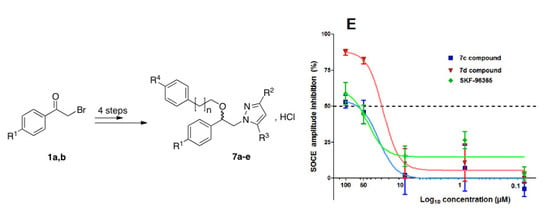
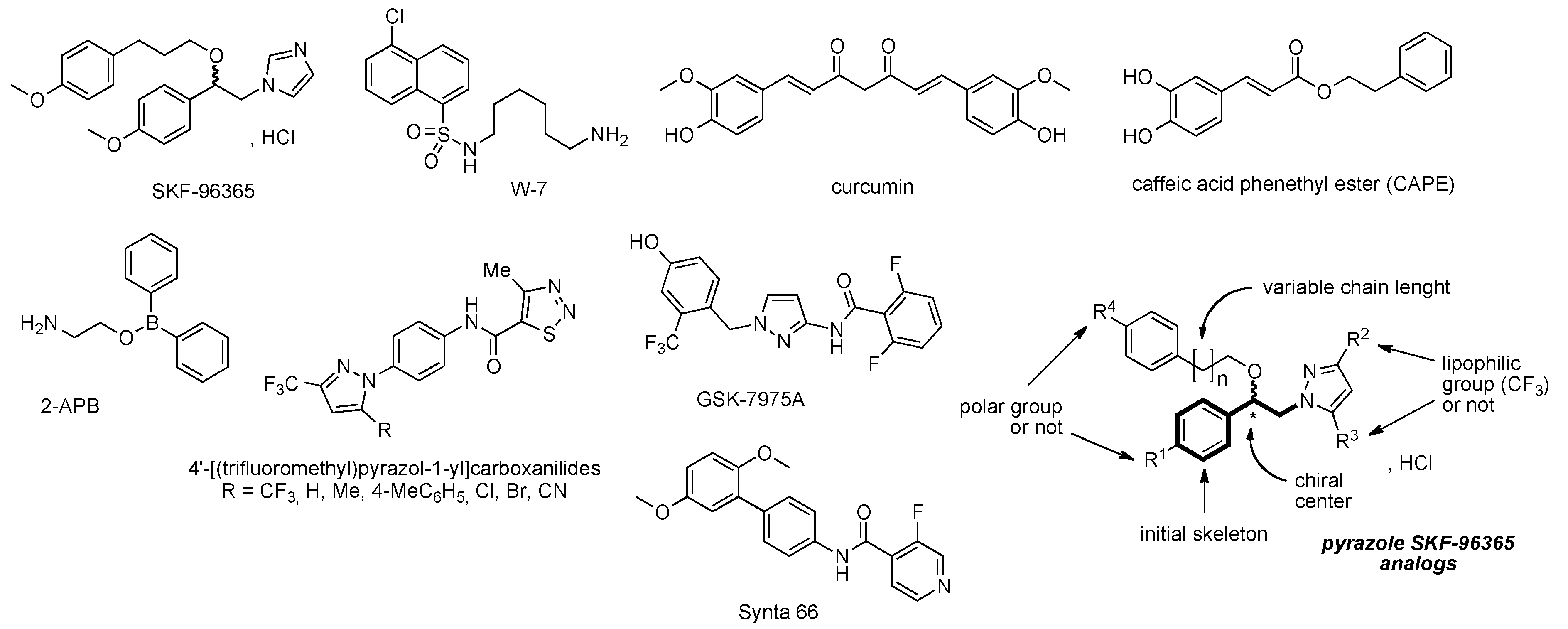
3.1. Chemistry Section
General Information. Preparative chromatography was realized on a Combi Flash Rf 200 psi UV ref. 208K20284 (Serlabo Technologies, Entraigues-sur-la-Sorgue, France) using pre-packed column of alumina gel 60 F 254 Merck equipped with a DAD UV/Vis 200–360 nm detector. Thin-layer chromatography (TLC) was accomplished on 0.2 mm precoated plates of silica gel 60 F-254 (Merck) with appropriate eluent. Visualization was made with ultraviolet light (254 and 365 nm) or with a fluorescence indicator. Solvents were evaporated with a BUCHI rotary evaporator (New Castle, PA, USA). All reagents and solvents were purchased from Acros Fisher (Illkirch, France), Sigma-Aldrich Chimie (St. Quentin Fallavier, France), and Fluka Chimie (Paris, France) and were used without further purification. 1H NMR spectra were recorded on Bruker AC 300 P (300 MHz) spectrometer and 13C NMR spectra on Bruker AC 300 P (75 MHz) spectrometer. Chemical shifts are expressed in parts per million downfield. Data are given in the following order: δ value, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint: quintuplet, m, multiplet; br, broad), number of protons, coupling constants J is given in Hertz. The high-resolution mass spectra (HRMS) were recorded in positive mode using direct Electrospray infusion, respectively on Waters Q-TOF 2 or on Thermo Fisher Scientific Q-Exactive spectrometers at the “Centre Régional de Mesures Physiques de l’Ouest” platform (CRMPO platform, ScanMAT UMS 2001 CNRS, Rennes, France). Melting points were determined on a Kofler melting point apparatus and were uncorrected. Optical rotations [αD] were measured on a Perkin-Elmer 214 polarimeter at room temperature (25 °C) and are recorded in units of deg cm−3 g−1 dm−1 (c in g cm−3 in MeOH) with a 1.0 cm cell. The ee- and de-values were determined by chiral HPLC analysis using Chiracel OJ-H column (250 × 4.60 mm) with UV detector at 220 nm using hexane/i-PrOH as mobile phase with appropriate composition and flow rate.
1-Phenyl-2-(1H-pyrazol-1-yl)ethan-1-one (3a). To a solution of 2-bromoacetophenone 1a (4 g, 20.1 mmol) in 20.8 mL of acetonitrile, pyrazole 2a (1.45 g, 21.3 mmol, 1.06 equiv.) was added in small portions under vigorous magnetic stirring (550 rpm) at room temperature, and mixing was pursued until complete dissolution of the reagents. To this homogeneous solution, K2CO3 (2.92 g, 21.1 mmol, 1.05 equiv.) was poured and the resulting suspension was stirred for 8 h at 25 °C and monitored by thin layer chromatography on 0.2 mm plates of silica gel 60 F-254 (Merck) using cyclohexane/AcOEt (1:1 v/v) as eluent. The reaction mixture was diluted with 20 mL of AcOEt, and the resulting solution was filtered on a Büchner funnel (porosity N°4) and the residual precipitate was washed with AcOEt (2 × 10 mL). The collected filtrate was transferred into a separating funnel. The organic phase was washed successively with deionized water (3 × 80 mL), brine (3 × 80 mL), and dried over magnesium sulfate. After filtration on a filter paper, the filtrate was concentrated in a rotary evaporator under reduced pressure and the oily residue was submitted to purification by preparative chromatography (Combi Flash Rf 200 psi apparatus with a DAD 200/360 nm detector) on pre-packed column of silica gel 60 F-254 (Merck) using a stepwise gradient of cyclohexane/AcOEt (0–50%) for elution. Pooling for 60 min and elimination of the solvent in vacuo gave 3.74 g (53% yield) of the pure desired compound 3a as yellowish powder. Mp = 96–97 °C. 1H NMR (DMSO-d6) δ = 5.84 (s, 2H, CH2, H-2), 6.31 (dd, J = 2.3, 1.9 Hz, 1H, H-4′), 7.48 (dd, 1H, J = 1.9, 0.7 Hz, H-3′), 7.52–7.63 (m, 2H, H-3″, H-5″, Ar), 7.66–7.77 (m, 2H, H-4″, H-5′, Ar), 7.97–8.09 (m, 2H, H-2″, H-6″). 13C NMR (DMSO-d6) δ = 57.7 (C-2), 105.6 (C-4′), 128.1 (C-3″, C-5″), 129.0 (C-2″, C-6″), 131.6 (C-5′), 134.0 (C-4″), 134.6 (C-1″), 139.0 (C-3′), 193.7 (C=O). ES+ HRMS, m/z = 209.0689 found (calculated for C11H10N2ONa [M + Na]+ requires 209.0691).
1-(4-Methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-one (3b). To a solution of 2-bromo-1-(4-methoxyphenyl)ethan-1-one 1b (4.6 g, 20.1 mmol) in 20.8 mL of acetonitrile, pyrazole 2a (2.74 g, 40.2 mmol, 2 equiv.) was added in small portions under vigorous magnetic stirring (550 rpm) at room temperature, and mixing was pursued until complete dissolution of the reagents. To this homogeneous solution, K2CO3 (5.56 g, 40.2 mmol, 2 equiv.) was poured and the resulting suspension was stirred for 12 h at 25 °C and monitored by thin layer chromatography on 0.2 mm plates of silica gel 60 F-254 (Merck) using cyclohexane/AcOEt (3:7 v/v) as eluent. The reaction mixture was diluted with 20 mL of AcOEt, and the resulting suspension was filtered on a Büchner funnel (porosity N°4) and the residual precipitate was washed with AcOEt (2 × 10 mL). The collected filtrate was transferred into a separating funnel. The organic phase was washed successively with deionized water (3 × 80 mL), brine (3 × 80 mL), and dried over magnesium sulfate. After filtration on a filter paper, the filtrate was concentrated in a rotary evaporator under reduced pressure and the oily residue was submitted to purification by preparative chromatography (Combi Flash Rf 200 psi apparatus with a DAD 200/360 nm detector) on pre-packed column of silica gel 60 F-254 (Merck) using a stepwise gradient of cyclohexane/AcOEt (0–70%) for elution. Pooling for 60 min and elimination of the solvent in vacuo gave 4.35 g (66% yield) of the pure desired compound 3b as yellowish needles. Mp = 102–103 °C. 1H NMR (DMSO-d6) δ = 3.86 (s, 3H, OCH3), 5.77 (s, 2H, CH2, H-2), 6.30 (t, 1H, J = 2.1 Hz, H-4′), 7.09 (d, 2H, J = 8.9 Hz, H-3″, H-5″, Ar), 7.47 (dd, 1H, J = 1.9, 0.7 Hz, H-3′), 7.72 (dd, 1H, J = 2.3, 0.7 Hz, H-5′), 8.01 (d, 2H, J = 8.9 Hz, H-2″, H-6″, Ar). 13C NMR (DMSO-d6) δ = 55.7 (OCH3), 57.3 (C-2), 105.5 (C-4′), 114.2 (C-3″, C-5″), 127.4 (C-5′), 130.4 (C-2″, C-6″), 131.6 (C-1″), 138.8 (C-3′), 163.6 (C-4″), 191.9 (C=O). ES+ HRMS, m/z = 239.0798 found (calculated for C12H12N2O2Na [M + Na]+ requires 239.0797).
2-(3-Trifluoromethyl-1H-pyrazol-1-yl)-1-(4-methoxyphenyl)ethan-1-one (3c). To a solution of 2-bromo-1-(4-methoxyphenyl)ethan-1-one 1b (0.5 g, 2.18 mmol) in 2.25 mL of acetonitrile, 3-trifluoromethylpyrazole 2b (0.89 g, 6.54 mmol, 3 equiv.) was added in small portions under vigorous magnetic stirring (550 rpm) at room temperature, and mixing was pursued until complete dissolution of the reagents. To this homogeneous solution, K2CO3 (0.905 g, 6.54 mmol, 3 equiv.) was poured and the resulting suspension was stirred for 12 h at 25 °C and monitored by thin layer chromatography on 0.2 mm plates of silica gel 60 F-254 (Merck) using cyclohexane/AcOEt (1:1 v/v) as eluent. The reaction mixture was diluted with 5 mL of AcOEt, and the resulting solution was filtered on a Büchner funnel (porosity N°4) and the residual precipitate was washed with 5 mL of AcOEt. The collected filtrate was transferred into a separating funnel. The organic phase was washed successively with deionized water (3 × 20 mL), brine (3 × 20 mL), and dried over magnesium sulfate. After filtration on a filter paper, the filtrate was concentrated in a rotary evaporator under reduced pressure and gave a solid residue. After addition of 40 mL of hexane and mixing for 4 h, the solid was filtered on a Büchner funnel (porosity N°4) then dried at 60 °C for 3 h and gave 0.62 g (68% yield) of the desired compound 3c as white powder. Mp = 146–147 °C. 1H NMR (DMSO-d6) δ = 3.87 (s, 3H, OCH3), 5.94 (s, 2H, CH2, H-2), 6.78 (dd, 1H, J = 2.4, 0.7 Hz, H-4′, Ar), 7.12 (d, 2H, J = 8.9 Hz, H-3″, H-5″, Ar), 7.95 (dq, 1H, J = 2.1, 1.0 Hz, H-5′, Ar), 8.02 (d, 2H, J = 8.9 Hz, H-2″, H-6″, Ar). 13C NMR (DMSO-d6) δ = 55.8 (OCH3), 58.1 (C-2), 104.4 (C-4′), 104.4 (C-4′), 114.2 (C-2″, C-6″), 114.3 (C-2″, C-6″), 127.1 (C-1″), 130.4 (C-3″, C-5″), 130.5 (C-3″, C-5″), 134.2 (C-5′), 134.3 (C-5′), 163.8 (C-4″), 191.0 (C-1). ES+ HRMS, m/z = 307.0671 found (calculated for C13H11N2O2F3Na [M + Na]+ requires 307.0670); 285.0862 found (calculated for C13H12N2O2F3 [M + H]+ requires 285.0851).
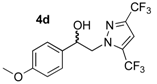
2-(3,5-Bis-trifluoromethyl-1H-pyrazol-1-yl)-1-(4-methoxyphenyl)ethan-1-one (3d). To a solution of 2-bromo-1-(4-methoxyphenyl)ethan-1-one 1b (0.5 g, 2.18 mmol) in 2.25 mL of acetonitrile, 3,5-bis-trifluoromethylpyrazole 2c (1.778 g, 8.72 mmol, 4 equiv.) was added in small portions under vigorous magnetic stirring (550 rpm) at room temperature, and mixing was pursued until complete dissolution of the reagents. To this homogeneous solution, K2CO3 (1.205 g, 8.72 mmol, 4 equiv.) was poured and the resulting suspension was stirred for 24 h at 25 °C and monitored by thin layer chromatography on 0.2 mm plates of silica gel 60 F-254 (Merck) using cyclohexane/AcOEt (1:1 v/v) as eluent. The reaction mixture was diluted with 5 mL of AcOEt, and the resulting solution was filtered on a Büchner funnel (porosity N°4) and the residual precipitate was washed with 5 mL of AcOEt. The collected filtrate was transferred into a separating funnel. The organic phase was washed successively with deionized water (3 × 20 mL), brine (3 × 20 mL), and dried over magnesium sulfate. After filtration on a filter paper, the filtrate was concentrated in a rotary evaporator under reduced pressure and gave a solid residue which was dried under high vacuum (10−2 Torr) at 25 °C for 2 h. The desired compound 3d was obtained in 69% yield as white powder. Mp = 114–115 °C. 1H NMR (DMSO-d6) δ = 3.88 (s, 3H, OCH3), 6.19 (s, 2H, CH2, H-2), 7.12 (d, 2H, J = 8.9 Hz, H-3″, H-5″, Ar), 7.69 (s, 1H, H-4′, Ar), 8.05 (d, 1H, J = 8.9 Hz, H-2″, H-6″, Ar). 13C NMR (DMSO-d6) δ = 55.8 (OCH3), 58.5 (C-2), 107.5 (C-4′), 114.3 (C-3″, C-5″), 126.3 (C-1″), 130.8 (C-2″, C-6″), 164.2 (C-4″), 189.9 (C=O). ES+ HRMS, m/z = 375.0544 found (calculated for C14H10N2O2F6Na [M + Na]+ requires 375.0544); 353.0734 found (calculated for C14H11N2O2F3 [M + H]+ requires 353.0734).
3.1.1. General Procedure for Reduction of 1-Phenyl-2-(1H-pyrazol-1-yl)ethan-1-one (3a–c) into 1-Phenyl-2-(1H-pyrazol-1-yl)ethan-1-ol (4a–c)
To a solution of 1-phenyl-2-(1H-pyrazol-1-yl)ethan-1-one 3 (5 mmol) in an appropriate volume of anhydrous methanol (9–40 mL), sodium borohydride NaBH4 (0.189 g, 5 mmol, 1 equiv.) was added in small portions at 0 °C (ice bath) under magnetic stirring (300 rpm). The resulting mixture was stirred (500 rpm) for an appropriate reaction time (5–7 h) at room temperature, and the reaction solution was monitored by thin layer chromatography (TLC) on 0.2 mm plates of silica gel 60 F-254 (Merck) using an appropriate mixture of solvents as eluent. The reaction mixture was concentrated in a rotary evaporator under reduced pressure and the resulting solid residue was washed with deionized water (3 × 45–3 × 47 mL) on a Büchner funnel. Then, the desired compounds 3a–c were dried at 60 °C for 3 h.
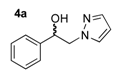
1-Phenyl-2-(1H-pyrazol-1-yl)ethan-1-ol (4a). According to the standard procedure, the compound 4a was prepared from 1-phenyl-2-(1H-pyrazol-1-yl)ethan-1-one 3a (0.93 g, 5 mmol) in 9.3 mL of anhydrous methanol after a reaction time of 5 h using CH2Cl2/MeOH 9:1 v/v as eluent for thin layer chromatography. Washing work-up was realized with 3 × 45 mL of deionized water and gave 0.94 g (82% yield) of the desired compound 4a as white powder. Mp = 132–134 °C. 1H NMR (DMSO-d6) δ = 4.14–4.30 (m, 2H, CH2, H-2), 4.83–5.01 (m, 1H, CH, H-1), 5.65 (br d, 1H, J = 4.7 Hz, OH), 6.18 (t, 1H, J = 2.0 Hz, H-4′), 7.19–7.39 (m, 5H, H-2″, H-3″, H-4″, H-5″, H-6″, Ar), 7.43 (dd, 1H, J = 1.9, 0.7 Hz, H-3′), 7.58 (dd, 1H, J = 2.3, 0.7 Hz, H-5′). 13C NMR (DMSO-d6) δ = 58.7 (C-2), 71.8 (C-1), 104.7 (C-4′), 126.1 (C-3″, C-5″), 127.4 (C-4″), 128.2 (C-2″, C-6″), 130.7 (C-5′), 138.5 (C-1″), 142.8 (C-3′). ES+ HRMS, m/z = 211.0848 found (calculated for C11H12N2O2Na [M + Na]+ requires 211.0847).
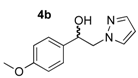
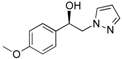
1-(4-Methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol (4b). According to the standard procedure, the compound 4b was prepared from 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-one 3b (1.08 g, 5 mmol) in 14.4 mL of anhydrous methanol after a reaction time of 7 h using CH2Cl2/MeOH 8:2 v/v as eluent for thin layer chromatography. Washing work-up was realized with 3 × 47 mL of deionized water and gave 1.091 g (86% yield) of the desired compound 4b as white crystals with de = 50% (hexane/i-PrOH 96:4 v/v as eluent, flow rate = 0.6 mL/min.), retention times tR(1) = 65.92 and tR(2) = 71.38 min. [αD] = 0.0 (c 1.0, MeOH). Mp = 116–118 °C. 1H NMR (DMSO-d6) δ = 3.73 (s, 3H, OCH3), 4.10–4.27 (m, 2H, CH2, H-2), 4.87 (dt, 1H, J = 7.5, 5.1 Hz, CH, H-1), 5.54 (br d, 1H, J = 4.6 Hz, OH), 6.17 (dd, 1H, J = 1.8 Hz, H-4′), 6.88 (d, 2H, J = 8.7 Hz, H-3″, H-5″, Ar), 7.15–7.29 (m, 2H, H-2″, H-6″, Ar), 7.42 (dd, 1H, J = 1.8, 0.7 Hz, H-3′), 7.57 (dd, 1H, J = 2.2, 0.8 Hz, H-5′). 13C NMR (DMSO-d6) δ = 55.0 (OCH3), 58.7 (C-2), 71.4 (C-1), 104.6 (C-4′), 113.5 (C-3″, C-5″), 127.2 (C-2″, C-6″), 130.5 (C-1″), 134.8 (C-5′), 138.4 (C-3′), 158.5 (C-4″). ES+ HRMS, m/z = 241.0954 found (calculated for C12H14N2O2Na [M + Na]+ requires 241.0953).
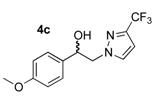
1-(4-Methoxyphenyl)-2-(3-trifluoromethyl-1H-pyrazol-1-yl)ethan-1-ol (4c). According to the standard procedure, the compound 4c was prepared from 1-(4-methoxyphenyl)-2-(3-trifluoromethyl-1H-pyrazol-1-yl)ethan-1-one 3c (1.42 g, 5 mmol) in 39 mL of anhydrous methanol after a reaction time of 7 h using hexane/AcOEt 1:1 v/v as eluent for thin layer chromatography. Washing work-up was realized with 3 × 47 mL of deionized water and gave 1.431 g (94% yield) of the desired compound 4c as white powder. Mp = 110–112 °C. 1H NMR (DMSO-d6) δ = 3.74 (s, 3H, OCH3), 4.27 (d, 2H, J = 6.5 Hz, CH2, H-2), 4.90 (t, 1H, J = 6.5 Hz, CH, H-1), 5.63 (br s, 1H, OH), 6.66 (d, 1H, J = 2.3 Hz, H-4′, Ar), 6.90 (d, 2H, J = 8.6 Hz, H-3″, H-5″, Ar), 7.28 (d, 2H, J = 8.3 Hz, H-2″, H-6″, Ar), 7.85 (s, 1H, H-5′, Ar). 13C NMR (DMSO-d6) δ = 55.0 (OCH3), 55.1 (OCH3), 59.3 (C-2), 71.0 (C-1), 103.7, (C-4′), 103.7 (C-4′), 113.6 (C-2″, C-6″), 113.6 (C-2″, C-6″), 127.2 (C-3″, C-5″), 127.3 (C-3″, C-5″), 133.1 (C-5′), 133.2 (C-5′), 134.2 (C-1″), 140.13 (q, J = 37.1 Hz, CF3), 158.7 (C-4″). ES+ HRMS, m/z = 309.0824 found (calculated for C13H12N2O2F3Na [M + Na]+ requires 309.0827); 269.0902 found (calculated for C13H11N2OF3 [M − H2O + H]+ requires 309.0827).
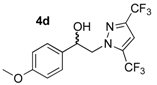
2-(3,5-Bis-trifluoromethyl-1H-pyrazol-1-yl)-1-(4-methoxyphenyl)ethan-1-ol (4d). To a solution of 2-(3,5-bis-trifluoromethyl-1H-pyrazol-1-yl)-1-(4-methoxyphenyl)ethan-1-one 3d (1.76 g, 5 mmol) in 31 mL of anhydrous methanol, sodium borohydride NaBH4 (0.378 g, 10 mmol, 2 equiv.) was added in small portions at 0 °C (ice bath) under magnetic stirring (300 rpm). The resulting mixture was stirred (500 rpm) for 7 h at room temperature and the reaction solution was monitored by thin layer chromatography on 0.2 mm plates of silica gel 60 F-254 (Merck) using hexane/AcOEt 1:1 v/v as eluent. After concentration of the reaction mixture in vacuo, 44 mL of deionized water was added in the crude oily residue, and extraction was conducted with AcOEt (3 × 44 mL) in a separating funnel. The combined extracts were washed with 44 mL of brine and dried over magnesium sulfate. After filtration on a filter paper, the filtrate was concentrated in a rotary evaporator under reduced pressure and gave an oily residue which was dried under high vacuum (10−2 Torr) at 25 °C for 2 h. The desired compound 4d (1.771 g) was obtained as yellowish mobile oil in 98% yield. 1H NMR (DMSO-d6) δ = 3.74 (s, 3H, OCH3), 4.29 (dd, 1H, J = 13.9, 8.9 Hz, CH, H-2), 4.40 (dd, 1H, J = 13.9, 4.4 Hz, CH, H-2), 5.01 (dt, 1H, J = 8.9, 4.4 Hz, CH, H-1), 5.73 (dd, 1H, J = 4.6, 0.7 Hz, OH), 6.92 (d, 2H, J = 8.7 Hz, H-3″, H-5″, Ar), 7.28 (d, 2H, J = 8.4 Hz, H-2″, H-6″, Ar), 7.50–7.54 (m, 1H, H-4′, Ar). 13C NMR (DMSO-d6) δ = 55.1 (OCH3), 58.8 (C-2), 70.6 (C-1), 106.8 (C-4′), 113.7 (C-3″, C-5″), 127.2 (C-2″, C-6″), 133.5 (C-1″), 158.9 (C-4″). ES+ HRMS, m/z = 377.0700 found (calculated for C14H12N2O2F6Na [M + Na]+ requires 377.0701); 337.0763 found (calculated for C14H11N2OF6 [M − H2O + H]+ requires 337.0776).
3.1.2. General Procedure for the Synthesis of (1R, 1S) 1-[β-(Phenylalkoxy)-phenethyl]-1H-pyrazolium Hydrochloride (7a–e)
Pellets of potassium hydroxide KOH (0.112 g, 2 mmol, 2 equiv.) were added to a solution of 1-phenyl-2-(1H-pyrazol-1-yl)ethan-1-ol 4 (1 mmol, 1 equiv.) in 3.76 mL of dry dimethylsulfoxide pa at room temperature. The resulting mixture was stirred vigorously (550 rpm) for 30 min until complete dissolution of KOH, then commercial arylalkyl halide 5 (1 mmol, 1 equiv.) was added in one portion in the reaction mixture. The reaction mixture was heated at 50 °C under magnetic stirring (300 rpm) for 48 h. After cooling down to room temperature, 18.7 mL of saturated brine was added to the reaction mixture and the resulting solution was transferred into a separating funnel. Extraction was conducted with 18.7 mL of diethyl ether Et2O, then the organic phase was dried over magnesium sulphate and filtered on a filter paper. The filtrate was concentrated in a rotary evaporator under reduced pressure. The oily crude residue containing1-[β-(phenylalkoxy)-phenethyl]-1H-pyrazole 6 was dissolved in appropriate volume of diethyl ether Et2O pa under a stream of argon with magnetic stirring (200 rpm). To this homogeneous solution, a commercial solution of 1 M HCl (1 equiv.) in ether was added dropwise rapidly. During mixing at room temperature, the initial pale yellow oil crystallized progressively on the circumference of the round flask and mixing (from 24 h to 5 days) was pursued until complete crystallization of the desired 1-[β-(phenylalkoxy)-phenethyl]-1H-pyrazolium hydrochloride 7. The resulting precipitate was collected by filtration on a Büchner funnel (porosity N°4) and washed with dry diethyl ether pa. The desired compound 7 was dried at room temperature for 4 h and stored in a dessicator.
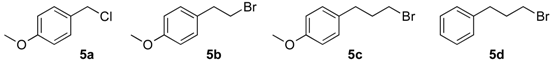
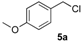
(1R, 1S) 1-[β-((4-Methoxybenzyl)oxy)-phenethyl]-1H-pyrazolium hydrochloride (7a). Using the standard procedure from 1-phenyl-2-(1H-pyrazol-1-yl)ethan-1-ol 4a (0.188 g, 1 mmol) and 4-methoxybenzyl chloride 5a (0.157 g, 1 mmol, 1 equiv.), the 1-[β-((4-methoxybenzyl)oxy)-phenethyl]-1H-pyrazole 6a (0.199 g, 0.65 mmol) was dissolved in 4.6 mL of dry diethyl ether pa under a stream of argon. The addition of 0.65 mL of 1M HCl (0.65 mmol, 1 equiv.) produced a viscous gum on the circumference of the round flask after mixing at room temperature for 5 days. Then, this reaction mixture was stored in a refrigerator at 4 °C for 2 weeks and produced a compact gum. This gum was then manually and carefully triturated in 4 mL of dry hexane and progressively produced a powder. The resulting suspension was stirred (200 rpm) at room temperature for 5 days, and produced a completely divided powder. The desired salt 7a was collected by filtration on a Büchner funnel (porosity N°4) and washed with 0.5 mL of dry diethyl ether pa. After drying at 25 °C for 4 h, the 1-[β-((4-methoxybenzyl)oxy)-phenethyl]-1H-pyrazolium hydrochloride 7a (35.72 mg, 10% yield) was obtained as white powder. Mp = 62–65 °C. 1H NMR (DMSO-d6) δ = 3.72 (s, 3H, OCH3), 4.09 (d, 1H, J = 11.6 Hz, CH, H-1″), 4.19–4.32 (m, 2H, CH, H-1″, H-1′), 4.40 (dd, 1H, J = 13.9, 8.5 Hz, CH, H-1′), 4.78 (dd, 1H, J = 8.4, 4.4 Hz, CH, H-2′), 6.22 (t, 1H, J = 2.0 Hz, H-4, Ar), 6.82 (d, 2H, J = 8.6 Hz, H-3b, H-5b, Ar), 6.99 (d, 2H, J = 8.6 Hz, H-2b, H-6b, Ar), 7.27–7.44 (m, 5H, H-2a, H-3a, H-4a, H-5a, H-6a), 7.46 (d, 1H, J = 1.8 Hz, H-3, Ar), 7.62 (d, 1H, J = 2.2 Hz, H-5, Ar). 13C NMR (DMSO-d6) δ = 55.1 (OCH3), 57.0 (C-1′), 69.7 (C-1″), 79.4 (C-2′), 104.9 (C-4), 113.6 (C-3a, C-5a), 126.8 (C-2a, C-6a), 128.1 (C-4a), 128.6 (C-2a, C-6a), 128.8 (C-3a, C-5a), 129.9 (C-1b), 130.9 (C-5), 138.6 (C-3), 139.0 (C-1a), 158.6 (C-4b). ES+ HRMS, m/z = 331.1418 found (calculated for C19H20N2O2Na [M + Na]+ requires 331.1417).
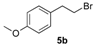
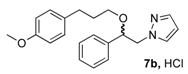
(1R, 1S) 1-[β-((3-(4-Methoxyphenyl)propyl)oxy)-phenethyl]-1H-pyrazolium hydrochloride (7b). Using the standard procedure from 1-phenyl-2-(1H-pyrazol-1-yl)ethan-1-ol 4a (0.188 g, 1 mmol) and 1-(3-bromopropyl)-4-methoxybenzene 5c (0.229 g, 1 mmol, 1 equiv.), the 1-[β-((3-phenylpropyl)oxy)-phenethyl]-1H-pyrazole 6b (0.151 g, 0.45 mmol) was dissolved in 3 mL of dry diethyl ether pa under a stream of argon. The addition of 0.45 mL of 1M HCl (0.45 mmol, 1 equiv.) produced a pale yellow viscous oil that crystallized progressively on the circumference of the round flask, and mixing (200 rpm, 72 h) was pursued until complete crystallization of the oily gum. The desired salt 7b was collected by filtration on a Büchner funnel (porosity N°4) and washed with 3 × 0.5 mL of dry diethyl ether pa. After drying at 25 °C for 4 h, the 1-[β-((3-(4-methoxyphenyl)propyl)oxy)-phenethyl]-1H-pyrazolium hydrochloride 7b (78.3 mg, 21% yield) was obtained as white powder. Mp = 76–80 °C. 1H NMR (DMSO-d6) δ = 1.62 (dt, 2H, J = 8.2, 6.2 Hz, CH2, H-2″), 2.36 (dd, 2H, J = 8.4, 6.7 Hz, CH2, H-3″), 3.05 (dt, 1H, J = 9.5, 6.1 Hz, CH, H-1″), 3.22 (dt, 1H, J = 9.5, 6.0 Hz, CH, H-1″), 3.69 (s, OCH3), 4.23 (dd, 1H, J = 13.9, 4.4 Hz, CH, H-1′), 4.37 (dd, 1H, J = 13.9, 8.7 Hz, CH, H-1′), 4.69 (dd, 1H, J = 8.7, 4.2 Hz, CH, H-2′), 6.23 (t, 1H, J = 2.1 Hz, H-4, Ar), 6.77 (d, 2H, J = 8.6 Hz, H-3b, H-5b, Ar), 6.94 (d, 2H, J = 8.6 Hz, H-2b, H-6b, Ar), 7.27–7.44 (m, 5H, H-2a, H-3a, H-4a, H-5a, H-6a, Ar), 7.46–7.50 (m, 1H, H-3), 7.68 (dd, 1H, J = 2.3, 0.7 Hz, H-5). 13C NMR (DMSO-d6) δ = 30.4 (C-3″), 31.1 (C-2″), 54.9 (OCH3), 57.2 (C-1′), 67.4 (C-1″), 80.1 (C-2′), 104.9 (C-4), 113.6 (C-3b, C-5b), 126.7 (C-2a, C-6a), 128.0 (C-4a), 128.5 (C-3a, C-5a), 129.2 (C-2b, C-6b), 131.0 (C-5), 133.4 (C-1b), 138.4 (C-3), 139.3 (C-1a), 157.3 (C-4b). ES+ HRMS, m/z = 359.1733 found (calculated for C21H24N2O2Na [M + Na]+ requires 359.1735); 337.1906 found (calculated for C21H25N2O2 [M + H]+ requires 337.1916).
(1R, 1S) 1-[β-((4-Methoxybenzyl)oxy)-(4-methoxyphenethyl]-1H-pyrazolium hydrochloride (7c). Using the standard procedure from 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (0.218 g, 1 mmol) and 4-methoxybenzyl chloride 5a (0.157 g, 1 mmol, 1 equiv.), the 1-[β-((4-methoxybenzyl)oxy)-(4-methoxyphenethyl]-1H-pyrazole 6c (0.231 g, 0.68 mmol) was dissolved in 4.57 mL of dry diethyl ether pa under a stream of argon. The addition of 0.68 mL of 1M HCl (0.68 mmol, 1 equiv.) produced a pale pink viscous oil that crystallized rapidly (30 min.) on the circumference of the round flask, and mixing (200 rpm, 72 h) was pursued until complete crystallization of the viscous gum into divided powder. The desired salt 7c was collected by filtration on a Büchner funnel (porosity N°4) and washed with 3 × 0.5 mL of dry diethyl ether pa. After drying at 25 °C for 4 h, the 1-[β-((4-methoxybenzyl)oxy)-(4-methoxyphenethyl]-1H-pyrazolium hydrochloride 7c (123.7 mg, 33% yield) was obtained as white powder. Mp = 112–113 °C. 1H NMR (DMSO-d6) δ = 3.72 (s, 3H, OCH3), 3.76 (s, 3H, OCH3), 4.06 (d, 1H, J = 11.6 Hz, CH, H-1″), 4.16–4.30 (m, 2H, CH, H-1′, H-1″), 4.40 (dd, 1H, J = 13.8, 8.5 Hz, CH, H-1′), 4.71 (dd, 1H, J = 8.4, 4.5 Hz, CH, H-2′), 6.22 (t, 1H, J = 2.0 Hz, H-4, Ar), 6.82 (d, 2H, J = 8.6 Hz, H-3a, H-5a, Ar), 6.89–7.03 (m, 4H, H-2a H-6a, H-3b, H-5b, Ar), 7.26 (d, 2H, J = 8.6 Hz, H-2b, H-6b, Ar), 7.47 (d, 1H, J = 1.9 Hz, H-3, Ar), 7.62 (d, 1H, J = 2.2 Hz, H-5, Ar). 13C NMR (DMSO-d6) δ = 55.1 (OCH3), 55.1 (OCH3), 57.0 (C-1′), 69.3 (C-1″), 78.9 (C-2′), 104.9 (C-4), 113.6 (C-3a, C-5a), 114.0 (C-3b, C-5b), 128.2 (C-2a C-6a), 128.8 (C-2b, C-6b), 130.0 5 (C-1a), 130.8 (C-1b), 130.9 (C-5), 138.5 (C-3), 158.6 (C-4a), 159.1 (C-4b). ES+ HRMS, m/z = 361.1526 found (calculated for C20H22N2O3Na [M + Na]+ requires 361.1528); 339.1715 found (calculated for C20H23N2O3 [M + H]+ requires 339.1709).
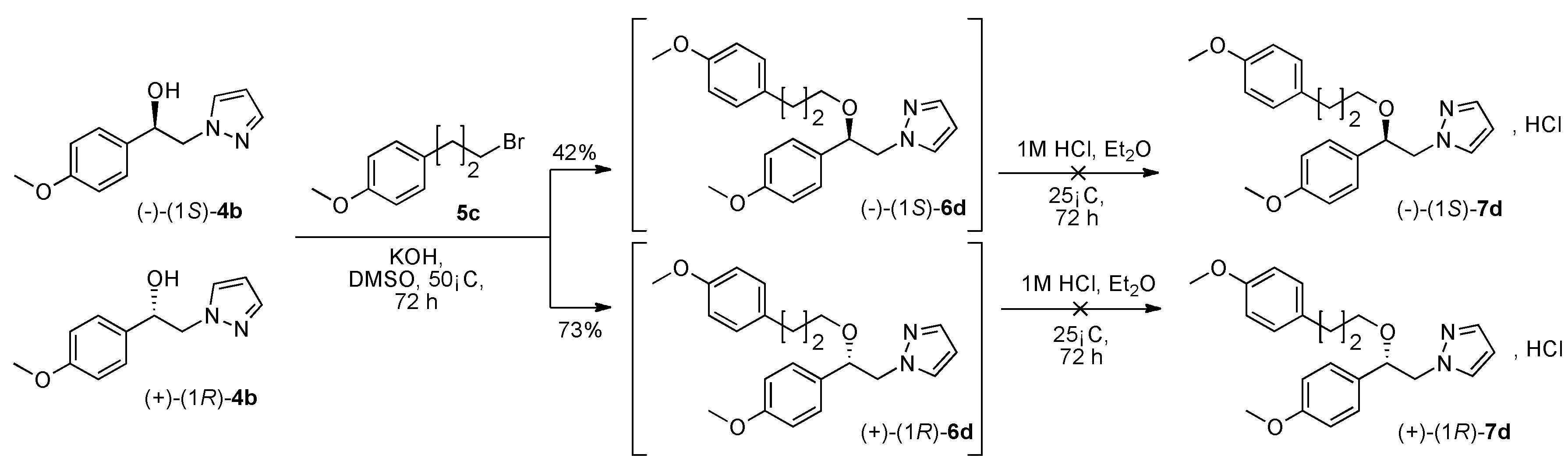
(1R, 1S) 1-[β-((4-Methoxyphenyl)-(3-(4-methoxyphenyl)propoxyethyl)]-1H-pyrazolium hydrochloride (7d). Using the standard procedure from 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (0.218 g, 1 mmol) and 1-(3-bromopropyl)-4-methoxybenzene 5c (0.229 g, 1 mmol, 1 equiv.), the 1-[β-((4-methoxyphenyl)-(3-(4-methoxyphenyl)propoxyethyl)]-1H-pyrazole 6d (0.156 g, 0.42 mmol) was dissolved in 2.84 mL of dry diethyl ether pa under a stream of argon. The addition of 0.42 mL of 1M HCl (0.42 mmol, 1 equiv.) produced a pale pink viscous oil that crystallized (12 h) on the circumference of the round flask, and mixing (200 rpm, 72 h) was pursued until complete crystallization of the viscous gum. The desired salt 7d was collected by filtration on a Büchner funnel (porosity N°4) and washed with 3 × 0.5 mL of dry diethyl ether pa. After drying at 25 °C for 4 h, the 1-[β-((4-methoxyphenyl)-(3-(4-methoxyphenyl)propoxyethyl)]-1H-pyrazolium hydrochloride 7d (72.53 mg, 18% yield) was obtained as white powder. Mp = 92–94 °C. 1H NMR (DMSO-d6) δ = 1.47–1.70 (m, 2H, CH2, H-2″), 2.36 (t, 2H, J = 7.5 Hz, CH2, H-3″), 3.03 (dt, 1H, J = 9.6, 6.2 Hz, CH, H-1″), 3.19 (dt, 1H, J = 9.5, 6.0 Hz, CH, H-1″), 3.69 (s, 3H, OCH3), 3.75 (s, 3H, OCH3), 4.18 (dd, 1H, J = 13.8, 4.3 Hz, CH, H-1′), 4.35 (dd, 1H, J = 13.8, 8.7 Hz, CH, H-1′), 4.62 (dd, 1H, J = 8.7, 4.2 Hz, CH, H-2′), 6.22 (t, 1H, J = 2.0 Hz, H-4, Ar), 6.77 (d, 2H, J = 8.6 Hz, H-3b, H-5b, Ar), 6.86–7.02 (m, 4H, H-3a, H-5a, H-2b, H-6b, Ar), 7.26 (d, 2H, J = 8.6 Hz, H-2a, H-6a, Ar), 7.47 (d, 1H, J = 1.8 Hz, H-3, Ar), 7.67 (d, 1H, J = 2.2 Hz, H-5, Ar). 13C NMR (DMSO-d6) δ = 30.4 (C-3″), 31.1 (C-2″), 54.9 (OCH3), 55.1 (OCH3), 57.2 (C-1′), 67.0 (C-1″), 79.7 (C-2′), 104.9 (C-4), 113.6 (C-3b, C-5b), 113.9 (C-3a, C-5a), 128.0 (C-2a, C-6a), 129.2 (C-2b, C-6b), 130.8 (C-3), 131.1 (C-1a), 133.4 (C-1b), 138.5 (C-5), 157.3 (C-4b), 159.0 (C-4a). ES+ HRMS, m/z = 389.1838 found (calculated for C22H26N2O3Na [M + Na]+ requires 389.1841).
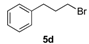
(1R, 1S) 1-[β-((4-Methoxyphenyl)-(3-phenylpropoxyethyl)]-1H-pyrazolium hydrochloride (7e). Using the standard procedure from 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (0.218 g, 1 mmol) and 1-bromo-3-phenylpropane 5d (0.199 g, 1 mmol, 1 equiv.), the 1-[β-((4-methoxyphenyl)-(3-phenylpropoxyethyl)]-1H-pyrazole 6e (0.124 g, 0.37 mmol) was dissolved in 2.5 mL of dry diethyl ether pa under a stream of argon. The addition of 0.37 mL of 1 M HCl (0.37 mmol, 1 equiv.) produced a translucent colourless viscous oil that crystallized progressively (24 h) on the circumference of the round flask, and mixing (200 rpm, 72 h) was pursued until complete crystallization in divided powder. The desired salt 7e was collected by filtration on a Büchner funnel (porosity N°4) and washed with 3 × 0.5 mL of dry diethyl ether pa. After drying at 25 °C for 4 h, the 1-[β-((4-methoxyphenyl)-(3-phenylpropoxyethyl)]-1H-pyrazolium hydrochloride 7d (82.04 mg, 22% yield) was obtained as white powder. Mp = 87–89 °C. 1H NMR (DMSO-d6) δ = 1.54–1.76 (m, 2H, CH2, H-2″), 2.37–2.46 (m, 2H, CH2, H-3″), 3.04 (dt, 1H, J = 9.6, 6.1 Hz, CH, H-1″), 3.21 (dt, 1H, J = 9.6, 6.0 Hz, CH, H-1″), 3.75 (s, 3H, OCH3), 4.18 (dd, 1H, J = 13.8, 4.3 Hz, CH, H-1′), 4.35 (dd, 1H, J = 13.8, 8.7 Hz, CH, H-1′), 4.63 (dd, 1H, J = 8.7, 4.3 Hz, CH, H-2′), 6.21 (t, 1H, J = 2.1 Hz, H-4, Ar), 6.92 (d, 2H, J = 8.7 Hz, H-3a, H-5a, Ar), 6.99–7.06 (m, 2H, H-2b, H-6b, Ar), 7.08–7.17 (m, 1H, H-4b, Ar), 7.17–7.31 (m, 4H, H-2a H-6a, H-3b, H-5b, Ar), 7.46 (dd, 1H, J = 1.9, 0.7 Hz, H-3, Ar), 7.66 (dd, 1H, J = 2.3, 0.7 Hz, H-5, Ar). 13C NMR (DMSO-d6) δ = 30.9 (C-2″), 31.4 (C-3″), 55.1 (OCH3), 57.2 (C-1′), 67.1 (C-1″), 79.7 (C-2′), 104.9 (C-4), 113.9 (C-3a, C-5a), 125.7 (C-4b), 128.1 (C-2a, C-6a), 128.2 (C-3b, C-5b), 128.4 (C-2b, C-6b), 130.8 (C-5), 131.2 (C-1a), 138.6 (C-3), 141.7 (C-1b), 159.1 (C-4a). ES+ HRMS, m/z = 359.1736 found (calculated for C21H24N2O2Na [M + Na]+ requires 359.1736).
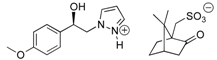
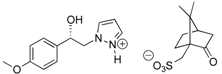
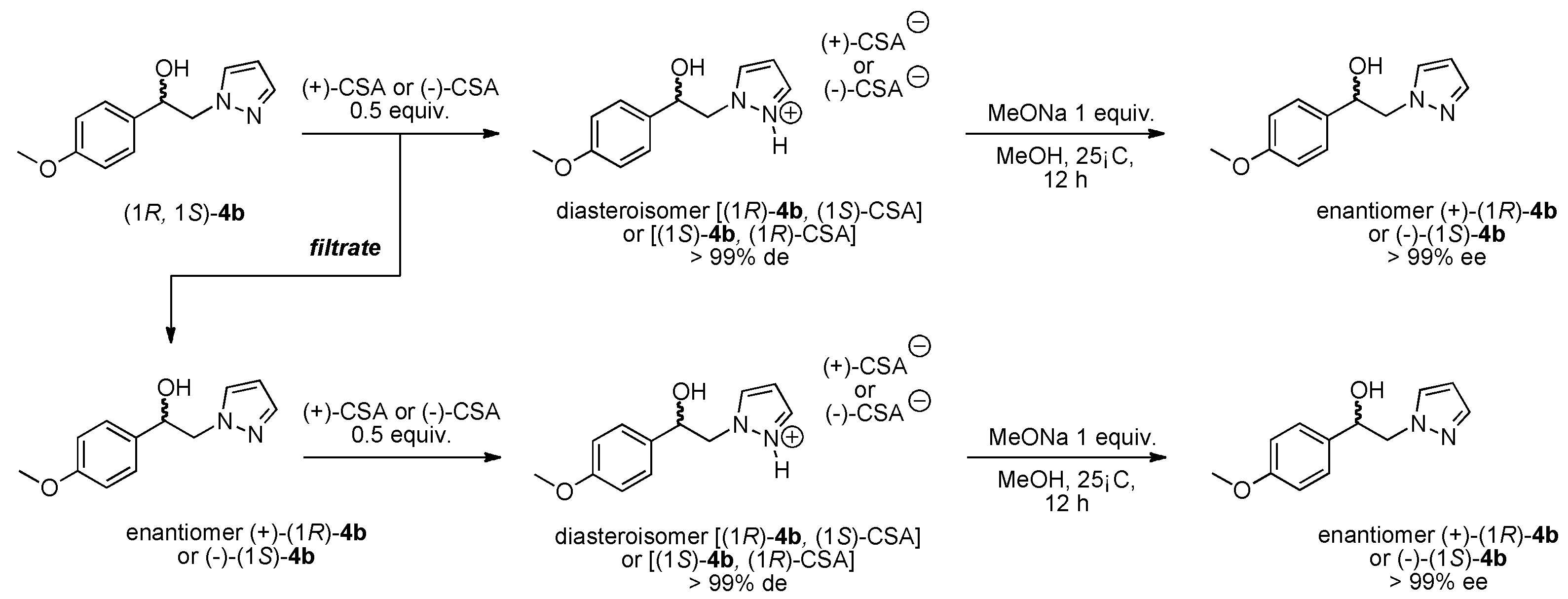
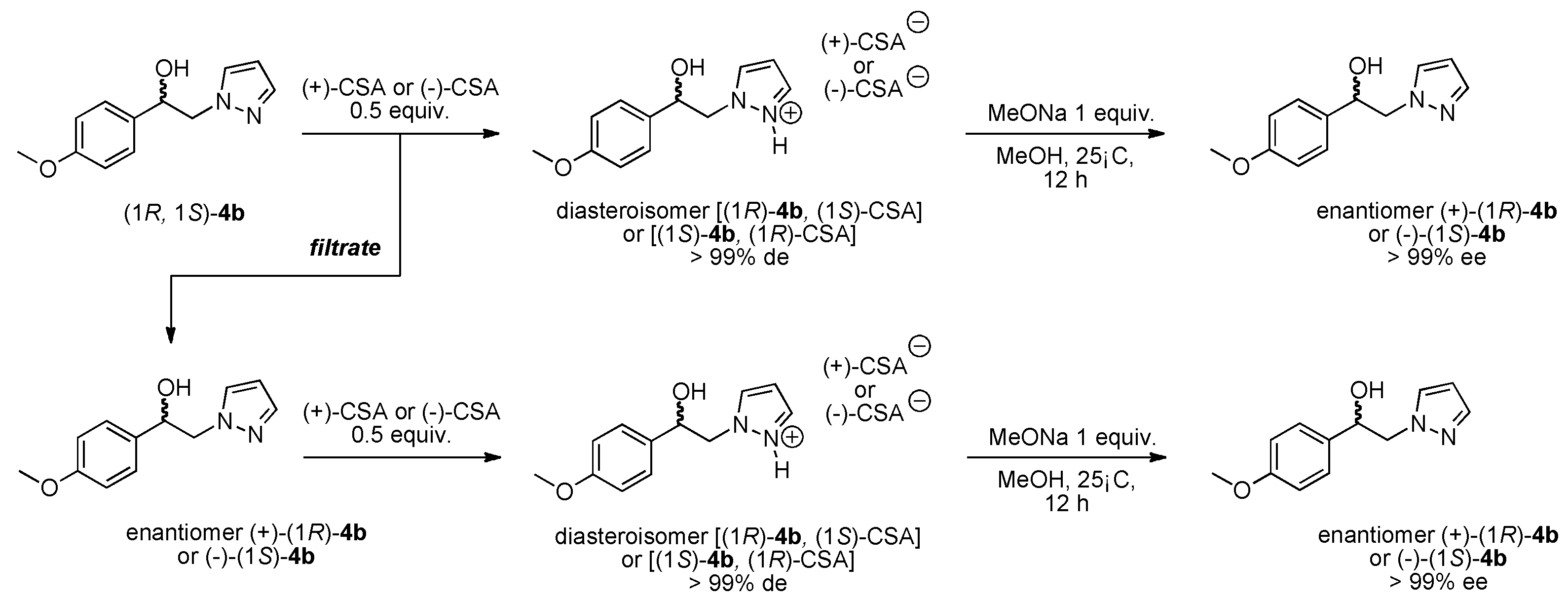
3.1.3. Resolution of (±)-(1R, 1S) 1-(4-Methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol (4b) with (−)-(1R)-10-Camphorsulfonic Acid and (+)-(1S)-10-Camphorsulfonic Acid by Two Convergent Methods of Half-Quantities
Method 1 from (−)-(1R)-CSA:
(−)-(1S) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol (4b): To a stirred solution of (±) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (0.5 g, 2.29 mmol) in 5 mL of dry acetone, a solution of (−)-(1R)-10-camphorsulfonic acid (0.266 g, 1.145 mmol) in 2 mL of dry acetone was added dropwise at room temperature for 5 min. Stirring (300 rpm) was pursued for 12 h at 25 °C. It is interesting to note that during the addition of the solution of (−)-(1R)-CSA, the formation of a white fine suspension of diastereomer appeared in the reaction mixture. This first fraction of diastereomer salt was collected by filtration on a Büchner funnel (porosity N°4), washed thoroughly with 4 × 0.3 mL of dry acetone, and dried in vacuum to give 0.363 g of the desired the salt (−)-(1S)-4b/(−)-(1R)-CSA (>85% de). This salt was recrystallized in dry acetone and afforded 0.271 g (52% isolated yield) of salt (−)-(1S)-4b/(−)-(1R)-CSA (>97% de). This salt was dissolved in 0.6 mL of dry acetone under magnetic stirring (300 rpm), and to this suspension, 0.02 equiv. of racemic (±) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b was added for diastereomeric enrichment. This suspension was stirred for 18 h at room temperature. The enriched salt was successively filtered on a Büchner funnel, washed with 0.5 mL of dry acetone, dried in vacuum, and recrystallized in dry acetone that produced 0.251 g (48% yield) of (−)-(1S)-4b/(−)-(1R)-CSA (>99% de) as white powder; [αD] = −6,0 (c 1.0, MeOH).
Starting from 200 mg of the pure salt (−)-(1S)-4b/(−)-(1R)-CSA, a mixed suspension was prepared in 2.5 mL of dry methanol and 1 equiv. of commercial MeONa was added in one portion. The resulting reaction mixture was stirred at room temperature for 12 h and then concentrated in a rotary evaporator under reduced pressure. The crude solid residue was washed thoroughly with 4 × 2 mL of deionized water. The insoluble compound was collected by filtration on a Büchner funnel (porosity N°4) and dried in vacuum, which produced 0.096 g (38% yield) of the desired (−)-(1S) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (99% ee); [αD] = −10,0 (c 1.0, MeOH) and Mp = 113–114 °C.
Recovery of the (+)-(1R) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b: The filtrate of the first fraction was submitted to concentration in a rotary evaporator under reduced pressure and afforded 0.345 g of a crude residue. To this solid residue, 14.5 mL of dry toluene was added and the resulting suspension was submitted to magnetic stirring for 4 h. This suspension was filtered through a Büchner funnel (porosity N°4) and the filtrate was concentrated in a rotary evaporator under reduced pressure and produced 0.190 g (0.87 mmol) of a white powder of (+)-(1R)-4b (65% ee). This white solid was dissolved in 3.8 mL of dry acetone under magnetic stirring and to the resulting solution, a solution of (+)-(1S)-CSA (0.2 g, 0.87 mmol, 1 equiv.) in 1.52 mL of dry acetone was added dropwise. During the addition of the (+)-(1S)-CSA solution, a fine suspension appeared in the mixture, which was stirred at room temperature for 12 h. The new diastereomer salt was collected by filtration then washed with 4 × 0.25 mL of dry acetone, dried in vacuum, which gave 0.249 g (63% yield) of the (+)-(1R)-4b/(+)-(1S)-CSA salt (96% de) as white powder. From this salt, diastereomeric enrichment was conducted with 0.04 equiv. of racemic (±) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b. The enriched suspension was stirred for 18 h at room temperature and the resulting enriched salt was successively submitted to filtration on a Büchner funnel, washing with 0.5 mL of dry acetone, drying in vacuum, and finally recrystallisation in dry acetone which gave 0.202 mg (51% yield) of (+)-(1R)-4b/(+)-(1S)-CSA (>99% de) as white needles; [αD] = +6,0 (c 1.0, MeOH).
Starting from 200 mg of the pure salt (+)-(1R)-4b/(+)-(1S)-CSA, a mixed suspension was prepared in 2.5 mL of dry methanol, and 1 equiv. of commercial MeONa was added in one portion. The resulting reaction mixture was stirred at room temperature for 12 h and then concentrated in a rotary evaporator under reduced pressure. The crude solid residue was washed thoroughly with 4 × 2 mL of deionized water. The insoluble compound was collected by filtration on a Büchner funnel (porosity N°4) and dried in vacuum which gave 0.078 g (41% yield) of the desired (+)-(1R) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (99% ee); [αD] = +10,0 (c 1.0, MeOH) and Mp = 110–111 °C.
Method 2 with (+)-(1S)-CSA:
(+)-(1R) 1-(4-Methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol (4b): To a stirred solution of (±) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (0.5 g, 2.29 mmol) in 5 mL of dry acetone, a solution of (+)-(1S)-10-camphorsulfonic acid (0.266 g, 1.145 mmol) in 2 mL of dry acetone was added dropwise at room temperature for 5 min. Stirring (300 rpm) was pursued for 12 h at 25 °C. It is interesting to note that during the addition of the solution of (+)-(1S)-CSA, the formation of a white fine suspension of diastereomer appeared in the reaction mixture. This first fraction of diastereomer salt was collected by filtration on a Büchner funnel (porosity N°4), washed thoroughly with 4 × 0.3 mL of dry acetone, and dried in vacuum to give 0.358 g of the desired the salt (+)-(1R)-4b/(+)-(1S)-CSA (>87% de). This salt was recrystallized in dry acetonitrile and afforded 0.280 g (54% isolated yield) of salt (+)-(1R)-4b/(+)-(1S)-CSA (>96% de). This salt was dissolved in 0.6 mL of dry acetone under magnetic stirring (300 rpm) and to this suspension was added 0.02 equiv. of racemic (±) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b for diastereomeric enrichment. This suspension was stirred for 18 h at room temperature. The enriched salt was successively filtered on a Büchner funnel, washed with 0.5 mL of dry acetone, dried in vacuum, and recrystallized in dry acetone that gave 0.229 g (44% yield) of (+)-(1R)-4b/(+)-(1S)-CSA (>99% de) as white powder; [αD] = +6,0 (c 1.0, MeOH).
Starting from 200 mg of the pure salt (+)-(1R)-4b/(+)-(1S)-CSA, a mixed suspension was prepared in 2.5 mL of dry methanol, and 1 equiv. of commercial MeONa was added in one portion. The resulting reaction mixture was stirred at room temperature for 12 h and then concentrated in a rotary evaporator under reduced pressure. The crude solid residue was washed thoroughly with 4 × 2 mL of deionized water. The insoluble compound was collected by filtration on a Büchner funnel (porosity N°4) and dried in vacuum, which gave 0.089 g (35% yield) of the desired (+)-(1R) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (99% ee); [αD] = +10,0 (c 1.0, MeOH) and Mp = 110–111 °C.
Recovery of the (−)-(1S) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b: The filtrate of the first fraction was submitted to concentration in a rotary evaporator under reduced pressure and afforded 0.366 g of a crude residue. To this solid residue, 14.5 mL of dry toluene was added and the resulting suspension was submitted to magnetic stirring for 4 h. This suspension was filtered through a Büchner funnel (porosity N°4) and the filtrate was concentrated in a rotary evaporator under reduced pressure and produced 0.207 g (0.95 mmol) of a white powder of (−)-(1S)-4b (74% ee). This white solid was dissolved in 4.2 mL of dry acetone under magnetic stirring and to the resulting solution, a solution of (−)-(1R)-CSA (0.219 g, 0.95 mmol, 1 equiv.) in 1.66 mL of dry acetone was added dropwise. During the addition of the (−)-(1R)-CSA solution, a fine suspension appeared in the mixture, which was stirred at room temperature for 12 h. The new diastereomer salt was collected by filtration then washed with 4 × 0.25 mL of dry acetone and dried in vacuum, which gave 0.285 g (66% yield) of the (−)-(1S)-4b/(−)-(1R)-CSA salt (98% de) as white powder. From this salt, diastereomeric enrichment was conducted with 0.01 equiv. of racemic (±) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b. The enriched suspension was stirred for 18 h at room temperature and the resulting enriched salt was successively submitted to filtration on a Büchner funnel, washing with 0.5 mL of dry acetone, drying in vacuum, and finally recrystallisation in dry acetone which gave 0.258 mg (60% yield) of (−)-(1S)-4b/(−)-(1R)-CSA (>99% de) as white needles; [αD] = −6,0 (c 1.0, MeOH).
Starting from 200 mg of the pure salt (−)-(1S)-4b/(−)-(1R)-CSA, a mixed suspension was prepared in 2.5 mL of dry methanol, and 1 equiv. of commercial MeONa was added in one portion. The resulting reaction mixture was stirred at room temperature for 12 h and then concentrated in a rotary evaporator under reduced pressure. The crude solid residue was washed thoroughly with 4 × 2 mL of deionized water. The insoluble compound was collected by filtration on a Büchner funnel (porosity N°4) and dried in vacuum which gave 0.090 g (43% yield) of the desired (−)-(1S) 1-(4-methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol 4b (>99% ee); [αD] = −10,0 (c 1.0, MeOH) and Mp = 113-114 °C.
(−)-(1S)-4b/(−)-(1R)-CSA salt: Yield = 48%. White powder, Mp = 168–170 °C. [αD] = −6.0 (c 1.0, MeOH) and >99% de (retention time tR = 34.01 min. using hexane/i-PrOH 94:6 v/v as eluent, flow rate = 0.8 mL/min.). 1H NMR (DMSO-d6) δ = 0.75 (s, 3H, C-7″CH3), 1.04 (s, 3H, C-7″CH3), 1.19–1.40 (m, 2H, CH2, H-2″, H-5″), 1.81 (d, 1H, J = 18.1 Hz, CH, H-4″), 1.80–1.92 (m, 1H, CH, H-5″), 1.95 (t, 1H, J = 4.5 Hz, CH, H-4″), 2.16–2.34 (m, 1H, CH, H-3″), 2.46 (d, 1H, J = 14.7 Hz, CH, CH2SO3H), 2.55–2.76 (m, 1H, CH, H-2″), 2.93 (d, 1H, J = 14.7 Hz, CH, CH2SO3H), 3.73 (s, 3H, OCH3), 4.10–4.32 (m, 2H, CH2, H-2), 4.87 (dd, 1H, J = 7.3, 5.5 Hz, CH, H-1), 6.20 (t, 1H, J = 2.1 Hz, H-4′, Ar), 6.87 (d, 2H, J = 8.7 Hz, H-3a, H-5a, Ar), 7.22 (d, 2H, J = 8.5 Hz, H-2a, H-6a, Ar), 7.47 (dd, 1H, J = 1.9, 0.7 Hz, H-3′, Ar), 7.61 (dd, 1H, J = 2.3, 0.7 Hz, H-5′, Ar). 13C NMR (DMSO-d6) δ = 19.5 (C-7″CH3), 20.0 (C-7″CH3), 24.2 (C-2″), 26.4 (C-5″), 42.1 (C-3″), 42.2 (C-4″), 46.8 (CH2SO3H), 47.1 (C-7″), 55.0 (OCH3), 58.1 (C-6″), 58.6 (C-2), 71.3 (C-1), 104.8 (C-4′), 113.5 (C-3a, C-5a), 127.2 (C-2a, C-6a), 130.9 (C-5′), 134.7 (C-1a), 138.1 (C-3′), 158.5 (C-4a), 216.0 (C=O). ES+ HRMS, m/z = 241.0946 found (calculated for C12H14N2O2Na [M – H + Na]+ requires 241.0947).
(+)-(1R)-4b/(+)-(1S)-CSA salt: Yield = 51%. White powder, Mp = 169-171 °C. [αD] = +6.0 (c 1.0, MeOH) and >99% de (retention time tR = 35.5 min. using hexane/i-PrOH 94:6 v/v as eluent, flow rate = 0.8 mL/min.). 1H NMR (DMSO-d6) δ = 0.75 (s, 3H, C-7″CH3), 1.04 (s, 3H, C-7″CH3), 1.17–1.42 (m, 2H, CH2, H-2″, H-5″), 1.81 (d, 1H, J = 18.1 Hz, CH, H-4″), 1.78–1.93 (m, 1H, CH, H-5″), 1.95 (t, 1H, J = 4.5 Hz, CH, H-4″), 2.25 (dt, 1H, J = 18.0, 4.0 Hz, CH, H-3″), 2.45 (d, 1H, J = 14.7 Hz, CH, CH2SO3H), 2.54-2.76 (m, 1H, CH, H-2″), 2.93 (d, 1H, J = 14.7 Hz, CH, CH2SO3H), 3.73 (s, 3H, OCH3), 4.08–4.32 (m, 2H, CH2, H-2), 4.87 (dd, 1H, J = 7.3, 5.5 Hz, CH, H-1), 6.20 (t, 1H, J = 2.1 Hz, H-4′, Ar), 6.87 (d, 2H, J = 8.7 Hz, H-3a, H-5a, Ar), 7.22 (d, 2H, J = 8.5 Hz, H-2a, H-6a, Ar), 7.43-7.41 (m, 1H, H-3′, Ar), 7.60 (dd, 1H, J = 2.3, 0.7 Hz, H-5′, Ar). 13C NMR (DMSO-d6) δ = 19.5 (C-7″CH3), 20.0 (C-7″CH3), 24.2 (C-2″), 26.4 (C-5″), 42.1 (C-3″), 42.2 (C-4″), 46.8 (CH2SO3H), 47.1 (C-7″), 55.0 (OCH3), 58.1 (C-6″), 58.6 (C-2), 71.3 (C-1), 104.8 (C-4′), 113.5 (C-3a, C-5a), 127.2 (C-2a, C-6a), 130.9 (C-5′), 134.7 (C-1a), 138.1 (C-3′), 158.5 (C-4a), 216.0 (C=O). ES+ HRMS, m/z = 241.0946 found (calculated for C12H14N2O2Na [M − H + Na]+ requires 241.0947).
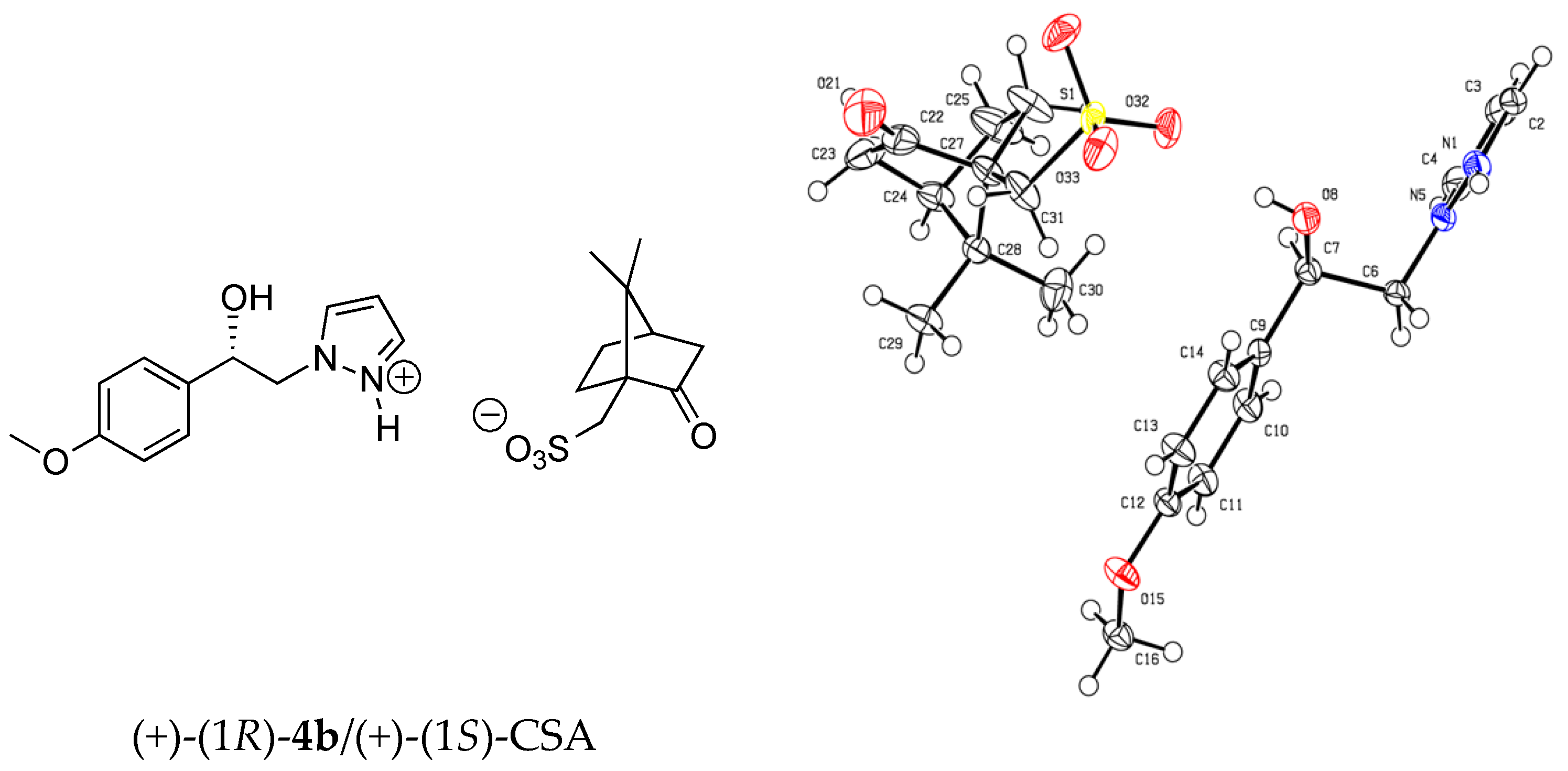
X-ray crystallographic data for (+)-(1
R)-
4b/(+)-(1
S)-CSA salt: (C
12H
15N
2O
2, C
10H
15O
4S);
M = 450.54. APEXII, Bruker-AXS diffractometer, Mo-Kα radiation (λ = 0.71073 Å),
T = 150(2) K; orthorhombic
P 21 21 21 (I.T.#19), a = 7.2781(5), b = 9.5344(5), c = 32.8134(19) Å,
V = 2277.0(2) Å
3.
Z = 4,
d = 1.314 g·cm
−3, μ = 0.182 mm
−1. The structure was solved by direct methods using the
SIR97 program [
29], and then refined with full-matrix least-square methods based on
F2 (
SHELXL-97) [
30] with the aid of the
WINGX [
31] program. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Except oxygen- and nitrogen-linked hydrogen atoms that were introduced in the structural model through Fourier difference maps analysis, H atoms were finally included in their calculated positions. A final refinement on
F2 with 4854 unique intensities and 289 parameters converged at ω
R(
F2) = 0.0932 (
R(
F) = 0.0415) for 4347 observed reflections with
I > 2σ(
I). Crystallographic data for the structure of (+)-(1
R)-
4b/(+)-(1
S)-CSA salt in this paper have been deposited in the Cambridge Crystallographic Data Centre as supplementary publication number CCDC 1580544. Copies of the data can be obtained, free of charge, on application to CCDC, 12 Union Road, Cambridge CB21EZ, UK (fax: +44(0)-1223-336033 or e-mail:
deposit@ccdc.cam.ac.uk).
(−)-(1S) 1-(4-Methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol (4b): Yield = 38%. White powder, Mp = 113–114 °C. [αD] = −10.0 (c 1.0, MeOH) and >99% de (retention time tR = 52.6 min. using hexane/i-PrOH 96:4 v/v as eluent, flow rate = 0.6 mL/min.). 1H NMR (DMSO-d6) δ = 3.73 (s, 3H, OCH3), 4.06–4.30 (m, 2H, H-2, CH2, H-2), 4.87 (q, 1H, J = 5.6 Hz, CH, H-1), 5.50 (br d, 1H, J = 4.5 Hz, OH), 6.16 (t, 1H, J = 2.0 Hz, H-4′, Ar), 6.87 (d, 2H, J = 8.6 Hz, H-3a, H-5a, Ar), 7.21 (d, 2H, J = 8.6 Hz, H-2a, H-6a, Ar), 7.41 (d, 1H, J = 1.8 Hz, H-3′, Ar), 7.56 (d, 1H, J = 2.2 Hz, H-5′, Ar). 13C NMR (DMSO-d6) δ = 55.0 (OCH3), 58.7 (C-2), 71.4 (C-1), 104.6 (C-4′), 113.5 (C-3a, C-5a), 127.2 (C-2a, C-6a), 130.5 C-5′), 134.8 (C-1a), 138.4 (C-3′), 158.5 (C-4a). ES+ HRMS, m/z = 241.0950 found (calculated for C12H14N2O2Na [M – H + Na]+ requires 241.0953).
(+)-(1R) 1-(4-Methoxyphenyl)-2-(1H-pyrazol-1-yl)ethan-1-ol (4b): Yield = 41%. White powder, Mp = 110–111 °C. [αD] = +10.0 (c 1.0, MeOH) and >99% de (retention time tR = 63.5 min. using hexane/i-PrOH 96:4 v/v as eluent, flow rate = 0.6 mL/min.). 1H NMR (DMSO-d6) δ = 3.73 (s, 3H, OCH3), 4.09–4.29 (m, 2H, H-2, CH2, H-2), 4.87 (q, 1H, J = 5.6 Hz, CH, H-1), 5.50 (br d, 1H, J = 4.5 Hz, OH), 6.16 (t, 1H, J = 2.0 Hz, H-4′, Ar), 6.87 (d, 2H, J = 8.6 Hz, H-3a, H-5a, Ar), 7.22 (d, 2H, J = 8.6 Hz, H-2a, H-6a, Ar), 7.41 (d, 1H, J = 1.8 Hz, H-3′, Ar), 7.56 (d, 1H, J = 2.2 Hz, H-5′, Ar). 13C NMR (DMSO-d6) δ = 55.0 (OCH3), 58.7 (C-2), 71.4 (C-1), 104.6 (C-4′), 113.5 (C-3a, C-5a), 127.2 (C-2a, C-6a), 130.5 C-5′), 134.8 (C-1a), 138.4 (C-3′), 158.5 (C-4a). ES+ HRMS, m/z = 241.0950 found (calculated for C12H14N2O2Na [M − H + Na]+ requires 241.0953).

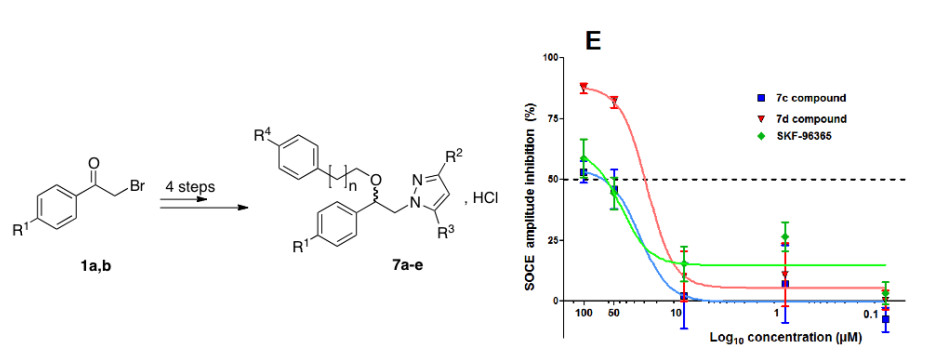
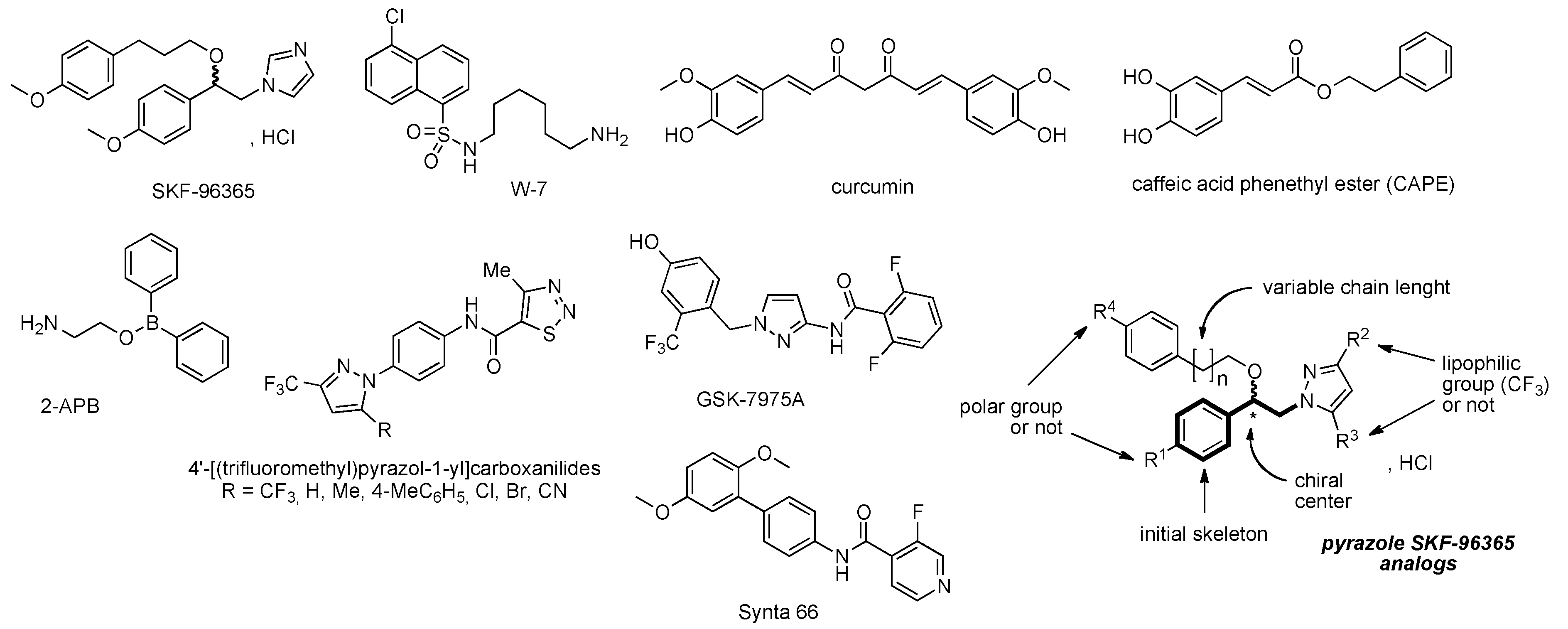
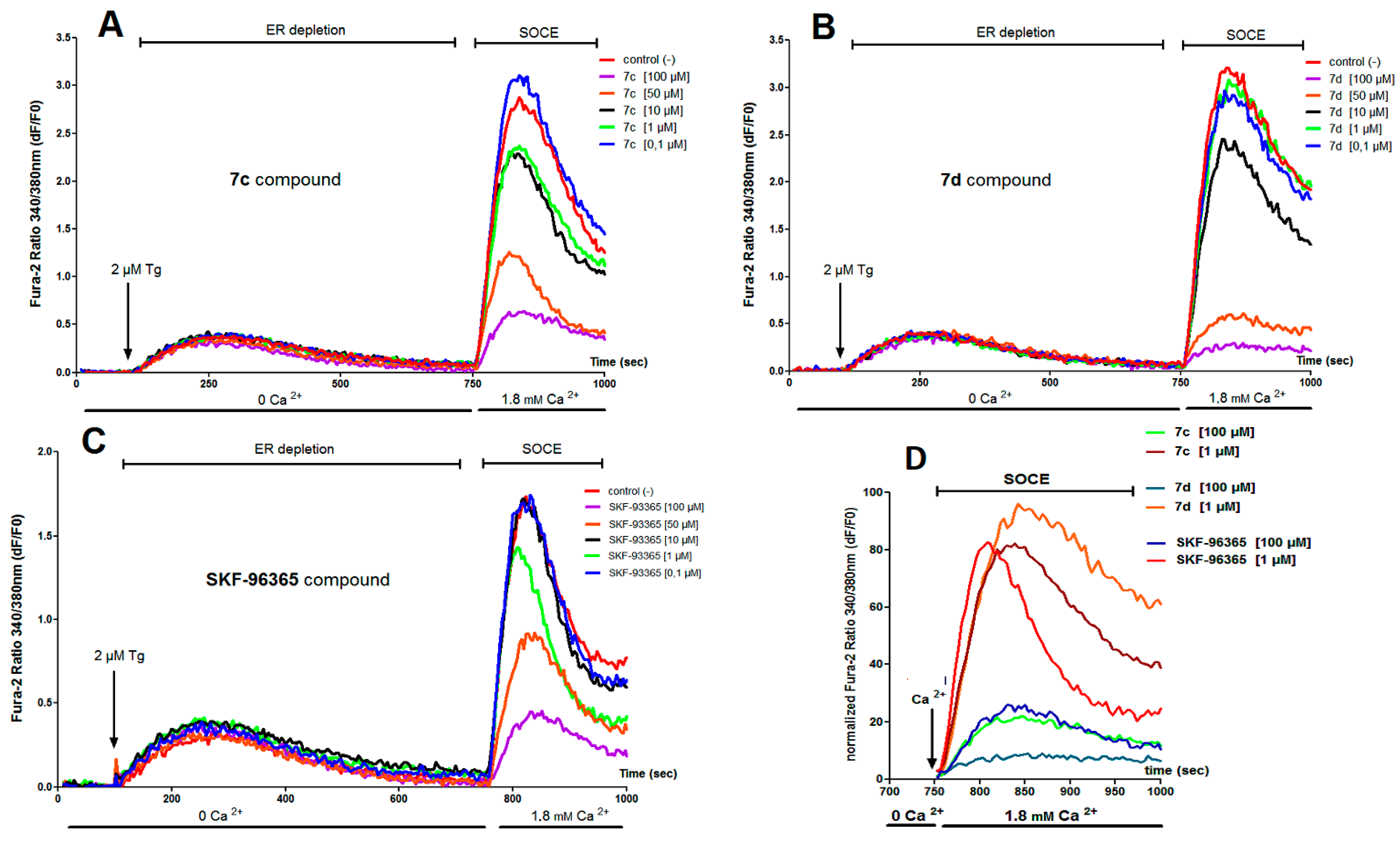
 ,
, 




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}