CrossRef]
- Penney, K.L.; Sinnott, J.A.; Fall, K.; Pawitan, Y.; Hoshida, Y.; Kraft, P.; Stark, J.R.; Fiorentino, M.; Perner, S.; Finn, S.; et al. mRNA expression signature of Gleason grade predicts lethal prostate cancer. J. Clin. Oncol. 2011, 29, 2391–2396. [Google Scholar] [CrossRef] [PubMed]
- Beroukhim, R.; Mermel, C.H.; Porter, D.; Wei, G.; Raychaudhuri, S.; Donovan, J.; Barretina, J.; Boehm, J.S.; Dobson, J.; Urashima, M.; et al. The landscape of somatic copy-number alteration across human cancers. Nature 2010, 463, 899–905. [Google Scholar] [CrossRef] [PubMed]
- Schoenborn, J.R.; Nelson, P.; Fang, M. Genomic profiling defines subtypes of prostate cancer with the potential for therapeutic stratification. Clin. Cancer Res. 2013, 19, 4058–4066. [Google Scholar] [CrossRef] [PubMed]
- Menezes, R.X.; Boetzer, M.; Sieswerda, M.; van Ommen, G.J.; Boer, J.M. Integrated analysis of DNA copy number and gene expression microarray data using gene sets. BMC Bioinform. 2009, 10, 203. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Zoppis, I.; Gariboldi, M.; Castiglioni, I.; Mauri, G.; Antoniotti, M. Combined analysis of chromosomal instabilities and gene expression for colon cancer progression inference. J. Clin. Bioinform. 2014, 4, 2. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Zoppis, I.; Mauri, G.; Ripamonti, M.; Gallivanone, F.; Salvatore, C.; Gilardi, M.C.; Castiglioni, I. Combination of gene expression and genome copy number alteration has a prognostic value for breast cancer. In Proceedings of the 35th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Osaka, Japan, 3–7 July 2013; IEEE: Piscataway, NJ, USA, 2013; pp. 608–611. [Google Scholar]
- Xu, Y.; Duanmu, H.; Chang, Z.; Zhang, S.; Li, Z.; Li, Z.; Liu, Y.; Li, K.; Qiu, F.; Li, X. The application of gene co-expression network reconstruction based on CNVs and gene expression microarray data in breast cancer. Mol. Biol. Rep. 2012, 39, 1627–1637. [Google Scholar] [CrossRef] [PubMed]
- Catto, J.W.; Alcaraz, A.; Bjartell, A.S.; De Vere White, R.; Evans, C.P.; Fussel, S.; Hamdy, F.C.; Kallioniemi, O.; Mengual, L.; Schlomm, T.; et al. MicroRNA in prostate, bladder, and kidney cancer: A systematic review. Eur. Urol. 2011, 59, 671–681. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.; Jung, M.; Mollenkopf, H.J.; Wagner, I.; Stephan, C.; Jentzmik, F.; Miller, K.; Lein, M.; Kristiansen, G.; Jung, K. Diagnostic and prognostic implications of microRNA profiling in prostate carcinoma. Int. J. Cancer 2010, 126, 1166–1176. [Google Scholar] [CrossRef] [PubMed]
- Martens-Uzunova, E.S.; Jalava, S.E.; Dits, N.F.; van Leenders, G.J.; Møller, S.; Trapman, J.; Bangma, C.H.; Litman, T.; Visakorpi, T.; Jenster, G. Diagnostic and prognostic signatures from the small non-coding RNA transcriptome in prostate cancer. Oncogene 2012, 31, 978–991. [Google Scholar] [CrossRef] [PubMed]
- Brase, J.C.; Johannes, M.; Schlomm, T.; Fälth, M.; Haese, A.; Steuber, T.; Beissbarth, T.; Kuner, R.; Sültmann, H. Circulating miRNAs are correlated with tumor progression in prostate cancer. Int. J. Cancer 2011, 128, 608–616. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, E.B.; Verbruggen, B.; Heijmans, B.T.; Beekman, M.; Kok, J.N.; Slagboom, P.E.; Reinders, M.J. Integrating protein-protein interaction networks with gene-gene co-expression networks improves gene signatures for classifying breast cancer metastasis. J. Integr. Bioinform. 2011, 8, 188. [Google Scholar] [CrossRef] [PubMed]
- Serrano, N.A.; Xu, C.; Liu, Y.; Wang, P.; Fan, W.; Upton, M.P.; Houck, J.R.; Lohavanichbutr, P.; Kao, M.; Zhao, L.P.; et al. Integrative analysis in oral squamous cell carcinoma reveals DNA copy number-associated miRNAs dysregulating target genes. Otolaryngol. Head Neck Surg. 2012, 147, 501–508. [Google Scholar] [CrossRef] [PubMed]
- Eo, H.S.; Heo, J.Y.; Choi, Y.; Hwang, Y.; Choi, H.S. A pathway-based classification of breast cancer integrating data on differentially expressed genes, copy number variations and microRNA target genes. Mol. Cells 2012, 34, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef]
- Kristensen, V.N.; Vaske, C.J.; Ursini-Siegel, J.; Van Loo, P.; Nordgard, S.H.; Sachidanandam, R.; Sørlie, T.; Wärnberg, F.; Haakensen, V.D.; Helland, Å.; et al. Integrated molecular profiles of invasive breast tumors and ductal carcinoma in situ (DCIS) reveal differential vascular and interleukin signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 2802–2807. [Google Scholar] [CrossRef] [PubMed]
- Blenkiron, C.; Goldstein, L.D.; Thorne, N.P.; Spiteri, I.; Chin, S.F.; Dunning, M.J.; Barbosa-Morais, N.L.; Teschendorff, A.E.; Green, A.R.; Ellis, I.O.; et al. MicroRNA expression profiling of human breast cancer identifies new markers of tumor subtype. Genome Biol. 2007, 8, R214. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Bertoli, G.; Ripamonti, M.; Mauri, G.; Zoppis, I.; Della Rosa, P.A.; Gilardi, M.C.; Castiglioni, I. Integration of mRNA expression profile, copy number alterations, and microRNA expression levels in breast cancer to improve grade definition. PLoS ONE 2014, 9, e97681. [Google Scholar] [CrossRef] [PubMed]
- Reid, J.F.; Gariboldi, M.; Sokolova, V.; Capobianco, P.; Lampis, A.; Perrone, F.; Signoroni, S.; Costa, A.; Leo, E.; Pilotti, S.; et al. Integrative approach for prioritizing cancer genes in sporadic colon cancer. Genes Chromosomes Cancer 2009, 48, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative genomic profiling of human prostate cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Knudson, A.G. Two genetic hits (more or less) to cancer. Nat. Rev. Cancer 2001, 1, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Meric-Bernstam, F. Heterogenic loss of BRCA in breast cancer: The “two-hit” hypothesis takes a hit. Ann. Surg. Oncol. 2007, 14, 2428–2429. [Google Scholar] [CrossRef] [PubMed]
- Konishi, H.; Mohseni, M.; Tamaki, A.; Garay, J.P.; Croessmann, S.; Karnan, S.; Ota, A.; Wong, H.Y.; Konishi, Y.; Karakas, B.; et al. Mutation of a single allele of the cancer susceptibility gene BRCA1 leads to genomic instability in human breast epithelial cells. Proc. Natl. Acad. Sci. USA 2011, 108, 17773–17778. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Soma-Nagae, T.; Migita, T.; Kinoshita, R.; Iwamoto, A.; Yuasa, T.; Yonese, J.; Ishikawa, Y.; Seimiya, H. TRIB1 supports prostate tumorigenesis and tumor-propagating cell survival by regulation of endoplasmic reticulum chaperone expression. Cancer Res. 2014, 74, 4888–4897. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, F.; Belloni, L.; Crafa, P.; Lazzaretti, M.; Remondini, D.; Ferretti, S.; Cortellini, P.; Corti, A.; Bettuzzi, S. A novel gene signature for molecular diagnosis of human prostate cancer by RT-qPCR. PLoS ONE 2008, 3, e3617. [Google Scholar] [CrossRef] [PubMed]
- Duhagon, M.A.; Hurt, E.M.; Sotelo-Silveira, J.R.; Zhang, X.; Farrar, W.L. Genomic profiling of tumor initiating prostatospheres. BMC Genom. 2010, 11, 324. [Google Scholar] [CrossRef] [PubMed]
- Özdemir, B.C.; Hensel, J.; Secondini, C.; Wetterwald, A.; Schwaninger, R.; Fleischmann, A.; Raffelsberger, W.; Poch, O.; Delorenzi, M.; Temanni, R.; et al. The molecular signature of the stroma response in prostate cancer-induced osteoblastic bone metastasis highlights expansion of hematopoietic and prostate epithelial stem cell niches. PLoS ONE 2014, 9, e114530. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.B.; Maia, A.T.; O’Reilly, M.; Ghoussaini, M.; Prathalingam, R.; Porter-Gill, P.; Ambs, S.; Prokunina-Olsson, L.; Carroll, J.; Ponder, B.A. A functional variant at a prostate cancer predisposition locus at 8q24 is associated with PVT1 expression. PLoS Genet. 2011, 7, e1002165. [Google Scholar] [CrossRef] [PubMed]
- Varisli, L. Identification of new genes downregulated in prostate cancer and investigation of their effects on prognosis. Genet. Test. Mol. Biomark. 2013, 17, 562–566. [Google Scholar] [CrossRef] [PubMed]
- Gong, M.; Dong, W.; Shi, Z.; Xu, Y.; Ni, W.; An, R. Genetic polymorphisms of GSTM1, GSTT1, and GSTP1 with prostate cancer risk: A meta-analysis of 57 studies. PLoS ONE 2012, 7, e50587. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Qian, J.; Slezak, J.M.; Lieber, M.M.; Bostwick, D.G.; Bergstralh, E.J.; Jenkins, R.B. Clinical significance of alterations of chromosome 8 in high-grade, advanced, nonmetastatic prostate carcinoma. J. Natl. Cancer Inst. 1999, 91, 1574–1580. [Google Scholar] [CrossRef] [PubMed]
- Cooney, K.A.; McCarthy, J.D.; Lange, E.; Huang, L.; Miesfeldt, S.; Montie, J.E.; Oesterling, J.E.; Sandler, H.M.; Lange, K. Prostate cancer susceptibility locus on chromosome 1q: A confirmatory study. J. Natl. Cancer Inst. 1997, 89, 955–959. [Google Scholar] [CrossRef] [PubMed]
- Tucci, P.; Agostini, M.; Grespi, F.; Markert, E.K.; Terrinoni, A.; Vousden, K.H.; Muller, P.A.; Dötsch, V.; Kehrloesser, S.; Sayan, B.S.; et al. Loss of p63 and its microRNA-205 target results in enhanced cell migration and metastasis in prostate cancer. Proc. Natl. Acad. Sci. USA 2012, 109, 15312–15317. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.; Tseng, G.C.; Yu, Y.P.; Gavel, T.; Nelson, J.; Wells, A.; Michalopoulos, G.; Kokkinakis, D.; Luo, J.H. CSR1 suppresses tumor growth and metastasis of prostate cancer. Am. J. Pathol. 2006, 168, 597–607. [Google Scholar] [CrossRef] [PubMed]
- Medina-Villaamil, V.; Martínez-Breijo, S.; Portela-Pereira, P.; Quindós-Varela, M.; Santamarina-Caínzos, I.; Antón-Aparicio, L.M.; Gómez-Veiga, F. Circulating MicroRNAs in blood of patients with prostate cancer. Actas Urológicas Españolas 2014, 38, 633–639. [Google Scholar] [CrossRef] [PubMed]
- Casanova-Salas, I.; Rubio-Briones, J.; Calatrava, A.; Mancarella, C.; Masiá, E.; Casanova, J.; Fernández-Serra, A.; Rubio, L.; Ramírez-Backhaus, M.; Armiñán, A.; et al. Identification of miR-187 and miR-182 as biomarkers of early diagnosis and prognosis in patients with prostate cancer treated with radical prostatectomy. J. Urol. 2014, 192, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Li, J.; Teng, Z.; Zhang, Z.; Xu, Y. Overexpressed microRNA-182 promotes proliferation and invasion in prostate cancer PC-3 cells by down-regulating N-myc downstream regulated gene 1 (NDRG1). PLoS ONE 2013, 8, e68982. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Li, W.; Yuan, L.; Mehta, R.G.; Kopelovich, L.; McCormick, D.L. Inhibition of proliferation and induction of autophagy by atorvastatin in PC3 prostate cancer cells correlate with downregulation of Bcl2 and upregulation of miR-182 and p21. PLoS ONE 2013, 8, e70442. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Du, W.W.; Li, H.; Liu, F.; Khorshidi, A.; Rutnam, Z.J.; Yang, B.B. Both mature miR-17-5p and passenger strand miR-17-3p target TIMP3 and induce prostate tumor growth and invasion. Nucleic Acids Res. 2013, 41, 9688–9704. [Google Scholar] [CrossRef] [PubMed]
- Lichner, Z.; Ding, Q.; Samaan, S.; Saleh, C.; Nasser, A.; Al-Haddad, S.; Samuel, J.N.; Fleshner, N.E.; Stephan, C.; Jung, K.; et al. miRNAs dysregulated in association with Gleason grade regulate extracellular matrix, cytoskeleton and androgen receptor pathways. J. Pathol. 2015, 237, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Kachakova, D.; Mitkova, A.; Popov, E.; Popov, I.; Vlahova, A.; Dikov, T.; Christova, S.; Mitev, V.; Slavov, C.; Kaneva, R. Combinations of serum prostate-specific antigen and plasma expression levels of let-7c, miR-30c, miR-141, and miR-375 as potential better diagnostic biomarkers for prostate cancer. DNA Cell Biol. 2015, 34, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Ouyang, Y.; Lu, M.; Wei, J.; Zhang, H. miR-141-3p regulates the expression of androgen receptor by targeting its 3′UTR in prostate cancer LNCaP cells. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi 2015, 31, 736–739. [Google Scholar] [PubMed]
- Steinberg, J.; Oyasu, R.; Lang, S.; Sintich, S.; Rademaker, A.; Lee, C.; Kozlowski, J.M.; Sensibar, J.A. Intracellular levels of SGP-2 (Clusterin) correlate with tumor grade in prostate cancer. Clin. Cancer Res. 1997, 3, 1707–1711. [Google Scholar] [PubMed]
- Sala, A.; Bettuzzi, S.; Pucci, S.; Chayka, O.; Dews, M.; Thomas-Tikhonenko, A. Regulation of CLU gene expression by oncogenes and epigenetic factors implications for tumorigenesis. Adv. Cancer Res. 2009, 105, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Rizzi, F.; Bettuzzi, S. The clusterin paradigm in prostate and breast carcinogenesis. Endocr. Relat. Cancer 2010, 17, R1–R17. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bonacini, M.; Coletta, M.; Ramazzina, I.; Naponelli, V.; Modernelli, A.; Davalli, P.; Bettuzzi, S.; Rizzi, F. Distinct promoters, subjected to epigenetic regulation, drive the expression of two clusterin mRNAs in prostate cancer cells. Biochim. Biophys. Acta 2015, 1849, 44–54. [Google Scholar] [CrossRef] [PubMed]
- Bettuzzi, S.; Davalli, P.; Davoli, S.; Chayka, O.; Rizzi, F.; Belloni, L.; Pellacani, D.; Fregni, G.; Astancolle, S.; Fassan, M.; et al. Genetic inactivation of ApoJ/clusterin: Effects on prostate tumourigenesis and metastatic spread. Oncogene 2009, 28, 4344–4352. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Lin, P.J.; Beraldi, E.; Zhang, F.; Kawai, Y.; Leong, J.; Katsumi, H.; Fazli, L.; Fraser, R.; Cullis, P.R.; et al. siRNA Lipid Nanoparticle Potently Silences Clusterin and Delays Progression When Combined with Androgen Receptor Cotargeting in Enzalutamide-Resistant Prostate Cancer. Clin. Cancer Res. 2015, 21, 4845–4855. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.J. Knockdown of clusterin expression increases the in vitro sensitivity of human prostate cancer cells to paclitaxel. J. Toxicol. Environ. Health A 2014, 77, 1443–1450. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.; Fu, X.; Sun, X.; Guo, P.; Li, M.; Dong, J.T. Different expression patterns and functions of acetylated and unacetylated Klf5 in the proliferation and differentiation of prostatic epithelial cells. PLoS ONE 2013, 8, e65538. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Zhang, Z.; Xia, S.; Xing, C.; Ci, X.; Li, X.; Zhao, R.; Tian, S.; Ma, G.; Zhu, Z.; et al. KLF5 activates microRNA 200 transcription to maintain epithelial characteristics and prevent induced epithelial-mesenchymal transition in epithelial cells. Mol. Cell. Biol. 2013, 33, 4919–4935. [Google Scholar] [CrossRef] [PubMed]
- Ci, X.; Xing, C.; Zhang, B.; Zhang, Z.; Ni, J.J.; Zhou, W.; Dong, J.T. KLF5 inhibits angiogenesis in PTEN-deficient prostate cancer by attenuating AKT activation and subsequent HIF1α accumulation. Mol. Cancer 2015, 14, 91. [Google Scholar] [CrossRef] [PubMed]
- Wu, R.; Wang, H.; Wang, J.; Wang, P.; Huang, F.; Xie, B.; Zhao, Y.; Li, S.; Zhou, J. EphA3, induced by PC-1/PrLZ, contributes to the malignant progression of prostate cancer. Oncol. Rep. 2014, 32, 2657–2665. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Uematsu, S.; Okamoto, T.; Matsuura, Y.; Sato, S.; Kumar, H.; Satoh, T.; Saitoh, T.; Takeda, K.; Ishii, K.J.; et al. Enhanced TLR-mediated NF-IL6 dependent gene expression by Trib1 deficiency. J. Exp. Med. 2007, 204, 2233–2239. [Google Scholar] [CrossRef] [PubMed]
- Sanford, D.C.; DeWille, J.W. C/EBPdelta is a downstream mediator of IL-6 induced growth inhibition of prostate cancer cells. Prostate 2005, 63, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Du, W.; Wang, Y.; Xu, C.; Wang, J.; Zhang, Y.; Wang, H.; Ju, J.; Zhao, L.; Wang, Z.; et al. Suppression of AKT expression by miR-153 produced anti-tumor activity in lung cancer. Int. J. Cancer 2015, 136, 1333–1340. [Google Scholar] [CrossRef] [PubMed]
- Shan, N.; Shen, L.; Wang, J.; He, D.; Duan, C. MiR-153 inhibits migration and invasion of human non-small-cell lung cancer by targeting ADAM19. Biochem. Biophys. Res. Commun. 2015, 456, 385–391. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Deng, Y.; Liu, Y.; Chen, X.; Yang, G.; Mu, Y.; Zhang, D.; Kang, J.; Wu, Z. MicroRNA-153 is tumor suppressive in glioblastoma stem cells. Mol. Biol. Rep. 2013, 40, 2789–2798. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Ruiz, M.; Cebada, J.; Delgado-López, G.; Sánchez-Vázquez, M.L.; Pérez-Santos, J.L. miR-153 silencing induces apoptosis in the MDA-MB-231 breast cancer cell line. Asian Pac. J. Cancer Prev. 2013, 14, 2983–2986. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; He, B.; He, J.; Mao, X. Upregulation of miR-153 promotes cell proliferation via downregulation of the PTEN tumor suppressor gene in human prostate cancer. Prostate 2013, 73, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Filella, X.; Foj, L. miRNAs as novel biomarkers in the management of prostate cancer. Clin. Chem. Lab. Med. 2017, 55, 715–736. [Google Scholar] [CrossRef] [PubMed]
- Matin, F.; Jeet, V.; Clements, J.A.; Yousef, G.M.; Batra, J. MicroRNA Theranostics in Prostate Cancer Precision Medicine. Clin. Chem. 2016, 62, 1318–1333. [Google Scholar] [CrossRef] [PubMed]
- Lee, S. Mistakes in validating the accuracy of a prediction classifier in high-dimensional but small-sample microarray data. Stat. Methods Med. Res. 2008, 17, 635–642. [Google Scholar] [CrossRef] [PubMed]
- Blute, M.L.; Bergstralh, E.J.; Iocca, A.; Scherer, B.; Zincke, H. Use of Gleason score, prostate specific antigen, seminal vesicle and margin status to predict biochemical failure after radical prostatectomy. J. Urol. 2001, 165, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Dinu, I.; Poudel, S.; Pyne, S. Gene-Set Reduction for Analysis of Major and Minor Gleason Scores Based on Differential Gene-Set Expressions and Biological Pathways in Prostate Cancer. Cancer Inform. 2017, 16. [Google Scholar] [CrossRef] [PubMed]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. Edger: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 50, 289–300. [Google Scholar]
- Dweep, H.; Sticht, C.; Pandey, P.; Gretz, N. miRWalk—Database: Prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 2011, 44, 839–847. [Google Scholar] [CrossRef] [PubMed]
- Mermel, C.H.; Schumacher, S.E.; Hill, B.; Meyerson, M.L.; Beroukhim, R.; Getz, G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011, 12, R41. [Google Scholar] [CrossRef] [PubMed]
- Warde-Farley, D.; Donaldson, S.L.; Comes, O.; Zuberi, K.; Badrawi, R.; Chao, P.; Franz, M.; Grouios, C.; Kazi, F.; Lopes, C.T.; et al. The GeneMANIA prediction server: Biological network integration for gene prioritization and predicting gene function. Nucleic Acids Res. 2010, W214–W220. [Google Scholar] [CrossRef] [PubMed]
- Cava, C.; Colaprico, A.; Bertoli, G.; Graudenzi, A.; Silva, T.C.; Olsen, C.; Noushmehr, H.; Bontempi, G.; Mauri, G.; Castiglioni, I. SpidermiR: An R/Bioconductor Package for Integrative Analysis with miRNA Data. Int. J. Mol. Sci. 2017, 18, 274. [Google Scholar] [CrossRef] [PubMed]
- Liaw, A.; Wiener, M. Classification and regression by randomforest. R. News 2002, 2, 18–22. [Google Scholar]
- Cheng, H.H.; Plets, M.; Li, H.; Higano, C.S.; Tangen, C.M.; Agarwal, N.; Vogelzang, N.J.; Hussain, M.; Thompson, I.M., Jr.; Tewari, M.; et al. Circulating microRNAs and treatment response in the Phase II SWOG S0925 study for patients with new metastatic hormone-sensitive prostate cancer. Prostate 2018, 78, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.M.; Mahon, K.L.; Spielman, C.; Gurney, H.; Mallesara, G.; Stockler, M.R.; Bastick, P.; Briscoe, K.; Marx, G.; Swarbrick, A.; et al. Phase 2 study of circulating microRNA biomarkers in castration-resistant prostate cancer. Br. J. Cancer 2017, 116, 1002–1011. [Google Scholar] [CrossRef] [PubMed]
- Freytag, S.O.; Stricker, H.; Movsas, B.; Kim, J.H. Prostate cancer gene therapy clinical trials. Mol. Ther. 2007, 15, 1042–1052. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Alteration | Gene | Position |
|---|---|---|
| Upregulated and amplified | AMY2B | 1p21.1 |
| Upregulated and amplified | CLEC18B | 16q23.1 |
| Upregulated and amplified | DPY19L2 | 12q14.2 |
| Upregulated and amplified | GUSBP3 | 5q13.2 |
| Upregulated and amplified | LOC157381 | 8q24.21 |
| Upregulated and amplified | LOC391322 | 22q11.23 |
| Upregulated and amplified | LOC728855 | 1q21.2 |
| Upregulated and amplified | MLIP | 6p12.1 |
| Upregulated and amplified | POU5F1B | 8q24.21 |
| Upregulated and amplified | PVT1 | 8q24.21 |
| Upregulated and amplified | SYCE1 | 10q26.3 |
| Upregulated and amplified | TARP | 7p14.1 |
| Upregulated and amplified | TRIB1 | 8q24.21 |
| Upregulated and amplified | ZDHHC11 | 5p15.33 |
| Downregulated and deleted | ADAMTSL3 | 15q25.2 |
| Downregulated and deleted | ADRA1A | 8p21.2 |
| Downregulated and deleted | CES1P1 | 16q12.2 |
| Downregulated and deleted | CHRFAM7A | 15q13.2 |
| Downregulated and deleted | CLU | 8p21.2 |
| Downregulated and deleted | DMBT1 | 10q26.13 |
| Downregulated and deleted | EPHA3 | 3p11.1 |
| Downregulated and deleted | ETS2 | 21q22.2 |
| Downregulated and deleted | EYA1 | 8q13.3 |
| Downregulated and deleted | FCGR3B | 1q23.3 |
| Downregulated and deleted | FILIP1 | 6q14.1 |
| Downregulated and deleted | FMN2 | 1q43 |
| Downregulated and deleted | GSTM1 | 1p13.3 |
| Downregulated and deleted | HSPA6 | 1q23.3 |
| Downregulated and deleted | HSPA7 | 1q23.3 |
| Downregulated and deleted | KLF5 | 13q22.1 |
| Downregulated and deleted | MPP2 | 17q21.31 |
| Downregulated and deleted | NAGS | 17q21.31 |
| Downregulated and deleted | PNMA2 | 8p21.2 |
| Downregulated and deleted | SCARA3 | 8p21.2 |
| Downregulated and deleted | SPG20 | 13q13.3 |
| Downregulated and deleted | THSD7B | 2q22.1 |
| Downregulated and deleted | TP63 | 3q28 |
| Downregulated and deleted | ZNF826P | 19p12 |
| Alteration | Gene Name | miRNA |
|---|---|---|
| Upregulated and amplified | TRIB1 | hsa-miR-10a |
| Upregulated and amplified | ZDHHC11 | hsa-miR-552 |
| Upregulated and amplified | DPY19L2 | hsa-miR-323-3p |
| Downregulated and deleted | CLU | hsa-miR-217 |
| Downregulated and deleted | SCARA3 | hsa-miR-182 |
| Downregulated and deleted | TP63 | hsa-miR-141, hsa-miR-217 |
| Downregulated and deleted | HSPA6 | hsa-miR-17 |
| Downregulated and deleted | EYA1 | hsa-miR-103 |
| Downregulated and deleted | MPP2 | hsa-miR-103 |
| Downregulated and deleted | FILIP1 | hsa-miR-129-5p |
| Downregulated and deleted | DMBT1 | hsa-miR-197 |
| Downregulated and deleted | NAGS | hsa-miR-506 |
| Downregulated and deleted | PNMA2 | hsa-miR-183, hsa-miR-217 |
| Downregulated and deleted | THSD7B | hsa-miR-183 |
| Downregulated and deleted | FCGR3B | hsa-miR-149 |
| Downregulated and deleted | KLF5 | hsa-miR-148a, hsa-miR-217, hsa-miR-182, hsa-miR-141 |
| Downregulated and deleted | FMN2 | hsa-miR-101 |
| Downregulated and deleted | ETS2 | hsa-miR-182 |
| Downregulated and deleted | SPG20 | hsa-miR-17 |
| Downregulated and deleted | EPHA3 | hsa-miR-182, hsa-miR-103, hsa-miR-197, hsa-miR-153, hsa-let-7f, hsa-miR-506, hsa-miR-454, hsa-miR-507, hsa-miR-196a, hsa-miR-489, hsa-miR-513a-3p, hsa-miR-1283, hsa-miR-103 |
| Downregulated and deleted | ADAMTSL3 | hsa-miR-103, hsa-miR-101, hsa-miR-19a/b, hsa-miR-491-3p, hsa-miR-19b, hsa-miR-129-5p, hsa-miR-142-5p, hsa-miR-155, hsa-miR-15b, hsa-miR-507, hsa-miR-512-5p, hsa-miR-513a-5p/3p, hsa-miR-103 |
| Samples Used to Generate PC Signature | Author | N. Genes | Common Genes with Our Signature |
|---|---|---|---|
| 3D spheroid cell culture model | Mashima et al. [43] | 1 | TRIB1 |
| 41 patients with no therapy | Rizzi et al. [44] | 8 | CLU |
| LNCaP cell line and three patient PC | Duhagon et al. [45] | 66 | KLF5 |
| Xenografts and cell line | Özdemir et al. [46] | 3 | EPHA3 |
| Entity 1 | Entity 2 | References to Create the Network |
|---|---|---|
| CEBPB | CEBPD | [Arijs-Rutgeerts-2009, Bahr-Bowler-2013, Mallon-McKay-2013, Roth-Zlotnik-2006, Salaverria-Siebert-2011, Wang-Maris-2006, Wu-Garvey-2007] |
| EFNA1 | EFNA4 | [Bild-Nevins-2006 B, Innocenti-Brown-2011] |
| EFNA3 | EFNA4 | [Innocenti-Brown-2011, Salaverria-Siebert-2011] |
| EFNA1 | EFNA3 | [Gysin-McMahon-2012, Innocenti-Brown-2011] |
| ALOX12 | EPHA3 | [Ramaswamy-Golub-2001, Wang-Maris-2006] |
| LYN | RRBP1 | [Alizadeh-Staudt-2000, Rieger-Chu-2004] |
| CEBPB | TRIB1 | [Bahr-Bowler-2013, Rieger-Chu-2004] |
| CLUL1 | CLU | [Mallon-McKay-2013] |
| EFNA1 | CEBPD | [Gysin-McMahon-2012] |
| MYBL2 | CLUL1 | [Cheok-Evans-2003] |
| TNC | CLU | [Ramaswamy-Golub-2001] |
| EFNA3 | EFNA5 | [Roth-Zlotnik-2006] |
| CEBPD | TRIB1 | [Bahr-Bowler-2013] |
| TNC | KLF5 | [Perou-Botstein-1999] |
| ALOX12 | CLU | [Burington-Shaughnessy-2008, Gysin-McMahon-2012] |
| EFNA1 | CLUL1 | [Rieger-Chu-2004] |
| EFNA1 | KLF5 | [Ramaswamy-Golub-2001, Salaverria-Siebert-2011] |
| RRBP1 | EFNA3 | [Arijs-Rutgeerts-2009] |
| LYN | EFNA5 | [Perou-Botstein-1999] |
| EFNA1 | FBXW7 | [Kang-Willman-2010] |
| NUCB2 | KLF5 | [Wang-Maris-2006] |
| EFNA1 | CEBPB | [Roth-Zlotnik-2006] |
| LYN | CLU | [Perou-Botstein-1999] |
| NUCB2 | RRBP1 | [Rieger-Chu-2004] |
| FBXW7 | EFNA5 | [Roth-Zlotnik-2006] |
| LYN | CEBPB | [Ramaswamy-Golub-2001] |
| TNC | CEBPB | [Arijs-Rutgeerts-2009] |
| EFNA1 | TNC | [Ramaswamy-Golub-2001] |
| EFNA4 | CLUL1 | [Gysin-McMahon-2012] |
| XRCC6 | RRBP1 | [Bahr-Bowler-2013] |
| EFNA3 | TRIB1 | [Wang-Maris-2006] |
| PC Patients | Controls | |
|---|---|---|
| Age | ||
| 43–50 | 26 | 5 |
| 51–60 | 135 | 18 |
| 61–70 | 165 | 25 |
| >70 | 18 | 4 |
| Gleason Score | ||
| 7 | 227 | |
| 8 | 43 | |
| 9 | 72 | |
| 10 | 2 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cava, C.; Bertoli, G.; Colaprico, A.; Bontempi, G.; Mauri, G.; Castiglioni, I. In-Silico Integration Approach to Identify a Key miRNA Regulating a Gene Network in Aggressive Prostate Cancer. Int. J. Mol. Sci. 2018, 19, 910. https://doi.org/10.3390/ijms19030910
Cava C, Bertoli G, Colaprico A, Bontempi G, Mauri G, Castiglioni I. In-Silico Integration Approach to Identify a Key miRNA Regulating a Gene Network in Aggressive Prostate Cancer. International Journal of Molecular Sciences. 2018; 19(3):910. https://doi.org/10.3390/ijms19030910
Chicago/Turabian StyleCava, Claudia, Gloria Bertoli, Antonio Colaprico, Gianluca Bontempi, Giancarlo Mauri, and Isabella Castiglioni. 2018. "In-Silico Integration Approach to Identify a Key miRNA Regulating a Gene Network in Aggressive Prostate Cancer" International Journal of Molecular Sciences 19, no. 3: 910. https://doi.org/10.3390/ijms19030910
APA StyleCava, C., Bertoli, G., Colaprico, A., Bontempi, G., Mauri, G., & Castiglioni, I. (2018). In-Silico Integration Approach to Identify a Key miRNA Regulating a Gene Network in Aggressive Prostate Cancer. International Journal of Molecular Sciences, 19(3), 910. https://doi.org/10.3390/ijms19030910