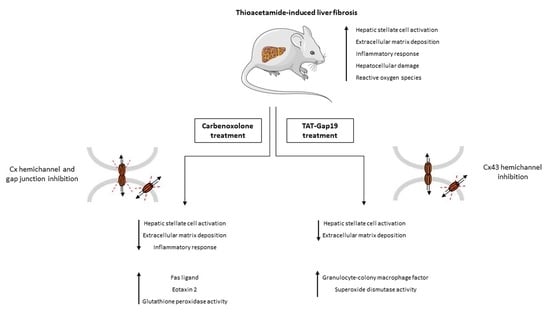
TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice

 , , ,
, , ,  , ,
, , 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
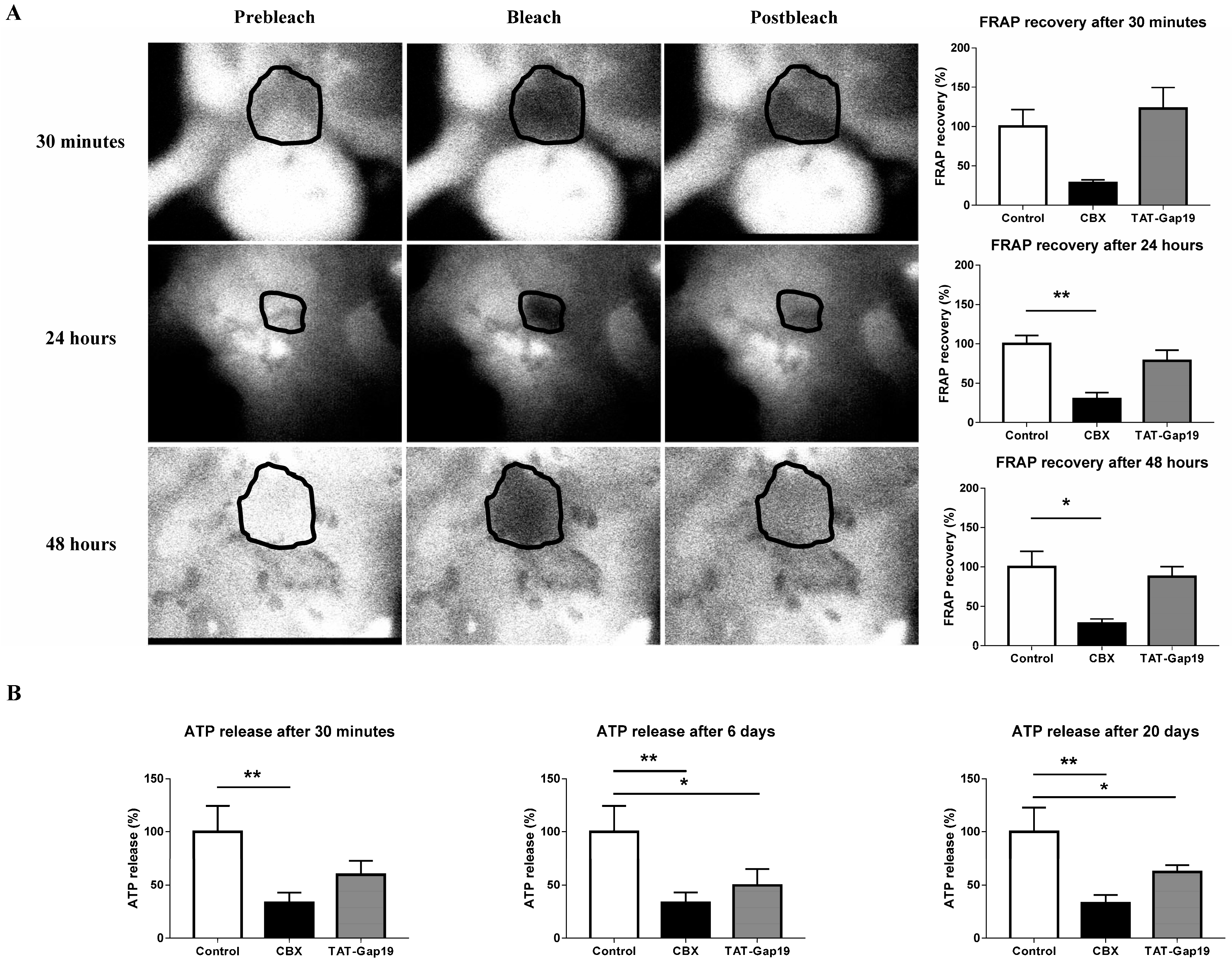
2.1. Effects of CBX and TAT-Gap19 on Gap Junctions and Hemichannels in Cultures of Primary Rat Hepatocytes
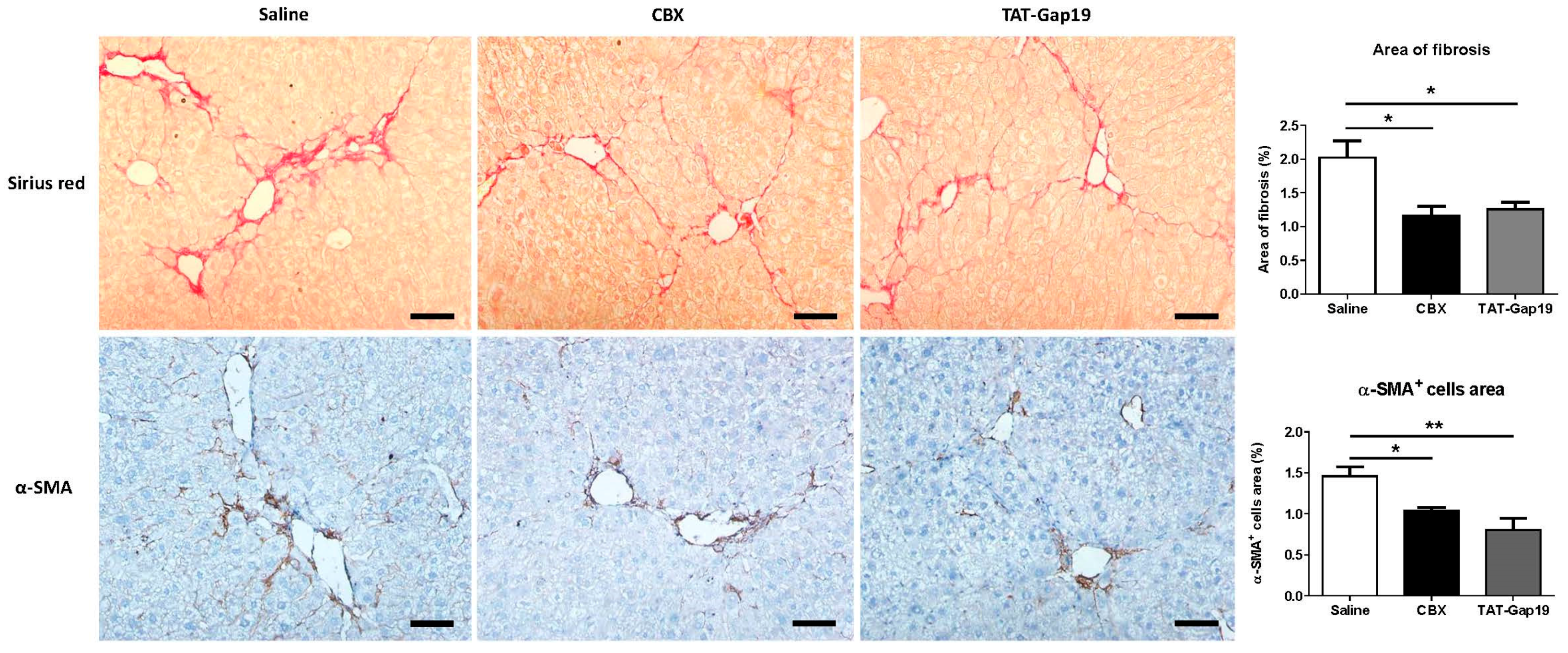
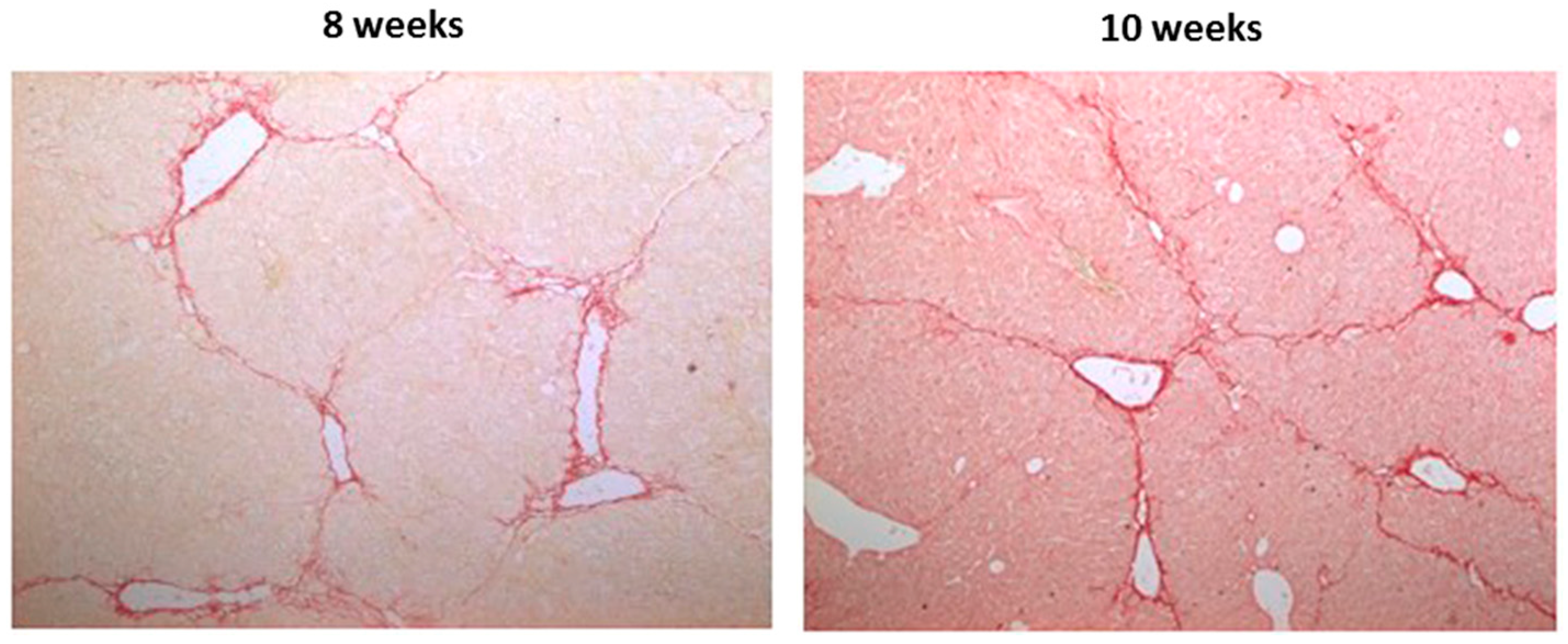
2.2. Effects of CBX and TAT-Gap19 on the Fibrotic Response after TAA-Induced Chronic Hepatic Injury in Mice
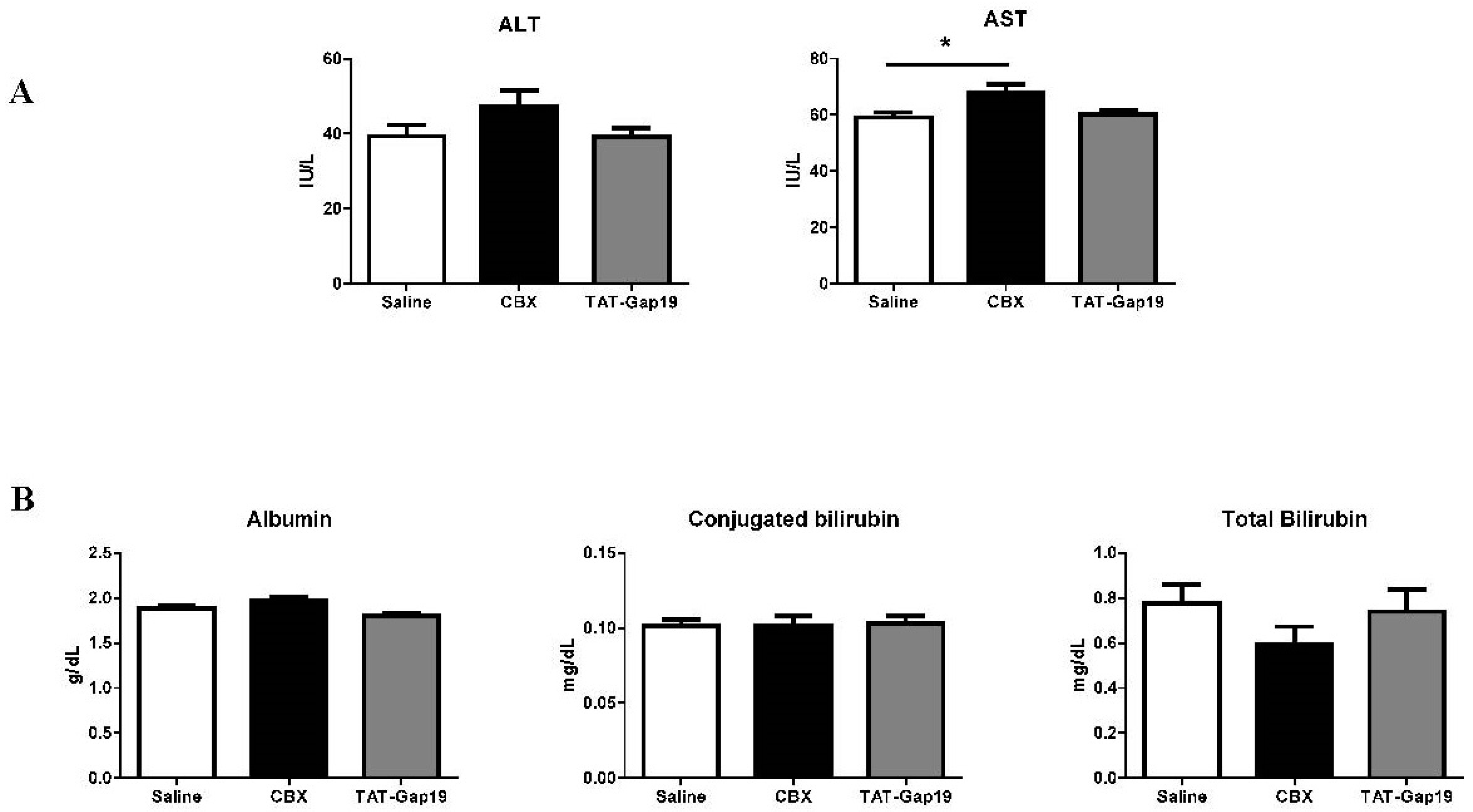
2.3. Effects of CBX and TAT-Gap19 on Biochemical Parameters after TAA-Induced Chronic Hepatic Injury in Mice
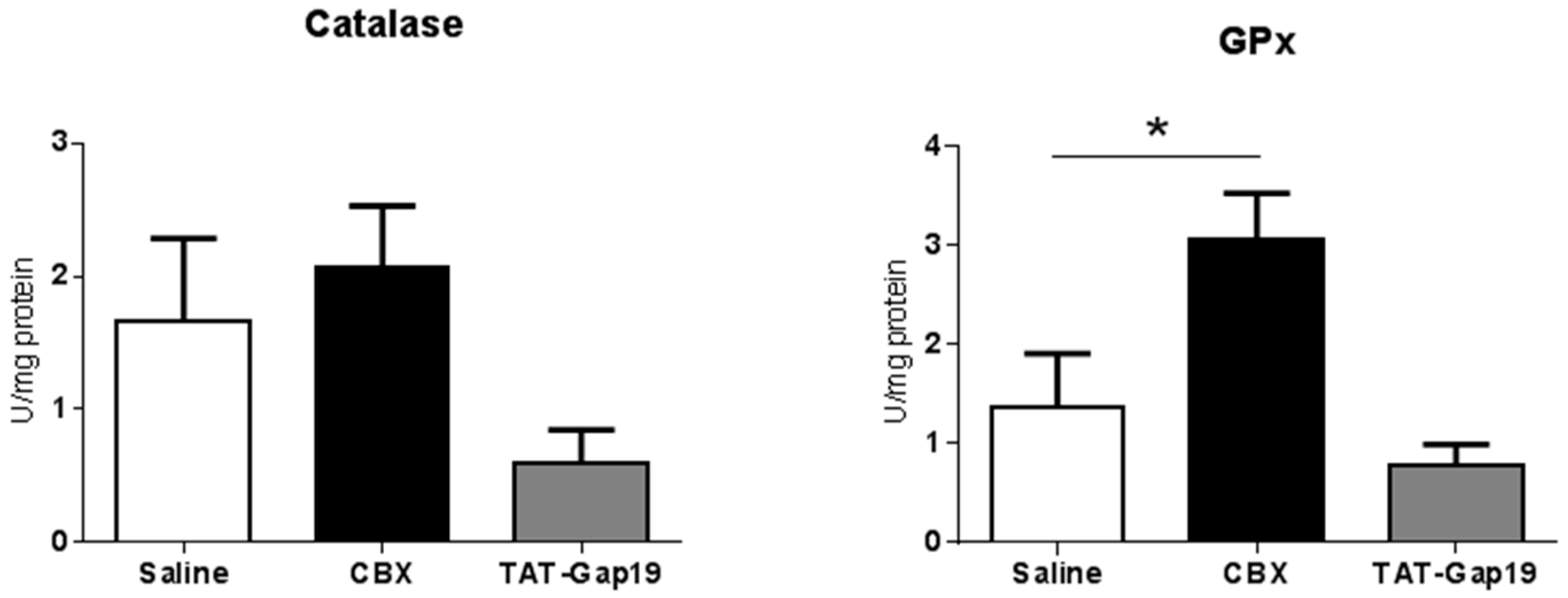
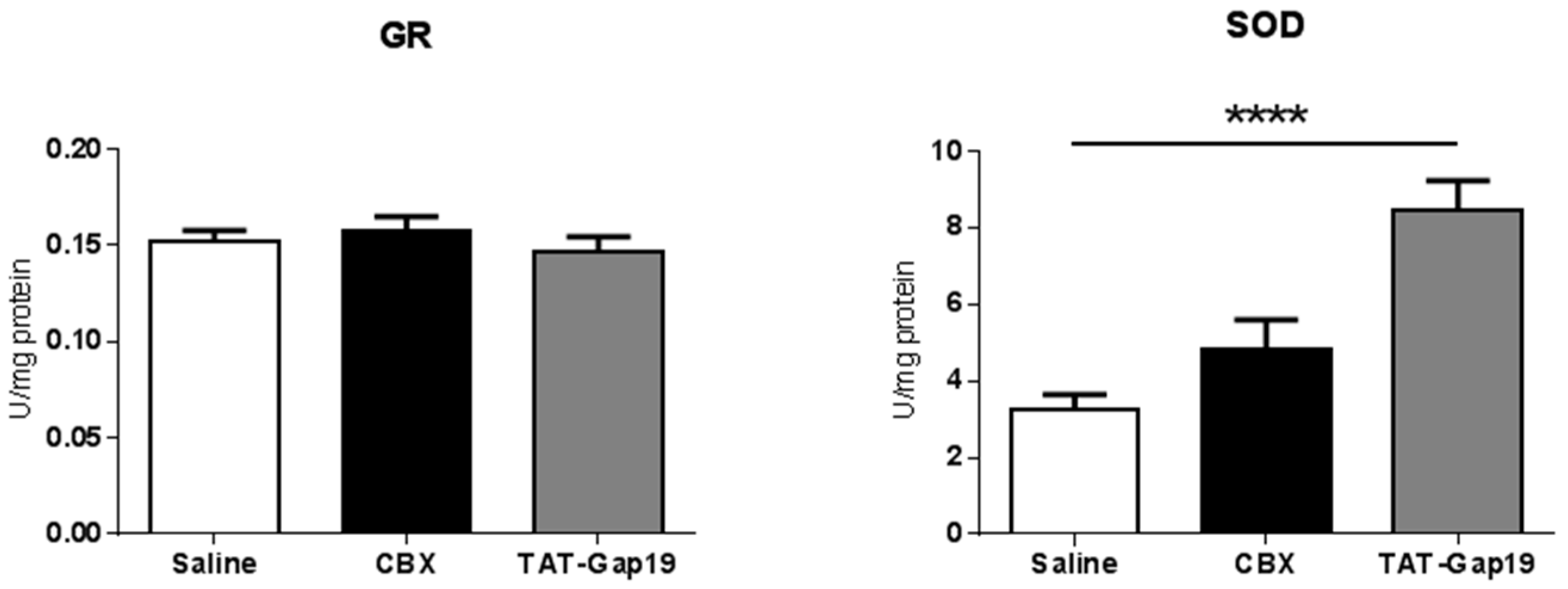
2.4. Effects of CBX and TAT-Gap19 on Anti-Oxidative Enzyme Activity after TAA-Induced Chronic Hepatic Injury in Mice
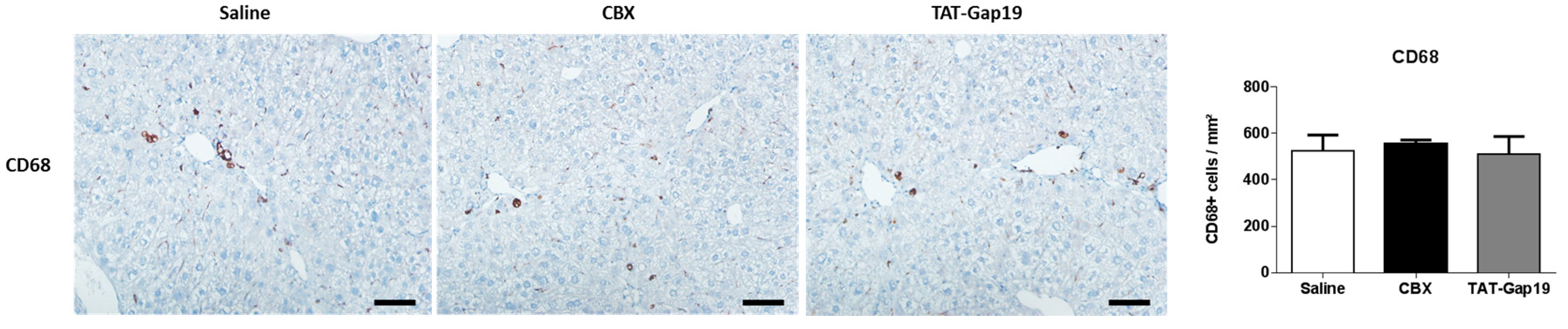
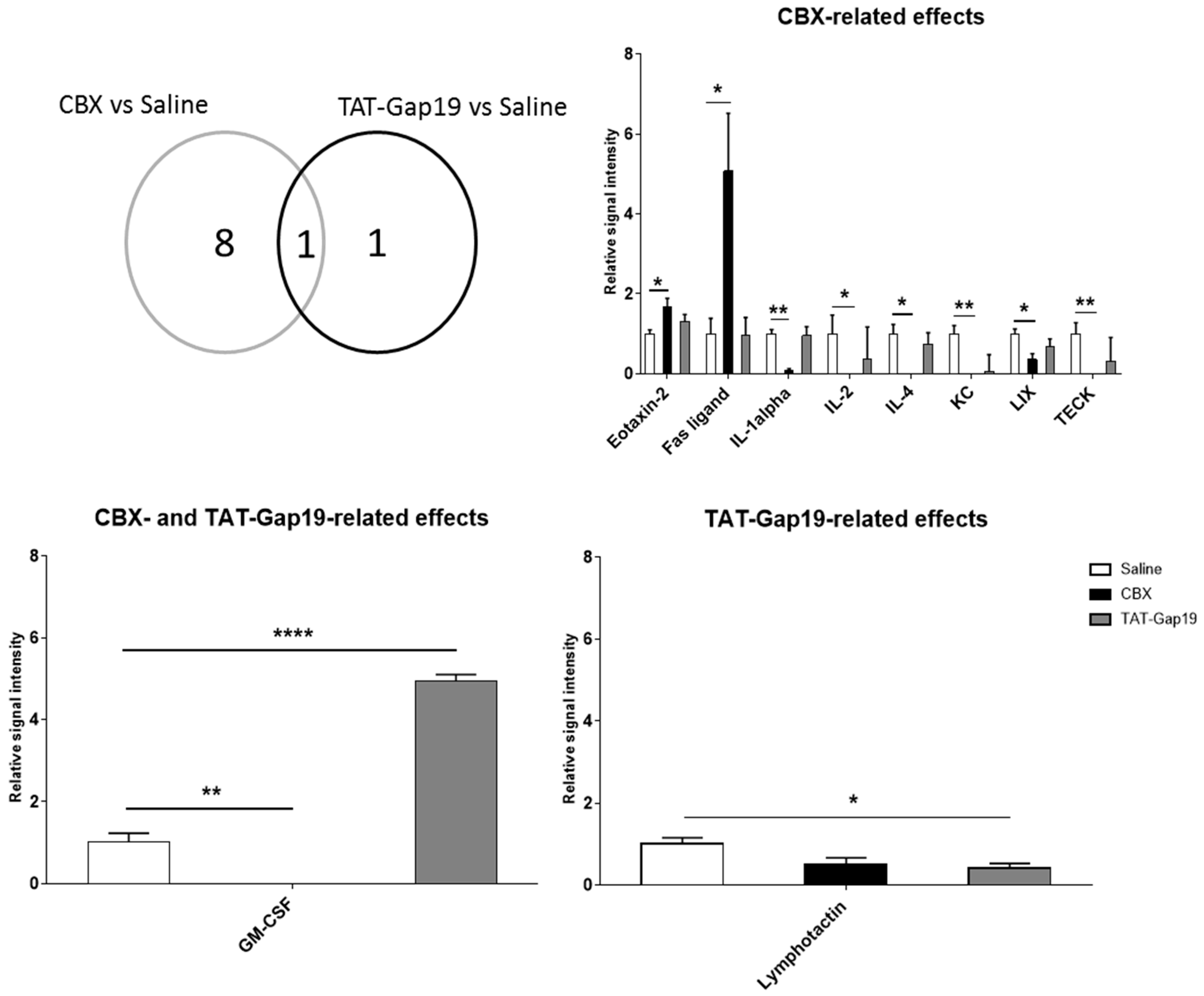
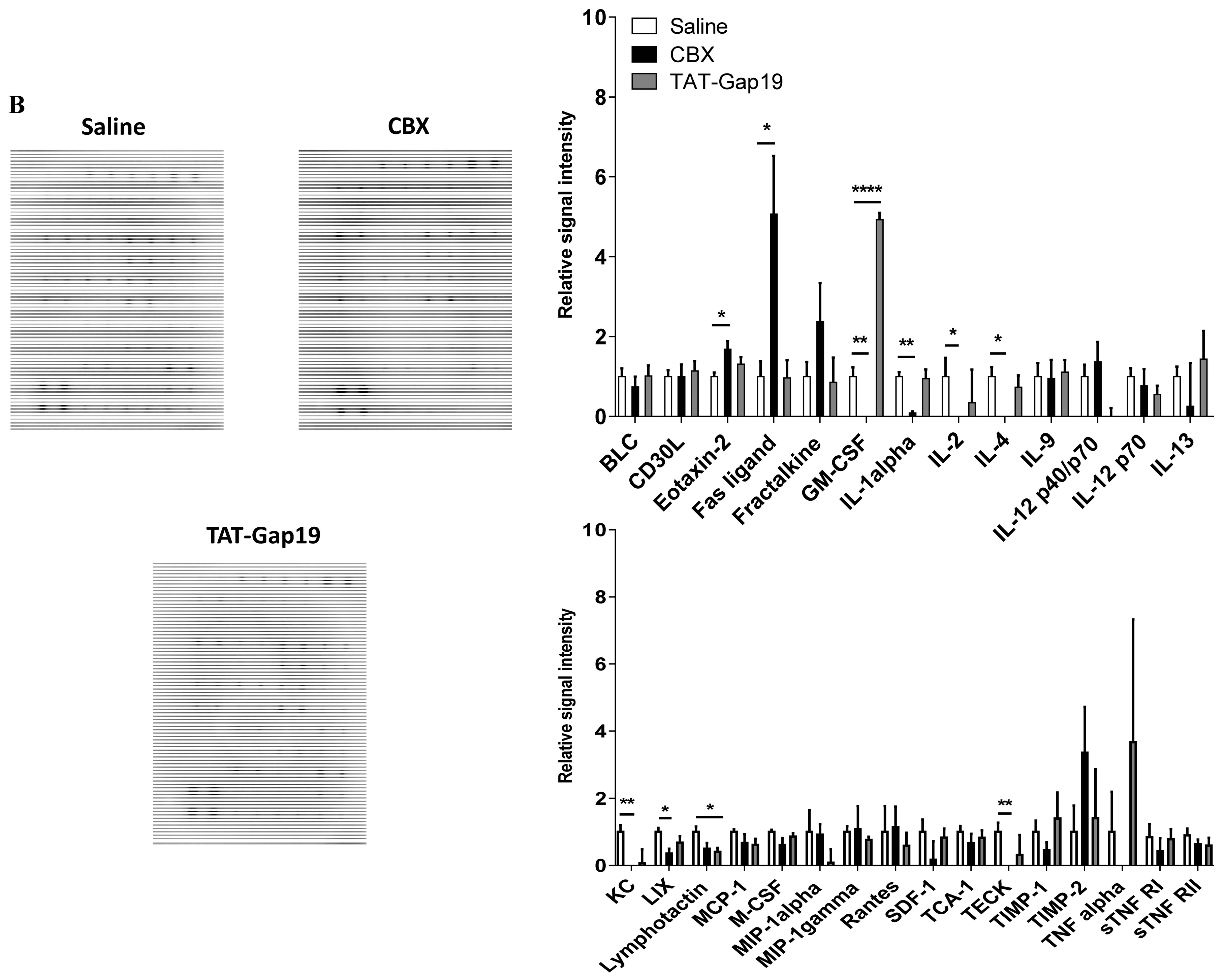
2.5. Effects of CBX and TAT-Gap19 on the Inflammatory Response after TAA-Induced Chronic Hepatic Injury in Mice
3. Discussion
4. Materials and Methods
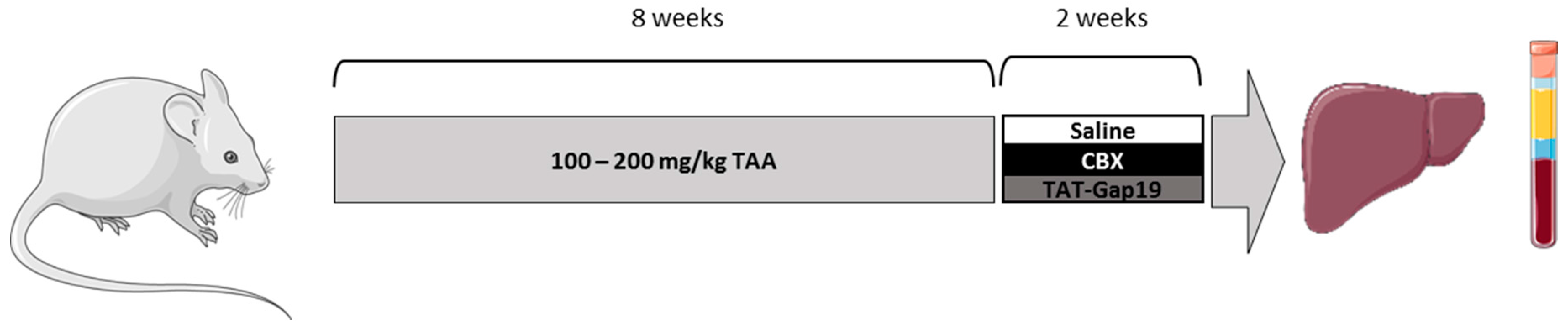
4.1. Animals and Treatment
4.2. Hepatocyte Rat Isolation and Cultivation
4.3. TAT-Gap19 and CBX
4.4. Fluorescence Recovery after Photobleaching
4.5. Measurement of Extracellular Adenosine-5′- Triphosphate
4.6. Histopathological Liver Examination and Collagen Analysis
4.7. Immunohistochemistry
4.8. Analysis of Serum Biochemical Parameters
4.9. Analysis of Hepatic Anti-Oxidant Enzymes
4.10. Analysis of Liver Inflammatory Markers
4.11. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ALT | Alanine aminotransferase |
| ANOVA | Analysis of variance |
| α-SMA | Alpha smooth muscle actin |
| AST | Aspartate aminotransferase |
| ATP | Adenosine-5’-triphosphate |
| CBX | Carbenoxolone |
| Cx | Connexin |
| DF | Divalent free |
| FRAP | Fluorescence recovery after photobleaching |
| GJ(s) | Gap junction(s) |
| GM-CSF | Granulocyte-macrophage colony stimulating factor |
| GPx | Glutathione peroxidase |
| GR | Glutathione reductase |
| HBSS | Hank’s balanced salt solution |
| HSC(s) | Hepatic stellate cell(s) |
| IL | Interleukin |
| KC | Growth-regulated alpha protein |
| LIX | Lipopolysaccharide-induced CXC chemokine |
| n | Number of biological repeats |
| N | Number of technical repeats |
| p | Probability |
| SEM | Standard error of the mean |
| SOD | Superoxide dismutase |
| TAA | Thioacetamide |
| TAT | Transactivator of transcription |
| TECK | Thymus-expressed chemokine |
Appendix A



References
- Bataller, R.; Brenner, D.A. Liver fibrosis. J. Clin. Investig. 2005, 115, 209–218. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Karlmark, K.R.; Weiskirchen, R.; Zimmermann, H.W.; Gassler, N.; Ginhoux, F.; Weber, C.; Merad, M.; Luedde, T.; Trautwein, C.; Tacke, F. Hepatic recruitment of the inflammatory gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009, 50, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Tsuchida, T.; Friedman, S.L. Mechanisms of hepatic stellate cell activation. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Henkens, T.; de Rop, E.; Fraczek, J.; Vanhaecke, T.; Rogiers, V. Biology and pathobiology of gap junctional channels in hepatocytes. Hepatology 2008, 47, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Flagg-Newton, J.; Simpson, I.; Loewenstein, W.R. Permeability of the cell-to-cell membrane channels in mammalian cell juncton. Science 1979, 205, 404–407. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, G.S.; Moreno, A.P.; Lampe, P.D. Gap junctions between cells expressing connexin 43 or 32 show inverse permselectivity to adenosine and APT. J. Biol. Chem. 2002, 277, 36725–36730. [Google Scholar] [CrossRef] [PubMed]
- Decrock, E.; Vinken, M.; de Vuyst, E.; Krysko, D.V.; D’Herde, K.; Vanhaecke, T.; Vandenabeele, P.; Rogiers, V.; Leybaert, L. Connexin-related signaling in cell death: To live or let die? Cell Death Differ. 2009, 16, 524–536. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Decrock, E.; Cogliati, B.; Oliveira, A.G.; Marques, P.E.; Dagli, M.L.; Menezes, G.B.; Mennecier, G.; Leybaert, L.; Vanhaecke, T.; et al. Connexin and pannexin (hemi)channels in the liver. Front. Physiol. 2014, 4, 405. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Decrock, E.; de Vuyst, E.; De Bock, M.; Vandenbroucke, R.E.; de Geest, B.G.; Demeester, J.; Sanders, N.N.; Vanhaecke, T.; Leybaert, L.; et al. Connexin32 hemichannels contribute to the apoptotic-to-necrotic transition during fas-mediated hepatocyte cell death. Cell Mol. Life Sci. 2010, 67, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; McGill, M.R.; da Silva, T.C.; Abels, C.; Lebofsky, M.; Maria Monteiro de Araújo, C.; Tiburcio, T.; Veloso Alves Pereira, I.; Willebrords, J.; Crespo Yanguas, S.; et al. Involvement of connexin43 in acetaminophen-induced liver injury. Biochim. Biophys. Acta 2016, 1862, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Eugenín, E.A.; González, H.E.; Sánchez, H.A.; Brañes, M.C.; Sáez, J.C. Inflammatory conditions induce gap junctional communication between rat kupffer cells both in vivo and in vitro. Cell. Immunol. 2007, 247, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Balasubramaniyan, V.; Dhar, D.K.; Warner, A.E.; Vivien Li, W.Y.; Amiri, A.F.; Bright, B.; Mookerjee, R.P.; Davies, N.A.; Becker, D.L.; Jalan, R. Importance of connexin-43 based gap junction in cirrhosis and acute-on-chronic liver failure. J. Hepatol. 2013, 58, 1194–1200. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Guerra, M.; González-Méndez, Y.; de Ganzo, Z.A.; Salido, E.; García-Pagán, J.C.; Abrante, B.; Malagón, A.M.; Bosch, J.; Quintero, E. Role of gap junctions modulating hepatic vascular tone in cirrhosis. Liver Int. 2014, 34, 859–868. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Pitchakarn, P.; Suzuki, S.; Chewonarin, T.; Tang, M.; Takahashi, S.; Naiki-Ito, A.; Sato, S.; Asamoto, M.; Shirai, T. Silencing of connexin 43 suppresses invasion, migration and lung metastasis of rat hepatocellular carcinoma cells. Cancer Sci. 2012, 103, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Crespo Yanguas, S.; Willebrords, J.; Maes, M.; da Silva, T.C.; Veloso Alves Pereira, I.; Cogliati, B.; Zaidan Dagli, M.L.; Vinken, M. Connexins and pannexins in liver damage. EXCLI J. 2016, 15, 177–186. [Google Scholar] [PubMed]
- Fischer, R.; Reinehr, R.; Lu, T.P.; Schönicke, A.; Warskulat, U.; Dienes, H.P.; Häussinger, D. Intercellular communication via gap junctions in activated rat hepatic stellate cells. Gastroenterology 2005, 128, 433–448. [Google Scholar] [CrossRef] [PubMed]
- González, H.E.; Eugenín, E.A.; Garcés, G.; Solís, N.; Pizarro, M.; Accatino, L.; Sáez, J.C. Regulation of hepatic connexins in cholestasis: Possible involvement of kupffer cells and inflammatory mediators. Am. J. Physiol. Gastrointest. Liver Physiol. 2002, 282, G991–G1001. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, B.; Da Silva, T.C.; Aloia, T.P.; Chaible, L.M.; Real-Lima, M.A.; Sanches, D.S.; Matsuzaki, P.; Hernandez-Blazquez, F.J.; Dagli, M.L. Morphological and molecular pathology of ccl4-induced hepatic fibrosis in connexin43-deficient mice. Microsc. Res. Tech. 2011, 74, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Maes, M.; Crespo Yanguas, S.; Vinken, M. Inhibitors of connexin and pannexin channels as potential therapeutics. Pharmacol. Ther. 2017, 180, 144–160. [Google Scholar] [CrossRef] [PubMed]
- Salameh, A.; Dhein, S. Pharmacology of gap junctions. New pharmacological targets for treatment of arrhythmia, seizure and cancer? Biochim. Biophys. Acta 2005, 1719, 36–58. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; De Vuyst, E.; Ponsaerts, R.; Boengler, K.; Palacios-Prado, N.; Wauman, J.; Lai, C.P.; De Bock, M.; Decrock, E.; Bol, M.; et al. Selective inhibition of cx43 hemichannels by gap19 and its impact on myocardial ischemia/reperfusion injury. Basic Res. Cardiol. 2013, 108, 309. [Google Scholar] [CrossRef] [PubMed]
- Abudara, V.; Bechberger, J.; Freitas-Andrade, M.; De Bock, M.; Wang, N.; Bultynck, G.; Naus, C.C.; Leybaert, L.; Giaume, C. The connexin43 mimetic peptide gap19 inhibits hemichannels without altering gap junctional communication in astrocytes. Front. Cell. Neurosci. 2014, 8, 306. [Google Scholar] [CrossRef] [PubMed]
- Vinken, M.; Decrock, E.; Vanhaecke, T.; Leybaert, L.; Rogiers, V. Connexin43 signaling contributes to spontaneous apoptosis in cultures of primary hepatocytes. Toxicol. Sci. 2012, 125, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Reif, S.; Aeed, H.; Shilo, Y.; Reich, R.; Kloog, Y.; Kweon, Y.O.; Bruck, R. Treatment of thioacetamide-induced liver cirrhosis by the ras antagonist, farnesylthiosalicylic acid. J. Hepatol. 2004, 41, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, C.R. Oxidative stress and hepatic stellate cells: A paradoxical relationship. Trends Cell. Mol. Biol. 2012, 7, 1–10. [Google Scholar] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Duffield, J.S.; Forbes, S.J.; Constandinou, C.M.; Clay, S.; Partolina, M.; Vuthoori, S.; Wu, S.; Lang, R.; Iredale, J.P. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J. Clin. Investig. 2005, 115, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Crespo Yanguas, S.; Willebrords, J.; Weemhoff, J.L.; da Silva, T.C.; Decrock, E.; Lebofsky, M.; Pereira, I.V.A.; Leybaert, L.; Farhood, A.; et al. Connexin hemichannel inhibition reduces acetaminophen-induced liver injury in mice. Toxicol. Lett. 2017, 278, 30–37. [Google Scholar] [CrossRef] [PubMed]
- Willebrords, J.; Cogliati, B.; Pereira, I.V.A.; da Silva, T.C.; Crespo Yanguas, S.; Maes, M.; Govoni, V.M.; Lima, A.; Felisbino, D.A.; Decrock, E.; et al. Inhibition of connexin hemichannels alleviates non-alcoholic steatohepatitis in mice. Sci. Rep. 2017, 7, 8268. [Google Scholar] [CrossRef] [PubMed]
- Evans, W.H.; Leybaert, L. Mimetic peptides as blockers of connexin channel-facilitated intercellular communication. Cell. Commun. Adhes. 2007, 14, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Tarzemany, R.; Jiang, G.; Jiang, J.X.; Larjava, H.; Häkkinen, L. Connexin 43 hemichannels regulate the expression of wound healing-associated genes in human gingival fibroblasts. Sci. Rep. 2017, 7, 14157. [Google Scholar] [CrossRef] [PubMed]
- Uyama, N.; Shimahara, Y.; Okuyama, H.; Kawada, N.; Kamo, S.; Ikeda, K.; Yamaoka, Y. Carbenoxolone inhibits DNA synthesis and collagen gene expression in rat hepatic stellate cells in culture. J. Hepatol. 2003, 39, 749–755. [Google Scholar] [CrossRef]
- Lee, S.H.; Zhao, Y.Z.; Park, E.J.; Che, X.H.; Seo, G.S.; Sohn, D.H. 2′,4′,6′-tris(methoxymethoxy) chalcone induces apoptosis by enhancing fas-ligand in activated hepatic stellate cells. Eur. J. Pharmacol. 2011, 658, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Lodder, J.; Denaës, T.; Chobert, M.N.; Wan, J.; El-Benna, J.; Pawlotsky, J.M.; Lotersztajn, S.; Teixeira-Clerc, F. Macrophage autophagy protects against liver fibrosis in mice. Autophagy 2015, 11, 1280–1292. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.S.; Nakamoto, N.; Ebinuma, H.; Usui, S.; Saeki, K.; Matsumoto, A.; Mikami, Y.; Sugiyama, K.; Tomita, K.; Kanai, T.; et al. C-c motif chemokine receptor 9 positive macrophages activate hepatic stellate cells and promote liver fibrosis in mice. Hepatology 2013, 58, 337–350. [Google Scholar] [CrossRef] [PubMed]
- Michalski, K.; Kawate, T. Carbenoxolone inhibits pannexin1 channels through interactions in the first extracellular loop. J. Gen. Physiol. 2016, 147, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Preisser, L.; Miot, C.; Le Guillou-Guillemette, H.; Beaumont, E.; Foucher, E.D.; Garo, E.; Blanchard, S.; Frémaux, I.; Croué, A.; Fouchard, I.; et al. Il-34 and macrophage colony-stimulating factor are overexpressed in hepatitis c virus fibrosis and induce profibrotic macrophages that promote collagen synthesis by hepatic stellate cells. Hepatology 2014, 60, 1879–1890. [Google Scholar] [CrossRef] [PubMed]
- Dunning, S.; Ur Rehman, A.; Tiebosch, M.H.; Hannivoort, R.A.; Haijer, F.W.; Woudenberg, J.; van den Heuvel, F.A.; Buist-Homan, M.; Faber, K.N.; Moshage, H. Glutathione and antioxidant enzymes serve complementary roles in protecting activated hepatic stellate cells against hydrogen peroxide-induced cell death. Biochim. Biophys. Acta 2013, 1832, 2027–2034. [Google Scholar] [CrossRef] [PubMed]
- Kar, R.; Riquelme, M.A.; Werner, S.; Jiang, J.X. Connexin 43 channels protect osteocytes against oxidative stress-induced cell death. J. Bone Miner. Res. 2013, 28, 1611–1621. [Google Scholar] [CrossRef] [PubMed]
- Guerra, R.R.; Trotta, M.R.; Parra, O.M.; Avanzo, J.L.; Bateman, A.; Aloia, T.P.; Dagli, M.L.; Hernandez-Blazquez, F.J. Modulation of extracellular matrix by nutritional hepatotrophic factors in thioacetamide-induced liver cirrhosis in the rat. Braz. J. Med. Biol. Res. 2009, 42, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Cogliati, B.; Crespo Yanguas, S.; Da Silva, T.C.; Aloia, T.P.; Nogueira, M.S.; Real-Lima, M.A.; Chaible, L.M.; Sanches, D.S.; Willebrords, J.; Maes, M.; et al. Connexin32 deficiency exacerbates carbon tetrachloride-induced hepatocellular injury and liver fibrosis in mice. Toxicol. Mech. Methods 2016, 26, 362–370. [Google Scholar] [CrossRef] [PubMed]








© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crespo Yanguas, S.; Da Silva, T.C.; Pereira, I.V.A.; Willebrords, J.; Maes, M.; Sayuri Nogueira, M.; Alves de Castro, I.; Leclercq, I.; Romualdo, G.R.; Barbisan, L.F.; et al. TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice. Int. J. Mol. Sci. 2018, 19, 817. https://doi.org/10.3390/ijms19030817
Crespo Yanguas S, Da Silva TC, Pereira IVA, Willebrords J, Maes M, Sayuri Nogueira M, Alves de Castro I, Leclercq I, Romualdo GR, Barbisan LF, et al. TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice. International Journal of Molecular Sciences. 2018; 19(3):817. https://doi.org/10.3390/ijms19030817
Chicago/Turabian StyleCrespo Yanguas, Sara, Tereza C. Da Silva, Isabel V. A. Pereira, Joost Willebrords, Michaël Maes, Marina Sayuri Nogueira, Inar Alves de Castro, Isabelle Leclercq, Guilherme R. Romualdo, Luís F. Barbisan, and et al. 2018. "TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice" International Journal of Molecular Sciences 19, no. 3: 817. https://doi.org/10.3390/ijms19030817
APA StyleCrespo Yanguas, S., Da Silva, T. C., Pereira, I. V. A., Willebrords, J., Maes, M., Sayuri Nogueira, M., Alves de Castro, I., Leclercq, I., Romualdo, G. R., Barbisan, L. F., Leybaert, L., Cogliati, B., & Vinken, M. (2018). TAT-Gap19 and Carbenoxolone Alleviate Liver Fibrosis in Mice. International Journal of Molecular Sciences, 19(3), 817. https://doi.org/10.3390/ijms19030817