CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS)


{kind=link}
{kind=link}
Abstract
1. Introduction
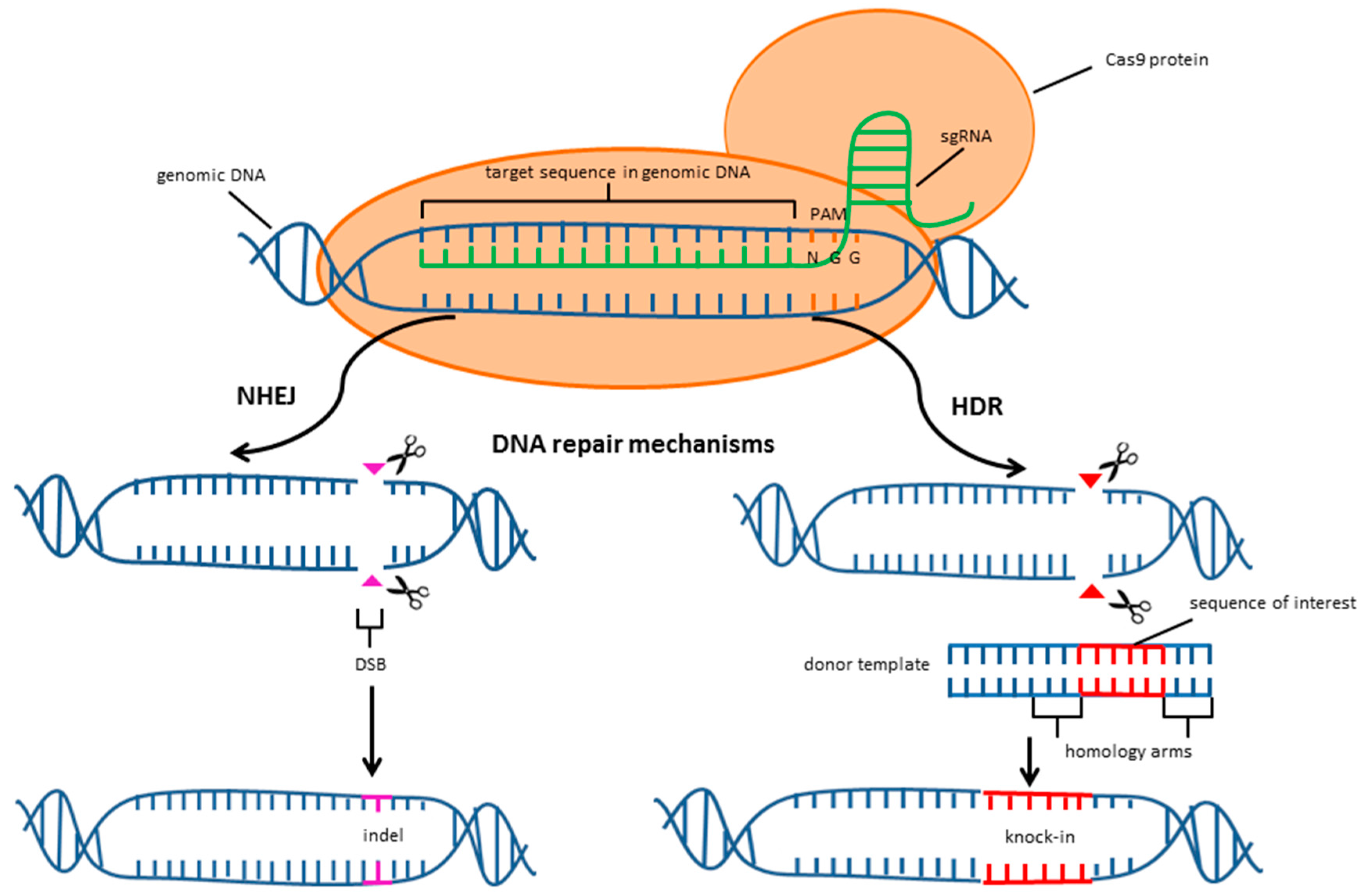
2. CRISPR/Cas9 System
3. Limitations of the CRISPR/Cas9 System
4. Improving the Specificity and Efficiency of the CRISPR/Cas9 System
5. The Application of CRISPR/Cas9 for Modeling Neurodegenerative Disease
6. The Application of the CRISPR/Cas9 System for Amyotrophic Lateral Sclerosis Therapy
7. Conclusions and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Urnov, F.D.; Rebar, E.J.; Holmes, M.C.; Zhang, H.S.; Gregory, P.D. Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 2010, 11, 636–646. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Gersbach, C.A. The Development of TALE Nucleases for Biotechnology. Methods Mol. Biol. 2016, 1338, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Ceasar, S.A.; Rajan, V.; Prykhozhij, S.V.; Berman, J.N.; Ignacimuthu, S. Insert, remove or replace: A highly advanced genome editing system using CRISPR/Cas9. Biochim. Biophys. Acta 2016, 1863, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Xiong, X.; Chen, M.; Lim, W.A.; Zhao, D.; Qi, L.S. CRISPR/Cas9 for Human Genome Engineering and Disease Research. Annu. Rev. Genom. Hum. Genet. 2016, 31, 131–154. [Google Scholar] [CrossRef] [PubMed]
- Agustín-Pavón, C.; Isalan, M. Synthetic biology and therapeutic strategies for the degenerating brain. Bioessays 2014, 36, 979–990. [Google Scholar] [CrossRef] [PubMed]
- Kanchiswamy, C.N.; Maffei, M.; Malnoy, M.; Velasco, R.; Kim, J.S. Fine-Tuning Next-Generation Genome Editing Tools. Trends Biotechnol. 2016, 34, 562–574. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, e2579–e2686. [Google Scholar] [CrossRef] [PubMed]
- Chira, S.; Gulei, D.; Hajitou, A.; Zimta, A.A.; Cordelier, P.; Berindan-Neagoe, I. CRISPR/Cas9: Transcending the Reality of Genome Editing. Mol. Ther.-Nucleic Acids 2017, 16, 211–222. [Google Scholar] [CrossRef] [PubMed]
- Kiernan, M.C.; Vucic, S.; Cheah, B.C.; Turner, M.R.; Eisen, A.; Hardiman, O.; Burrell, J.R.; Zoing, M.C. Amyotrophic lateral sclerosis. Lancet 2011, 377, 942–955. [Google Scholar] [CrossRef]
- Ishino, Y.; Shinagawa, H.; Makino, K.; Amemura, M.; Nakata, A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product. J. Bacteriol. 1987, 169, 5429–5433. [Google Scholar] [CrossRef] [PubMed]
- Bhaya, D.; Davison, M.; Barrangou, R. CRISPR-Cas systems in bacteria and archaea: Versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 2011, 45, 273–397. [Google Scholar] [CrossRef] [PubMed]
- Wiedenheft, B.; Sternberg, S.H.; Doudna, J.A. RNA-guided genetic silencing systems in bacteria and archaea. Nature 2012, 482, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Sapranauskas, R.; Gasiunas, G.; Fremaux, C.; Barrangou, R.; Horvath, P.; Siksnys, V. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Res. 2011, 39, 9275–9282. [Google Scholar] [CrossRef] [PubMed]
- Mojica, F.J.; Díez-Villaseñor, C.; García-Martínez, J.; Almendros, C. Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology 2009, 155 Pt 3, 733–740. [Google Scholar] [CrossRef] [PubMed]
- Sander, J.D.; Joung, J.K. CRISPR-Cas systems for editing, regulating and targeting genomes. Nat. Biotechnol. 2014, 32, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Saleh-Gohari, N.; Helleday, T. Conservative homologous recombination preferentially repairs DNA double-strand breaks in the S phase of the cell cycle in human cells. Nucleic Acids Res. 2004, 32, 3683–3688. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Staahl, B.T.; Alla, R.K.; Doudna, J.A. Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 2014, 3, e04766. [Google Scholar] [CrossRef] [PubMed]
- Miyaoka, Y.; Berman, J.R.; Cooper, S.B.; Mayerl, S.J.; Chan, A.H.; Zhang, B.; Karlin-Neumann, G.A.; Conklin, B.R. Systematic quantification of HDR and NHEJ reveals effects of locus, nuclease, and cell type on genome-editing. Sci. Rep. 2016, 6, 23549. [Google Scholar] [CrossRef] [PubMed]
- Ratz, M.; Testa, I.; Hell, S.W.; Jakobs, S. CRISPR/Cas9-mediated endogenous protein tagging for RESOLFT super-resolution microscopy of living human cells. Sci. Rep. 2015, 5, 9592. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, T.; Dougan, S.K.; Truttmann, M.C.; Bilate, A.M.; Ingram, J.R.; Ploegh, H.L. Increasing the efficiency of precise genome editing with CRISPR-Cas9 by inhibition of nonhomologous end joining. Nat. Biotechnol. 2015, 33, 538–542. [Google Scholar] [CrossRef] [PubMed]
- Mali, P.; Yang, L.; Esvelt, K.M.; Aach, J.; Guell, M.; DiCarlo, J.E.; Norville, J.E.; Church, G.M. RNA-guided human genome engineering via Cas9. Science 2013, 339, 823–826. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Foden, J.A.; Khayter, C.; Maeder, M.L.; Reyon, D.; Joung, J.K.; Sander, J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013, 31, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; et al. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Dang, Y.; Abraham, S.; Ma, H.; Zhang, J.; Guo, H.; Cai, Y.; Mikkelsen, J.G.; Wu, H.; Shankar, P.; et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016, 23, 627–633. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed]
- Gwiazda, K.S.; Grier, A.E.; Sahni, J.; Burleigh, S.M.; Martin, U.; Yang, J.G.; Popp, N.A.; Krutein, M.C.; Khan, I.F.; Jacoby, K.; et al. High Efficiency CRISPR/Cas9-mediated Gene Editing in Primary Human T-cells Using Mutant Adenoviral E4orf6/E1b55k “Helper” Proteins. Mol. Ther. 2016, 24, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Samulski, R.J.; Muzyczka, N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu. Rev. Virol. 2014, 1, 427–451. [Google Scholar] [CrossRef] [PubMed]
- Vannucci, L.; Lai, M.; Chiuppesi, F.; Ceccherini-Nelli, L.; Pistello, M. Viral vectors: A look back and ahead on gene transfer technology. New Microbiol. 2013, 36, 1–22. [Google Scholar] [PubMed]
- Kim, S.; Kim, D.; Cho, S.W.; Kim, J.; Kim, J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014, 24, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Cottle, R.N.; Lee, C.M.; Archer, D.; Bao, G. Controlled delivery of β-globin-targeting TALENs and CRISPR/Cas9 into mammalian cells for genome editing using microinjection. Sci. Rep. 2015, 5, 16031. [Google Scholar] [CrossRef] [PubMed]
- Yu, X.; Liang, X.; Xie, H.; Kumar, S.; Ravinder, N.; Potter, J.; de Mollerat du Jeu, X.; Chesnut, J.D. Improved delivery of Cas9 protein/gRNA complexes using lipofectamine CRISPRMAX. Biotechnol. Lett. 2016, 38, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Song, L.; Liu, X.; Yang, X.; Li, X.; He, T.; Wang, N.; Yang, S.; Yu, C.; Yin, T.; et al. Artificial Virus Delivers CRISPR-Cas9 System for Genome Editing of Cells in Mice. ACS Nano 2017, 11, 95–111. [Google Scholar] [CrossRef] [PubMed]
- Bessis, N.; GarciaCozar, F.J.; Boissier, M.C. Immune responses to gene therapy vectors: Influence on vector function and effector mechanisms. Gene Ther. 2004, 11 (Suppl. 1), S10–S17. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Cradick, T.J.; Brown, M.T.; Deshmukh, H.; Ranjan, P.; Sarode, N.; Wile, B.M.; Vertino, P.M.; Stewart, F.J.; Bao, G. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Res. 2014, 42, 7473–7485. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Wu, H.; Wang, Y.; Liu, X.; Chen, L.; Li, Q.; Cui, C.; Liu, X.; Zhang, J.; Zhang, Y. Single Cas9 nickase induced generation of NRAMP1 knockin cattle with reduced off-target effects. Genome Biol. 2017, 18, 13. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Sander, J.D.; Reyon, D.; Cascio, V.M.; Joung, J.K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat. Biotechnol. 2014, 32, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Guilinger, J.P.; Thompson, D.B.; Liu, D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014, 32, 577–582. [Google Scholar] [CrossRef] [PubMed]
- Wyvekens, N.; Topkar, V.V.; Khayter, C.; Joung, J.K.; Tsai, S.Q. Dimeric CRISPR RNA-Guided FokI-dCas9 Nucleases Directed by Truncated gRNAs for Highly Specific Genome Editing. Hum. Gene Ther. 2015, 26, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Savonenko, A.; Schilling, G.; Wang, J.; Xu, G.; Borchelt, D.R. Transgenic mouse models of neurodegenerative disease: Opportunities for therapeutic development. Curr. Neurol. Neurosci. Rep. 2002, 2, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Van Den Bosch, L. Genetic Rodent Models of Amyotrophic Lateral Sclerosis. BioMed Res. Int. 2011, 2011, 48765. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, F.M.; Camargos, E.R.; de Souza, L.C.; Teixeira, A.L. Animal models of neurodegenerative diseases. Rev. Bras. Psiquiatr. 2013, 35 (Suppl. 2), S82–S91. [Google Scholar] [CrossRef] [PubMed]
- Tu, Z.; Yang, W.; Yan, S.; Guo, X.; Li, X.J. CRISPR/Cas9: A powerful genetic engineering tool for establishing large animal models of neurodegenerative diseases. Mol. Neurodegener. 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Cao, C.; Huang, J.; Yao, J.; Hai, T.; Zheng, Q.; Wang, X.; Zhang, H.; Qin, G.; Cheng, J.; et al. One-step generation of triple gene-targeted pigs using CRISPR/Cas9 system. Sci. Rep. 2016, 6, 20620. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 2014, 156, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Gordon, P.H. Amyotrophic lateral sclerosis: An update for 2013 clinical features, pathophysiology, management and therapeutic trials. Aging Dis. 2013, 4, 295–310. [Google Scholar] [CrossRef] [PubMed]
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Vande Velde, C.; Bouchard, J.P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F.; et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, T.J., Jr.; Bosco, D.A.; Leclerc, A.L.; Tamrazian, E.; Vanderburg, C.R.; Russ, C.; Davis, A.; Gilchrist, J.; Kasarskis, E.J.; Munsat, T.; et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009, 323, 1205–1208. [Google Scholar] [CrossRef] [PubMed]
- Cruts, M.; Engelborghs, S.; van der Zee, J.; Broeckhoven, C.V. C9orf72-Related Amyotrophic Lateral Sclerosis and Frontotemporal Dementia. In GeneReviews® [Internet]; Pagon, R.A., Adam, M.P., Ardinger, H.H., Wallace, S.E., Amemiya, A., Bean, L.J.H., Bird, T.D., Ledbetter, N., Mefford, H.C., Smith, R.J.H., et al., Eds.; University of Washington: Seattle, WA, USA, 2015; pp. 1993–2017. [Google Scholar]
- Armstrong, G.A.; Liao, M.; You, Z.; Lissouba, A.; Chen, B.E.; Drapeau, P. Homology Directed Knockin of Point Mutations in the Zebrafish tardbp and fus Genes in ALS Using the CRISPR/Cas9 System. PLoS ONE 2016, 11, e0150188. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.T.; Bolcun-Filas, E.; Grass, D.S.; Lutz, C.; Murray, S.; Shultz, L.; Rosenthal, N. Of mice and CRISPR. The post-CRISPR future of the mouse as a model system for the human condition. EMBO Rep. 2017, e201643717. [Google Scholar] [CrossRef]
- Sullivan, P.M.; Zhou, X.; Robins, A.M.; Paushter, D.H.; Kim, D.; Smolka, M.B.; Hu, F. The ALS/FTLD associated protein C9orf72 associates with SMCR8 and WDR41 to regulate the autophagy-lysosome pathway. Acta Neuropathol. Commun. 2016, 4. [Google Scholar] [CrossRef] [PubMed]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.; Hu, H.; Duan, W.; Liu, Y.; Tan, G.; Li, Z.; Liu, Y.; Deng, B.; Song, X.; Wang, W.; et al. Intramuscular Delivery of scAAV9-hIGF1 Prolongs Survival in the hSOD1G93A ALS Mouse Model via Upregulation of D-Amino Acid Oxidase. Mol. Neurobiol. 2016, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Mutihac, R.; Ababneh, N.; Scaber, J.; Wade-Martins, R.; Cowley, S.; Talbot, K. Modelling amyotrophic lateral sclerosis (ALS) using mutant and CAS9/CRISPR-corrected motor neurons from patients with C9ORF72 mutations reveals disease-specific cellular phenotypes. J. Neurol. Sci. 2015, 357 (Suppl. 1), e48. [Google Scholar] [CrossRef]
- Takahashi, M.; Suzuki, M.; Fukuoka, M.; Fujikake, N.; Watanabe, S.; Murata, M.; Wada, K.; Nagai, Y.; Hohjoh, H. Normalization of Overexpressed α-Synuclein Causing Parkinson’s Disease B. Mol. Ther. Nucleic Acids 2015, 4, e241. [Google Scholar] [CrossRef] [PubMed]
- Kaplitt, M.G.; During, M.J. GAD gene therapy for Parkinson’s disease. In Translational Neuroscience; Kaplitt, M.G., During, M.J., Eds.; Springer: New York, NY, USA, 2016; pp. 89–98. [Google Scholar]
- Ralph, G.S.; Radcliffe, P.A.; Day, D.M.; Carthy, J.M.; Leroux, M.A.; Lee, D.C.; Wong, L.F.; Bilsland, L.G.; Greensmith, L.; Kingsman, S.M.; et al. Silencing mutant SOD1 using RNAi protects against neurodegeneration and extends survival in an ALS model. Nat. Med. 2005, 11, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Toro Cabreja, G.C.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H., Jr.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Gaj, T.; Ojala, D.S.; Ekman, F.K.; Byrne, L.C.; Limsirichai, P.; Schaffer, D.V. In vivo genome editing improves motor function and extends survival in a mouse model of ALS. Sci. Adv. 2017, 3, eaar3952. [Google Scholar] [CrossRef] [PubMed]
- ALS News Today. Available online: https://alsnewstoday.com/2016/11/16/als-association-grant-boosts-als-one-aids-gene-therapy-research-at-umass-medical-school (accessed on 18 December 2017).
- Rogers, M.L. Neurotrophic Therapy for ALS/MND. In Handbook of Neurotoxicity; Kostrzewa, R.M., Ed.; Springer: Berlin, Germany, 2014; pp. 1755–1785. [Google Scholar]
- Weissmiller, A.M.; Wu, C. Current advances in using neurotrophic factors to treat neurodegenerative disorders. Transl. Neurodegener. 2012, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Van den Akker, G.; van Beuningen, H.; Blaney Davidson, E.; van der Kraan, P. CRISPR/CAS9 mediated genome engineering of human mesenchymal stem cells. Osteoarthr. Cartel. 2016, 24, S231. [Google Scholar] [CrossRef][Green Version]
- The ALS Research Forum. Neurotrophic Factors in ALS: A Winning Combination? Available online: http://www.alsresearchforum.org/neurotrophic-factors-in-als-a-winning-combination/ (accessed on 18 December 2017).


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kruminis-Kaszkiel, E.; Juranek, J.; Maksymowicz, W.; Wojtkiewicz, J. CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS). Int. J. Mol. Sci. 2018, 19, 906. https://doi.org/10.3390/ijms19030906
Kruminis-Kaszkiel E, Juranek J, Maksymowicz W, Wojtkiewicz J. CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS). International Journal of Molecular Sciences. 2018; 19(3):906. https://doi.org/10.3390/ijms19030906
Chicago/Turabian StyleKruminis-Kaszkiel, Ewa, Judyta Juranek, Wojciech Maksymowicz, and Joanna Wojtkiewicz. 2018. "CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS)" International Journal of Molecular Sciences 19, no. 3: 906. https://doi.org/10.3390/ijms19030906
APA StyleKruminis-Kaszkiel, E., Juranek, J., Maksymowicz, W., & Wojtkiewicz, J. (2018). CRISPR/Cas9 Technology as an Emerging Tool for Targeting Amyotrophic Lateral Sclerosis (ALS). International Journal of Molecular Sciences, 19(3), 906. https://doi.org/10.3390/ijms19030906