The Balance of Th17 versus Treg Cells in Autoimmunity


{kind=link}
{kind=link}
Abstract
1. Introduction
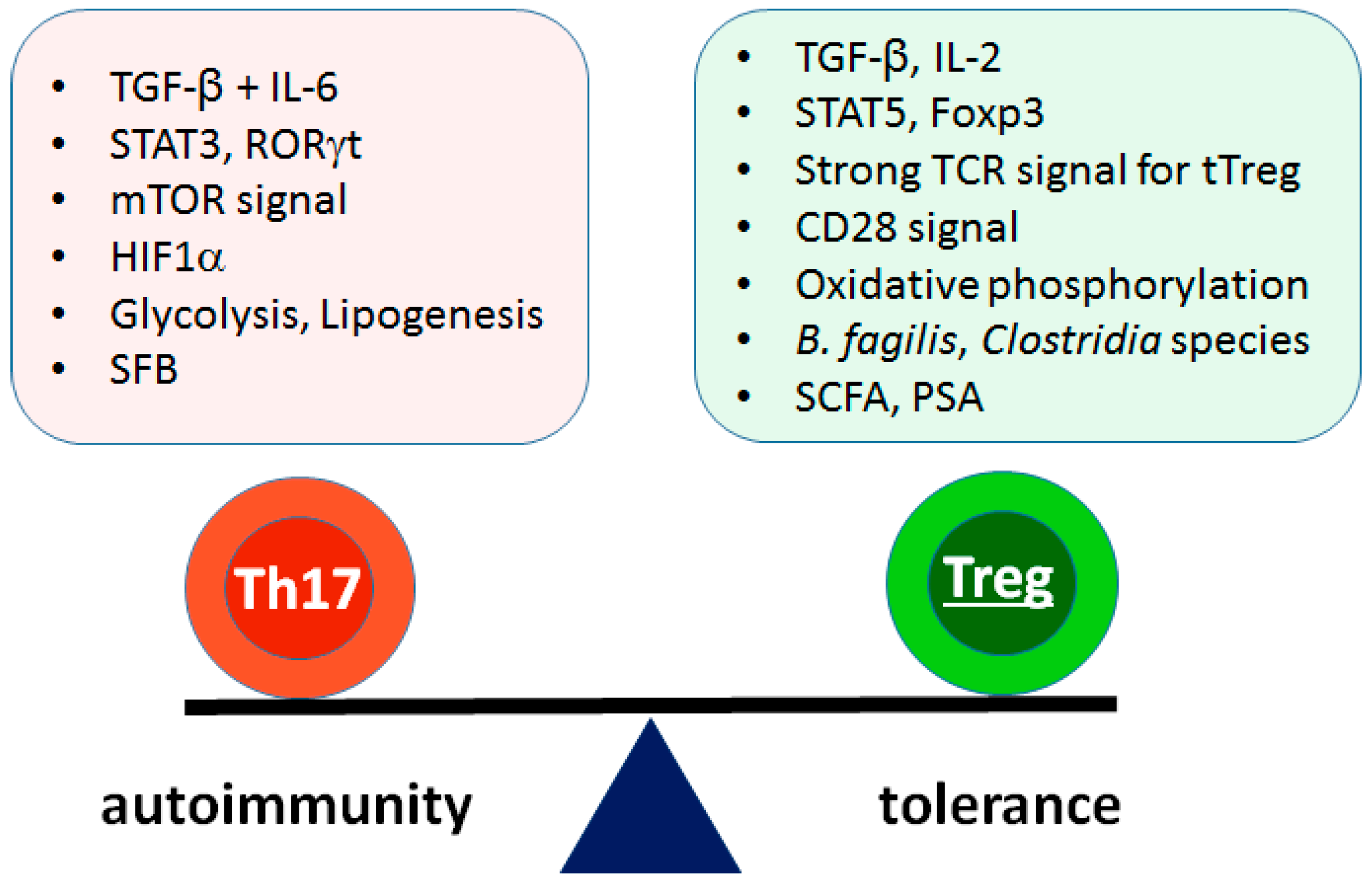
2. The Th17/Treg Balance in Autoimmunity
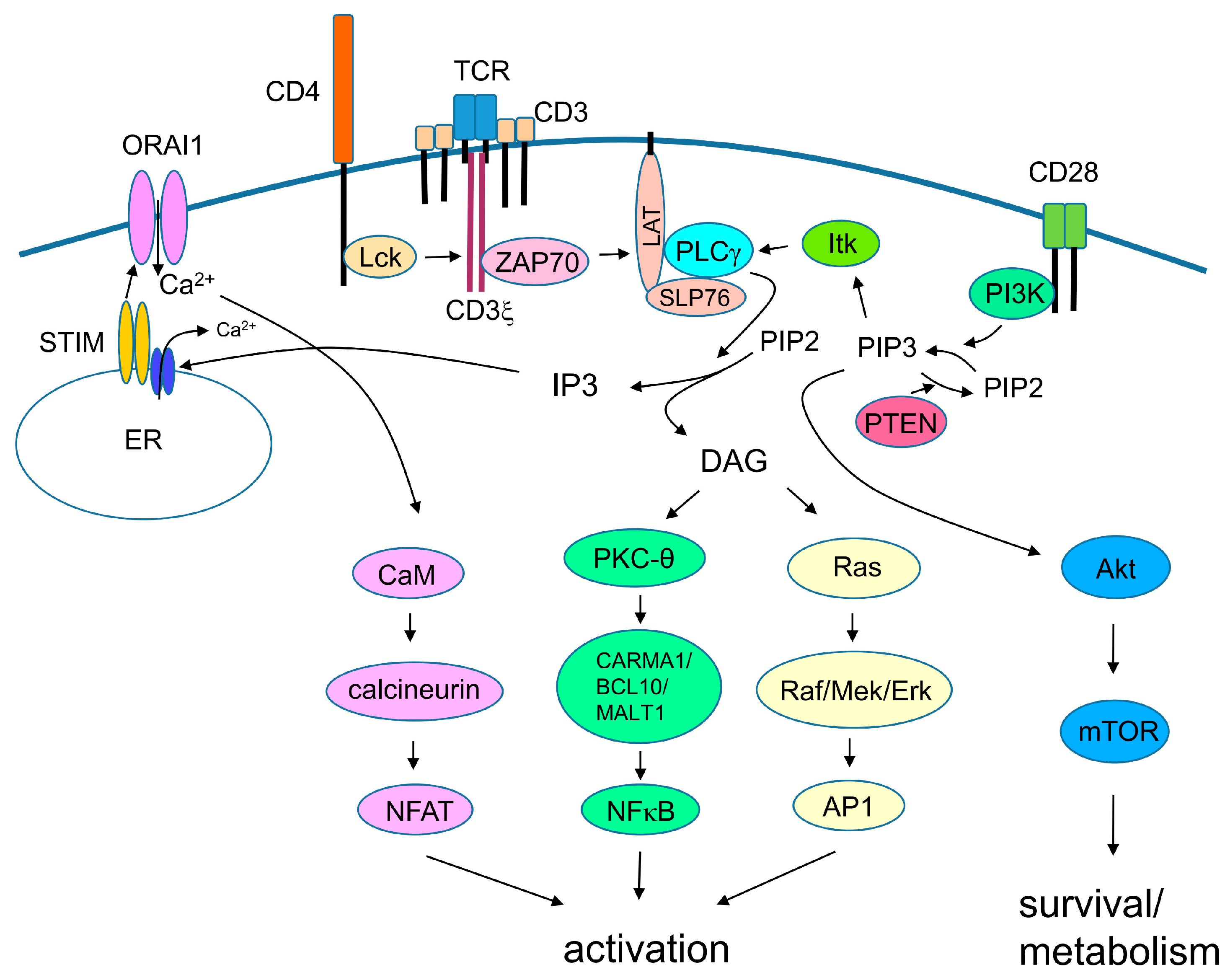
3. TCR Signaling
4. Costimulatory Signals
5. Cytokine Signaling
6. Metabolic Pathways
7. Microbiota
8. Plasticity of Th17 and Treg Cells
9. Autoimmune Diseases Caused by Dysregulation of the Th17/Treg Balance
10. Conclusions and Perspective
Acknowledgments
Conflicts of Interest
References
- Zhu, J.; Paul, W.E. Peripheral CD4+ T-cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev. 2010, 238, 247–262. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Zhou, L.; Littman, D.R. Transcriptional regulation of Th17 cell differentiation. Semin. Immunol. 2007, 19, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFβ in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-β induces development of the Th17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Littman, D.R.; Rudensky, A.Y. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010, 140, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORγt directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, M.; Nakatsukasa, H.; Okada, M.; Lu, Q.; Yoshimura, A. Induced regulatory T cells: Their development, stability, and applications. Trends Immunol. 2016, 37, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Carrier, Y.; Yuan, J.; Kuchroo, V.K.; Weiner, H.L. Th3 cells in peripheral tolerance. I. Induction of Foxp3-positive regulatory T cells by Th3 cells derived from TGFβ T cell-transgenic mice. J. Immunol. 2007, 178, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Roncarolo, M.G.; Gregori, S.; Bacchetta, R.; Battaglia, M. Tr1 cells and the counter-regulation of immunity: Natural mechanisms and therapeutic applications. Curr. Top. Microbiol. Immunol. 2014, 380, 39–68. [Google Scholar] [PubMed]
- Nocentini, G.; Cari, L.; Migliorati, G.; Riccardi, C. The role of GITR single-positive cells in immune homeostasis. Immun. Inflamm. Dis. 2017, 5, 4–6. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Rudensky, A.Y. T cell receptor signalling in the control of regulatory T cell differentiation and function. Nat. Rev. Immunol. 2016, 16, 220–233. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, S.; Maeda, S.; Hashimoto, M.; Fujimori, C.; Ito, Y.; Teradaira, S.; Hirota, K.; Yoshitomi, H.; Katakai, T.; Shimizu, A.; et al. Graded attenuation of TCR signaling elicits distinct autoimmune diseases by altering thymic T cell selection and regulatory T cell function. J. Immunol. 2010, 185, 2295–2305. [Google Scholar] [CrossRef] [PubMed]
- Siggs, O.M.; Miosge, L.A.; Yates, A.L.; Kucharska, E.M.; Sheahan, D.; Brdicka, T.; Weiss, A.; Liston, A.; Goodnow, C.C. Opposing functions of the T cell receptor kinase ZAP-70 in immunity and tolerance differentially titrate in response to nucleotide substitutions. Immunity 2007, 27, 912–926. [Google Scholar] [CrossRef] [PubMed]
- Hsu, L.Y.; Tan, Y.X.; Xiao, Z.; Malissen, M.; Weiss, A. A hypomorphic allele of ZAP-70 reveals a distinct thymic threshold for autoimmune disease versus autoimmune reactivity. J. Exp. Med. 2009, 206, 2527–2541. [Google Scholar] [CrossRef] [PubMed]
- Fu, G.; Chen, Y.; Yu, M.; Podd, A.; Schuman, J.; He, Y.; Di, L.; Yassai, M.; Haribhai, D.; North, P.E.; et al. Phospholipase Cγ1 is essential for T cell development, activation, and tolerance. J. Exp. Med. 2010, 207, 309–318. [Google Scholar] [CrossRef] [PubMed]
- Oh-Hora, M.; Yamashita, M.; Hogan, P.G.; Sharma, S.; Lamperti, E.; Chung, W.; Prakriya, M.; Feske, S.; Rao, A. Dual functions for the endoplasmic reticulum calcium sensors STIM1 and STIM2 in T cell activation and tolerance. Nat. Immunol. 2008, 9, 432–443. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.M.; Zou, T.; Joshi, R.P.; Leichner, T.M.; Pimentel, M.A.; Sommers, C.L.; Kambayashi, T. Diacylglycerol kinase ζ limits the generation of natural regulatory T cells. Sci. Signal. 2013, 6, ra101. [Google Scholar] [CrossRef] [PubMed]
- Joshi, R.P.; Schmidt, A.M.; Das, J.; Pytel, D.; Riese, M.J.; Lester, M.; Diehl, J.A.; Behrens, E.M.; Kambayashi, T.; Koretzky, G.A. The zeta isoform of diacylglycerol kinase plays a predominant role in regulatory T cell development and TCR-mediated ras signaling. Sci. Signal. 2013, 6, ra102. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Priatel, J.J.; Chow, M.T.; Teh, H.S. Preferential development of CD4 and CD8 T regulatory cells in RasGRP1-deficient mice. J. Immunol. 2008, 180, 5973–5982. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, J.E.; Costello, P.S.; Nicolas, R.H.; Robinson, N.J.; Stamp, G.; Powrie, F.; Treisman, R. Raf signaling but not the ERK effector SAP-1 is required for regulatory T cell development. J. Immunol. 2007, 179, 6836–6844. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Supprian, M.; Tian, J.; Grant, E.P.; Pasparakis, M.; Maehr, R.; Ovaa, H.; Ploegh, H.L.; Coyle, A.J.; Rajewsky, K. Differential dependence of CD4+CD25+ regulatory and natural killer-like T cells on signals leading to NF-κB activation. Proc. Natl. Acad. Sci. USA 2004, 101, 4566–4571. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Manicassamy, S.; Vasu, C.; Kumar, A.; Shang, W.; Sun, Z. Differential requirement of PKC-θ in the development and function of natural regulatory T cells. Mol. Immunol. 2008, 46, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Medoff, B.D.; Sandall, B.P.; Landry, A.; Nagahama, K.; Mizoguchi, A.; Luster, A.D.; Xavier, R.J. Differential requirement for CARMA1 in agonist-selected T-cell development. Eur. J. Immunol. 2009, 39, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Barnes, M.J.; Krebs, P.; Harris, N.; Eidenschenk, C.; Gonzalez-Quintial, R.; Arnold, C.N.; Crozat, K.; Sovath, S.; Moresco, E.M.; Theofilopoulos, A.N.; et al. Commitment to the regulatory T cell lineage requires CARMA1 in the thymus but not in the periphery. PLoS Biol. 2009, 7, e51. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.; Song, K.D.; Lesourne, R.; Lee, J.; Pinkhasov, J.; Li, L.; El-Khoury, D.; Love, P.E. Reduced TCR signaling potential impairs negative selection but does not result in autoimmune disease. J. Exp. Med. 2012, 209, 1781–1795. [Google Scholar] [CrossRef] [PubMed]
- Long, M.; Park, S.G.; Strickland, I.; Hayden, M.S.; Ghosh, S. Nuclear factor-κB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 2009, 31, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of Foxp3+ regulatory T cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef] [PubMed]
- Vaeth, M.; Schliesser, U.; Muller, G.; Reissig, S.; Satoh, K.; Tuettenberg, A.; Jonuleit, H.; Waisman, A.; Muller, M.R.; Serfling, E.; et al. Dependence on nuclear factor of activated T-cells (NFAT) levels discriminates conventional T cells from Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA 2012, 109, 16258–16263. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Rodriguez, J.; Wohlfert, E.A.; Handon, R.; Meylan, F.; Wu, J.Z.; Anderson, S.M.; Kirby, M.R.; Belkaid, Y.; Schwartzberg, P.L. Itk-mediated integration of T cell receptor and cytokine signaling regulates the balance between Th17 and regulatory T cells. J. Exp. Med. 2014, 211, 529–543. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Jang, S.W.; Lee, W.; Kim, K.; Sohn, H.; Hwang, S.S.; Lee, G.R. PTEN drives Th17 cell differentiation by preventing IL-2 production. J. Exp. Med. 2017, 214, 3381–3398. [Google Scholar] [CrossRef] [PubMed]
- Chuck, M.I.; Zhu, M.; Shen, S.; Zhang, W. The role of the LAT-PLC-γ1 interaction in T regulatory cell function. J. Immunol. 2010, 184, 2476–2486. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Xiao, Y.; Hu, H.; Jin, J.; Yu, J.; Zhou, X.; Wu, X.; Johnson, H.M.; Akira, S.; Pasparakis, M.; et al. Ubc13 maintains the suppressive function of regulatory T cells and prevents their conversion into effector-like T cells. Nat. Immunol. 2012, 13, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Liao, W.; Luo, C.T.; Yin, N.; Huse, M.; Kim, M.V.; Peng, M.; Chan, P.; Ma, Q.; Mo, Y.; et al. Novel Foxo1-dependent transcriptional programs control Treg cell function. Nature 2012, 491, 554–559. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Jin, H.S.; Lopez, J.; Lee, J.; Liao, L.; Elly, C.; Liu, Y.C. SHARPIN controls regulatory T cells by negatively modulating the T cell antigen receptor complex. Nat. Immunol. 2016, 17, 286–296. [Google Scholar] [CrossRef] [PubMed]
- Jang, S.W.; Hwang, S.S.; Kim, H.S.; Lee, K.O.; Kim, M.K.; Lee, W.; Kim, K.; Lee, G.R. Casein kinase 2 is a critical determinant of the balance of Th17 and Treg cell differentiation. Exp. Mol. Med. 2017, 49, e375. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.A.; Yang, W.; Yan, Z.; Liu, Y.; Rowse, A.L.; Weinmann, A.S.; Qin, H.; Benveniste, E.N. Protein kinase CK2 controls the fate between Th17 cell and regulatory T cell differentiation. J. Immunol. 2017, 198, 4244–4254. [Google Scholar] [CrossRef] [PubMed]
- Ulges, A.; Witsch, E.J.; Pramanik, G.; Klein, M.; Birkner, K.; Buhler, U.; Wasser, B.; Luessi, F.; Stergiou, N.; Dietzen, S.; et al. Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc. Natl. Acad. Sci. USA 2016, 113, 10145–10150. [Google Scholar] [CrossRef] [PubMed]
- Apostolidis, S.A.; Rodriguez-Rodriguez, N.; Suarez-Fueyo, A.; Dioufa, N.; Ozcan, E.; Crispin, J.C.; Tsokos, M.G.; Tsokos, G.C. Phosphatase PP2A is requisite for the function of regulatory T cells. Nat. Immunol. 2016, 17, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Esensten, J.H.; Helou, Y.A.; Chopra, G.; Weiss, A.; Bluestone, J.A. CD28 costimulation: From mechanism to therapy. Immunity 2016, 44, 973–988. [Google Scholar] [CrossRef] [PubMed]
- Salomon, B.; Lenschow, D.J.; Rhee, L.; Ashourian, N.; Singh, B.; Sharpe, A.; Bluestone, J.A. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000, 12, 431–440. [Google Scholar] [CrossRef]
- Gogishvili, T.; Luhder, F.; Goebbels, S.; Beer-Hammer, S.; Pfeffer, K.; Hunig, T. Cell-intrinsic and -extrinsic control of Treg-cell homeostasis and function revealed by induced CD28 deletion. Eur. J. Immunol. 2013, 43, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Huynh, A.; Whitcher, G.; Chang, J.; Maltzman, J.S.; Turka, L.A. An obligate cell-intrinsic function for CD28 in Tregs. J. Clin. Investig. 2013, 123, 580–593. [Google Scholar] [CrossRef] [PubMed]
- De Kouchkovsky, D.; Esensten, J.H.; Rosenthal, W.L.; Morar, M.M.; Bluestone, J.A.; Jeker, L.T. MicroRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 1594–1605. [Google Scholar] [CrossRef] [PubMed]
- Tai, X.; Cowan, M.; Feigenbaum, L.; Singer, A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat. Immunol. 2005, 6, 152–162. [Google Scholar] [CrossRef] [PubMed]
- Mahmud, S.A.; Manlove, L.S.; Schmitz, H.M.; Xing, Y.; Wang, Y.; Owen, D.L.; Schenkel, J.M.; Boomer, J.S.; Green, J.M.; Yagita, H.; et al. Costimulation via the tumor-necrosis factor receptor superfamily couples TCR signal strength to the thymic differentiation of regulatory T cells. Nat. Immunol. 2014, 15, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; Iclozan, C.; Suh, W.K.; Anasetti, C.; Yu, X.Z. CD28 controls differentiation of regulatory T cells from naive CD4 T cells. J. Immunol. 2008, 181, 2285–2291. [Google Scholar] [CrossRef] [PubMed]
- Benson, M.J.; Pino-Lagos, K.; Rosemblatt, M.; Noelle, R.J. All-trans retinoic acid mediates enhanced T reg cell growth, differentiation, and gut homing in the face of high levels of co-stimulation. J. Exp. Med. 2007, 204, 1765–1774. [Google Scholar] [CrossRef] [PubMed]
- Semple, K.; Nguyen, A.; Yu, Y.; Wang, H.; Anasetti, C.; Yu, X.Z. Strong CD28 costimulation suppresses induction of regulatory T cells from naive precursors through Lck signaling. Blood 2011, 117, 3096–3103. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Vignali, D.A. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef] [PubMed]
- Baumeister, S.H.; Freeman, G.J.; Dranoff, G.; Sharpe, A.H. Coinhibitory Pathways in Immunotherapy for Cancer. Annu. Rev. Immunol. 2016, 34, 539–573. [Google Scholar] [CrossRef] [PubMed]
- Wykes, M.N.; Lewin, S.R. Immune checkpoint blockade in infectious diseases. Nat. Rev. Immunol. 2018, 18, 91. [Google Scholar] [CrossRef] [PubMed]
- Manel, N.; Unutmaz, D.; Littman, D.R. The differentiation of human Th-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat. Immunol. 2008, 9, 641–649. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Panopoulos, A.D.; Nurieva, R.; Chang, S.H.; Wang, D.; Watowich, S.S.; Dong, C. STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J. Biol. Chem. 2007, 282, 9358–9363. [Google Scholar] [CrossRef] [PubMed]
- Durant, L.; Watford, W.T.; Ramos, H.L.; Laurence, A.; Vahedi, G.; Wei, L.; Takahashi, H.; Sun, H.W.; Kanno, Y.; Powrie, F.; et al. Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 2010, 32, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs Th-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef] [PubMed]
- Volpe, E.; Servant, N.; Zollinger, R.; Bogiatzi, S.I.; Hupe, P.; Barillot, E.; Soumelis, V. A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat. Immunol. 2008, 9, 650–657. [Google Scholar] [CrossRef] [PubMed]
- McGeachy, M.J.; Bak-Jensen, K.S.; Chen, Y.; Tato, C.M.; Blumenschein, W.; McClanahan, T.; Cua, D.J. TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain Th-17 cell-mediated pathology. Nat. Immunol. 2007, 8, 1390–1397. [Google Scholar] [CrossRef] [PubMed]
- Korn, T.; Bettelli, E.; Gao, W.; Awasthi, A.; Jager, A.; Strom, T.B.; Oukka, M.; Kuchroo, V.K. IL-21 initiates an alternative pathway to induce proinflammatory Th17 cells. Nature 2007, 448, 484–487. [Google Scholar] [CrossRef] [PubMed]
- Nurieva, R.; Yang, X.O.; Martinez, G.; Zhang, Y.; Panopoulos, A.D.; Ma, L.; Schluns, K.; Tian, Q.; Watowich, S.S.; Jetten, A.M.; et al. Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 2007, 448, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Moustakas, A.; Souchelnytskyi, S.; Heldin, C.H. Smad regulation in TGF-β signal transduction. J. Cell Sci. 2001, 114 Pt 24, 4359–4369. [Google Scholar] [PubMed]
- Almeida, A.R.; Legrand, N.; Papiernik, M.; Freitas, A.A. Homeostasis of peripheral CD4+ T cells: IL-2R alpha and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J. Immunol. 2002, 169, 4850–4860. [Google Scholar] [CrossRef] [PubMed]
- Malek, T.R.; Yu, A.; Vincek, V.; Scibelli, P.; Kong, L. CD4 regulatory T cells prevent lethal autoimmunity in IL-2Rβ-deficient mice. Implications for the nonredundant function of IL-2. Immunity 2002, 17, 167–178. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Li, M.O.; Flavell, R.A. TGF-β: A master of all T cell trades. Cell 2008, 134, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Lin, J.X.; Leonard, W.J. Interleukin-2 at the crossroads of effector responses, tolerance, and immunotherapy. Immunity 2013, 38, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Barbi, J.; Pardoll, D.; Pan, F. Metabolic control of the Treg/Th17 axis. Immunol. Rev. 2013, 252, 52–77. [Google Scholar] [CrossRef] [PubMed]
- MacIver, N.J.; Michalek, R.D.; Rathmell, J.C. Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 2013, 31, 259–283. [Google Scholar] [CrossRef] [PubMed]
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Chi, H. Regulation and function of mTOR signalling in T cell fate decisions. Nat. Rev. Immunol. 2012, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Huddleston, S.J.; Fraser, J.M.; Khoruts, A. De novo induction of antigen-specific CD4+CD25+Foxp3+ regulatory T cells in vivo following systemic antigen administration accompanied by blockade of mTOR. J. Leukoc. Biol. 2008, 83, 1230–1239. [Google Scholar] [CrossRef] [PubMed]
- Brandler, W.M.; Antaki, D.; Gujral, M.; Noor, A.; Rosanio, G.; Chapman, T.R.; Barrera, D.J.; Lin, G.N.; Malhotra, D.; Watts, A.C.; et al. Frequency and complexity of de novo structural mutation in autism. Am. J. Hum. Genet. 2016, 98, 667–679. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.D.; Pollizzi, K.N.; Heikamp, E.B.; Horton, M.R. Regulation of immune responses by mTOR. Annu. Rev. Immunol. 2012, 30, 39–68. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.W.; Chung, B.H.; Kim, B.M.; Cho, M.L.; Yang, C.W. The effect of mammalian target of rapamycin inhibition on T helper type 17 and regulatory T cell differentiation in vitro and in vivo in kidney transplant recipients. Immunology 2015, 144, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; Bahre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef] [PubMed]
- Dang, E.V.; Barbi, J.; Yang, H.Y.; Jinasena, D.; Yu, H.; Zheng, Y.; Bordman, Z.; Fu, J.; Kim, Y.; Yen, H.R.; et al. Control of Th17/Treg balance by hypoxia-inducible factor 1. Cell 2011, 146, 772–784. [Google Scholar] [CrossRef] [PubMed]
- Omenetti, S.; Pizarro, T.T. The Treg/Th17 axis: A dynamic balance regulated by the gut microbiome. Front. Immunol. 2015, 6, 639. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Atarashi, K.; Manel, N.; Brodie, E.L.; Shima, T.; Karaoz, U.; Wei, D.; Goldfarb, K.C.; Santee, C.A.; Lynch, S.V.; et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 2009, 139, 485–498. [Google Scholar] [CrossRef] [PubMed]
- Gaboriau-Routhiau, V.; Rakotobe, S.; Lecuyer, E.; Mulder, I.; Lan, A.; Bridonneau, C.; Rochet, V.; Pisi, A.; De Paepe, M.; Brandi, G.; et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity 2009, 31, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.K.; Menezes, J.S.; Umesaki, Y.; Mazmanian, S.K. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2011, 108, 4615–4622. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.J.; Ivanov, I.I.; Darce, J.; Hattori, K.; Shima, T.; Umesaki, Y.; Littman, D.R.; Benoist, C.; Mathis, D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity 2010, 32, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Panea, C.; Nakato, G.; Cebula, A.; Lee, C.; Diez, M.G.; Laufer, T.M.; Ignatowicz, L.; Ivanov, I.I. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity 2014, 40, 594–607. [Google Scholar] [CrossRef] [PubMed]
- Lecuyer, E.; Rakotobe, S.; Lengline-Garnier, H.; Lebreton, C.; Picard, M.; Juste, C.; Fritzen, R.; Eberl, G.; McCoy, K.D.; Macpherson, A.J.; et al. Segmented filamentous bacterium uses secondary and tertiary lymphoid tissues to induce gut IgA and specific T helper 17 cell responses. Immunity 2014, 40, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.I.; Frutos Rde, L.; Manel, N.; Yoshinaga, K.; Rifkin, D.B.; Sartor, R.B.; Finlay, B.B.; Littman, D.R. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 2008, 4, 337–349. [Google Scholar] [CrossRef] [PubMed]
- Shih, V.F.; Cox, J.; Kljavin, N.M.; Dengler, H.S.; Reichelt, M.; Kumar, P.; Rangell, L.; Kolls, J.K.; Diehl, L.; Ouyang, W.; et al. Homeostatic IL-23 receptor signaling limits Th17 response through IL-22-mediated containment of commensal microbiota. Proc. Natl. Acad. Sci. USA 2014, 111, 13942–13947. [Google Scholar] [CrossRef] [PubMed]
- Sano, T.; Huang, W.; Hall, J.A.; Yang, Y.; Chen, A.; Gavzy, S.J.; Lee, J.Y.; Ziel, J.W.; Miraldi, E.R.; Domingos, A.I.; et al. An IL-23R/IL-22 circuit regulates epithelial serum amyloid a to promote local effector Th17 responses. Cell 2015, 163, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Mazmanian, S.K.; Round, J.L.; Kasper, D.L. A microbial symbiosis factor prevents intestinal inflammatory disease. Nature 2008, 453, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Round, J.L.; Mazmanian, S.K. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. Proc. Natl. Acad. Sci. USA 2010, 107, 12204–12209. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.; Campbell, J.I.; King, T.P.; Grant, G.; Jansson, E.A.; Coutts, A.G.; Pettersson, S.; Conway, S. Commensal anaerobic gut bacteria attenuate inflammation by regulating nuclear-cytoplasmic shuttling of PPAR-γ and RelA. Nat. Immunol. 2004, 5, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Shima, T.; Imaoka, A.; Kuwahara, T.; Momose, Y.; Cheng, G.; Yamasaki, S.; Saito, T.; Ohba, Y.; et al. Induction of colonic regulatory T cells by indigenous Clostridium species. Science 2011, 331, 337–341. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Geuking, M.B.; Cahenzli, J.; Lawson, M.A.; Ng, D.C.; Slack, E.; Hapfelmeier, S.; McCoy, K.D.; Macpherson, A.J. Intestinal bacterial colonization induces mutualistic regulatory T cell responses. Immunity 2011, 34, 794–806. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.M.; Howitt, M.R.; Panikov, N.; Michaud, M.; Gallini, C.A.; Bohlooly, Y.M.; Glickman, J.N.; Garrett, W.S. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 2013, 341, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.V.; Xiang, W.V.; Kwak, C.; Yang, Y.; Lin, X.W.; Ota, M.; Sarpel, U.; Rifkin, D.B.; Xu, R.; Littman, D.R. GPR15-mediated homing controls immune homeostasis in the large intestine mucosa. Science 2013, 340, 1456–1459. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [PubMed]
- Michalak, A.; Mosinska, P.; Fichna, J. Common links between metabolic syndrome and inflammatory bowel disease: Current overview and future perspectives. Pharmacol. Rep. PR 2016, 68, 837–846. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.L.; Elson, C.O.; Hatton, R.D.; Weaver, C.T. Reciprocal interactions of the intestinal microbiota and immune system. Nature 2012, 489, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Zhou, L.; Chong, M.M.; Littman, D.R. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, S.; Vignali, D.A.; Rudensky, A.Y.; Niec, R.E.; Waldmann, H. The plasticity and stability of regulatory T cells. Nat. Rev. Immunol. 2013, 13, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Bailey-Bucktrout, S.L.; Jeker, L.T.; Penaranda, C.; Martinez-Llordella, M.; Ashby, M.; Nakayama, M.; Rosenthal, W.; Bluestone, J.A. Instability of the transcription factor Foxp3 leads to the generation of pathogenic memory T cells in vivo. Nat. Immunol. 2009, 10, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-hora, M.; Kodama, T.; Tanaka, S.; Bluestone, J.A.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2014, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Rubtsov, Y.P.; Niec, R.E.; Josefowicz, S.; Li, L.; Darce, J.; Mathis, D.; Benoist, C.; Rudensky, A.Y. Stability of the regulatory T cell lineage in vivo. Science 2010, 329, 1667–1671. [Google Scholar] [CrossRef] [PubMed]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; de Zoete, M.R.; Licona-Limon, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Huehn, J.; Beyer, M. Epigenetic and transcriptional control of Foxp3+ regulatory T cells. Semin. Immunol. 2015, 27, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Li, B. The functional stability of FOXP3 and RORγt in Treg and Th17 and their therapeutic applications. Adv. Protein Chem. Struct. Biol. 2017, 107, 155–189. [Google Scholar] [PubMed]
- Geng, J.; Yu, S.; Zhao, H.; Sun, X.; Li, X.; Wang, P.; Xiong, X.; Hong, L.; Xie, C.; Gao, J.; et al. The transcriptional coactivator TAZ regulates reciprocal differentiation of TH17 cells and Treg cells. Nat. Immunol. 2017, 18, 800–812. [Google Scholar] [CrossRef] [PubMed]
- Roychoudhuri, R.; Hirahara, K.; Mousavi, K.; Clever, D.; Klebanoff, C.A.; Bonelli, M.; Sciume, G.; Zare, H.; Vahedi, G.; Dema, B.; et al. BACH2 represses effector programs to stabilize Treg-mediated immune homeostasis. Nature 2013, 498, 506–510. [Google Scholar] [CrossRef] [PubMed]
- Hwang, S.S.; Jang, S.W.; Kim, M.K.; Kim, L.K.; Kim, B.S.; Kim, H.S.; Kim, K.; Lee, W.; Flavell, R.A.; Lee, G.R. YY1 inhibits differentiation and function of regulatory T cells by blocking Foxp3 expression and activity. Nat. Commun. 2016, 7, 10789. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.; Kim, H.S.; Hwang, S.S.; Lee, G.R. The transcription factor Batf3 inhibits the differentiation of regulatory T cells in the periphery. Exp. Mol. Med. 2017, 49, e393. [Google Scholar] [CrossRef] [PubMed]
- Bettelli, E.; Korn, T.; Oukka, M.; Kuchroo, V.K. Induction and effector functions of Th17 cells. Nature 2008, 453, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Durand, J.M.; Buchs, N.; Fossiez, F.; Page, G.; Frappart, L.; Miossec, P. Human interleukin-17: A T cell-derived proinflammatory cytokine produced by the rheumatoid synovium. Arthritis Rheum. 1999, 42, 963–970. [Google Scholar] [CrossRef]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef] [PubMed]
- Noack, M.; Miossec, P. Th17 and regulatory T cell balance in autoimmune and inflammatory diseases. Autoimmun. Rev. 2014, 13, 668–677. [Google Scholar] [CrossRef] [PubMed]
- Fasching, P.; Stradner, M.; Graninger, W.; Dejaco, C.; Fessler, J. Therapeutic potential of targeting the Th17/Treg axis in autoimmune disorders. Molecules 2017, 22, 134. [Google Scholar] [CrossRef] [PubMed]
- Samson, M.; Audia, S.; Janikashvili, N.; Ciudad, M.; Trad, M.; Fraszczak, J.; Ornetti, P.; Maillefert, J.F.; Miossec, P.; Bonnotte, B. Brief report: Inhibition of interleukin-6 function corrects Th17/Treg cell imbalance in patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 2499–2503. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Hashizume, M.; Kaneko, Y.; Yoshimoto, K.; Nishina, N.; Takeuchi, T. Peripheral blood CD4+CD25+CD127low regulatory T cells are significantly increased by tocilizumab treatment in patients with rheumatoid arthritis: Increase in regulatory T cells correlates with clinical response. Arthritis Res. Ther. 2015, 17, 10. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Wu, Z.; Li, L.; Liu, X. Interleukin-6 IL-6 receptor antagonist protects against rheumatoid arthritis. Med. Sci. Monit. Int. Med. J. Exp. Clin. Res. 2016, 22, 2113–2118. [Google Scholar] [CrossRef]
- Lee, J.; Baek, S.; Lee, D.G.; Park, M.K.; Cho, M.L.; Park, S.H.; Kwok, S.K. Digoxin ameliorates autoimmune arthritis via suppression of Th17 differentiation. Int. Immunopharmacol. 2015, 26, 103–111. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, G.R. The Balance of Th17 versus Treg Cells in Autoimmunity. Int. J. Mol. Sci. 2018, 19, 730. https://doi.org/10.3390/ijms19030730
Lee GR. The Balance of Th17 versus Treg Cells in Autoimmunity. International Journal of Molecular Sciences. 2018; 19(3):730. https://doi.org/10.3390/ijms19030730
Chicago/Turabian StyleLee, Gap Ryol. 2018. "The Balance of Th17 versus Treg Cells in Autoimmunity" International Journal of Molecular Sciences 19, no. 3: 730. https://doi.org/10.3390/ijms19030730
APA StyleLee, G. R. (2018). The Balance of Th17 versus Treg Cells in Autoimmunity. International Journal of Molecular Sciences, 19(3), 730. https://doi.org/10.3390/ijms19030730