Altered Expression of Ganglioside Metabolizing Enzymes Results in GM3 Ganglioside Accumulation in Cerebellar Cells of a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis


 ,
, 

Abstract
1. Introduction
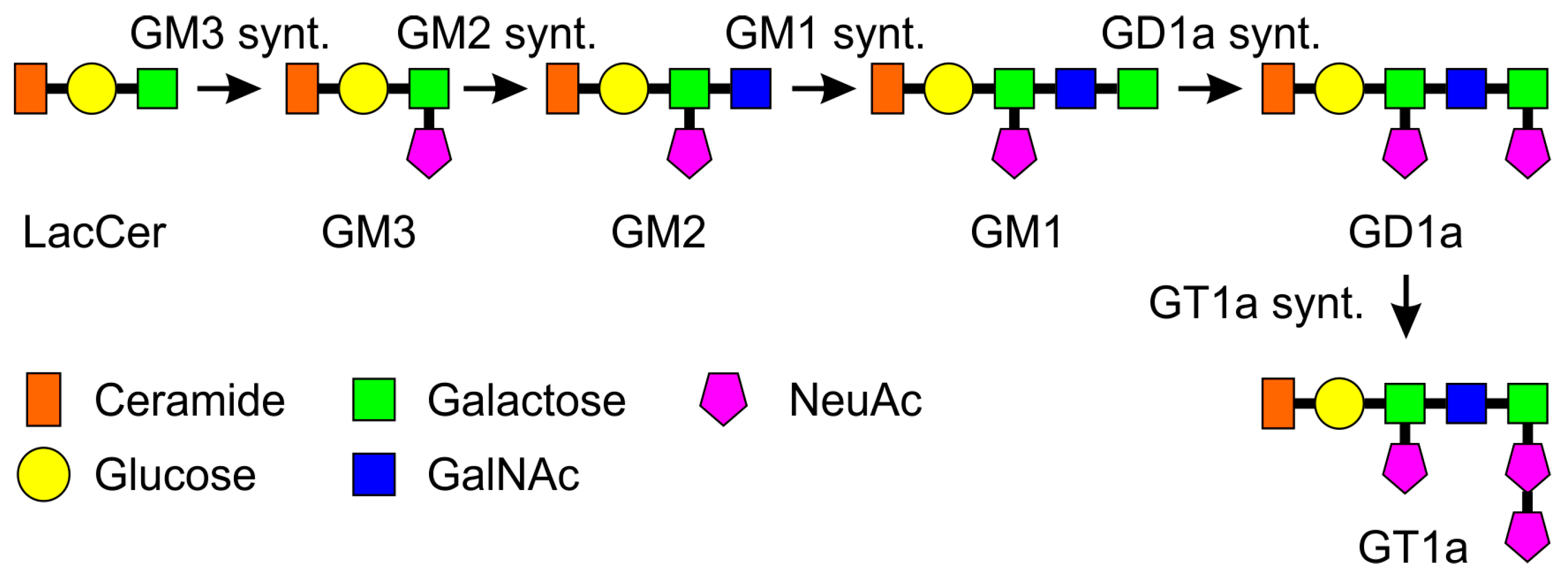
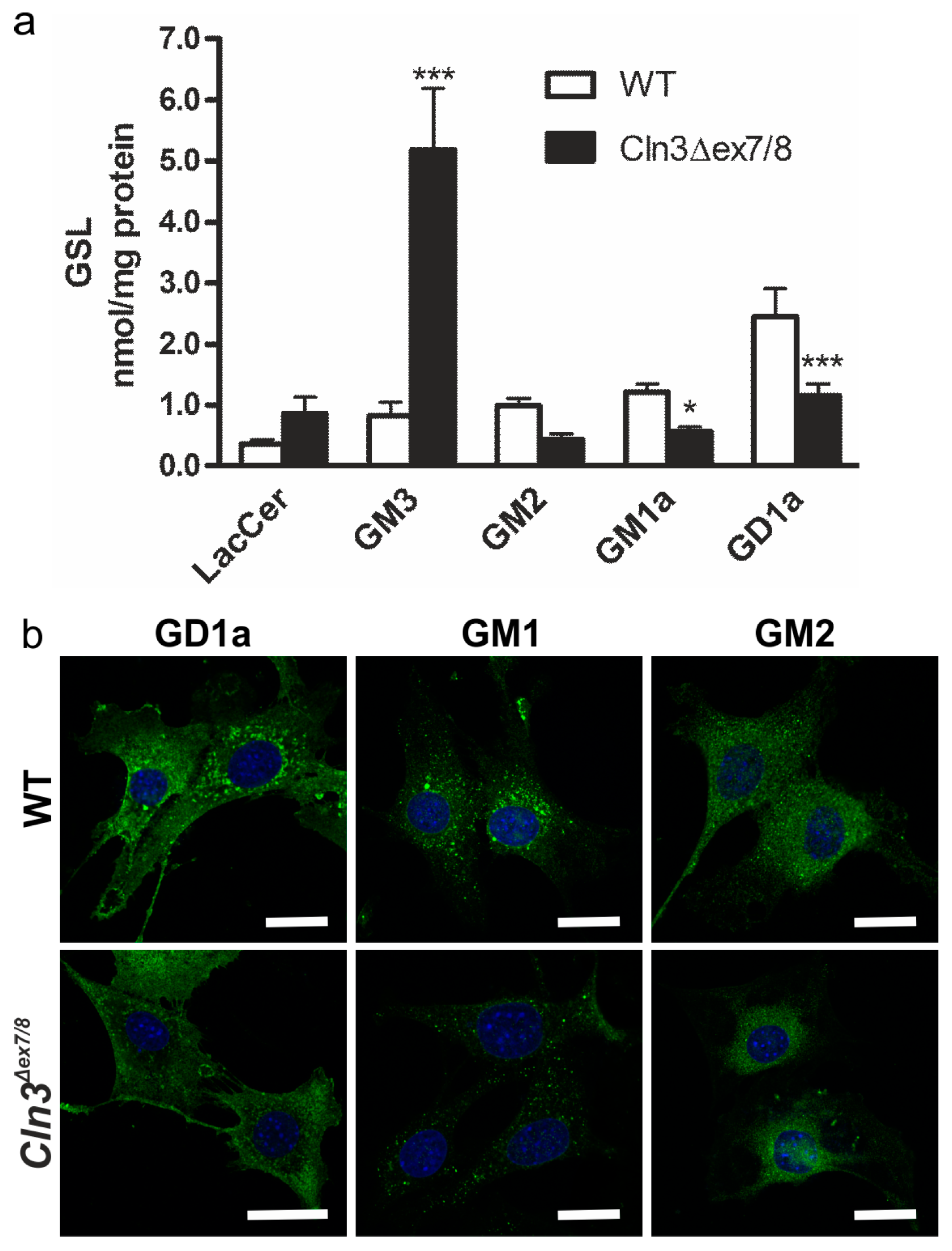
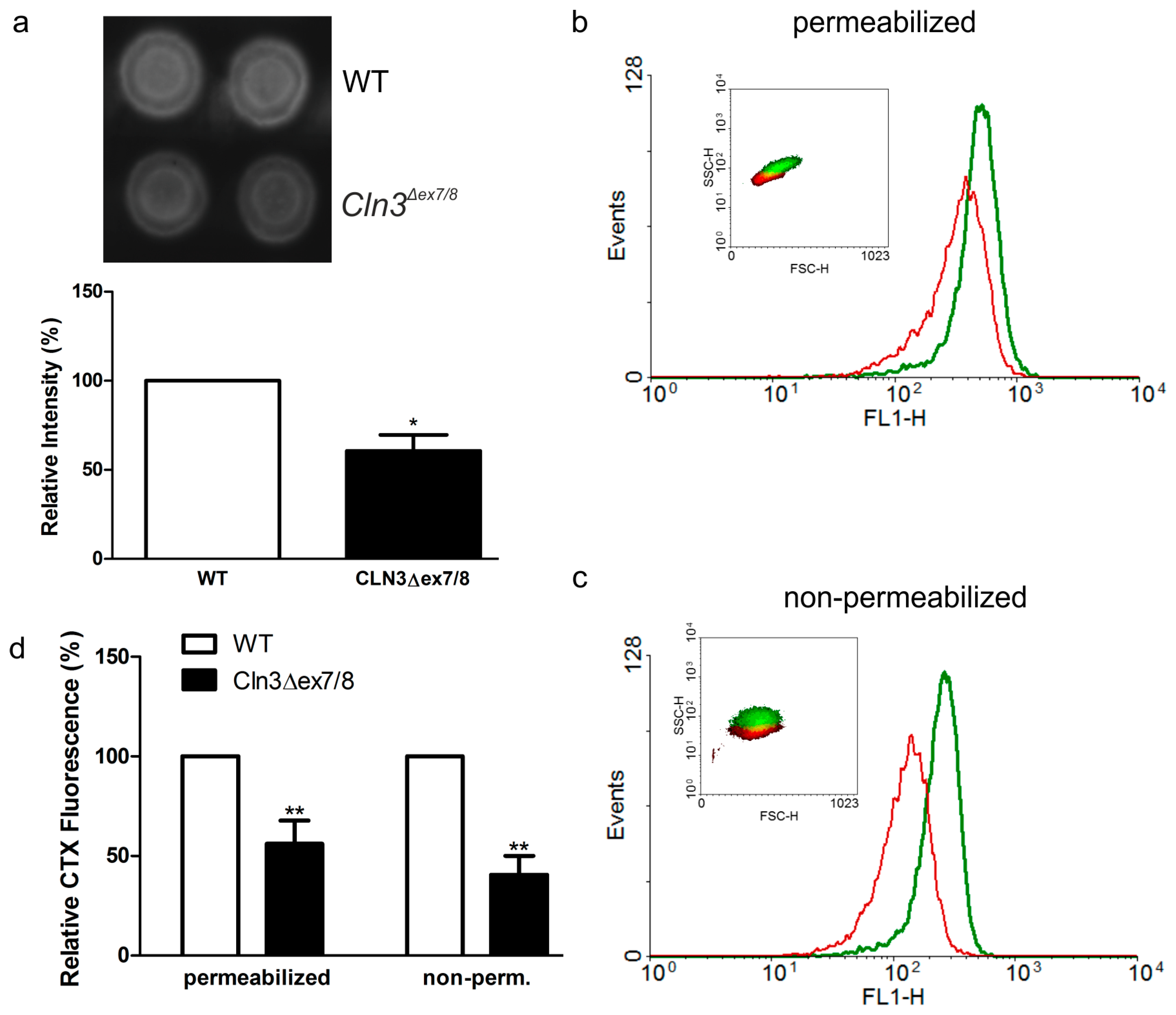
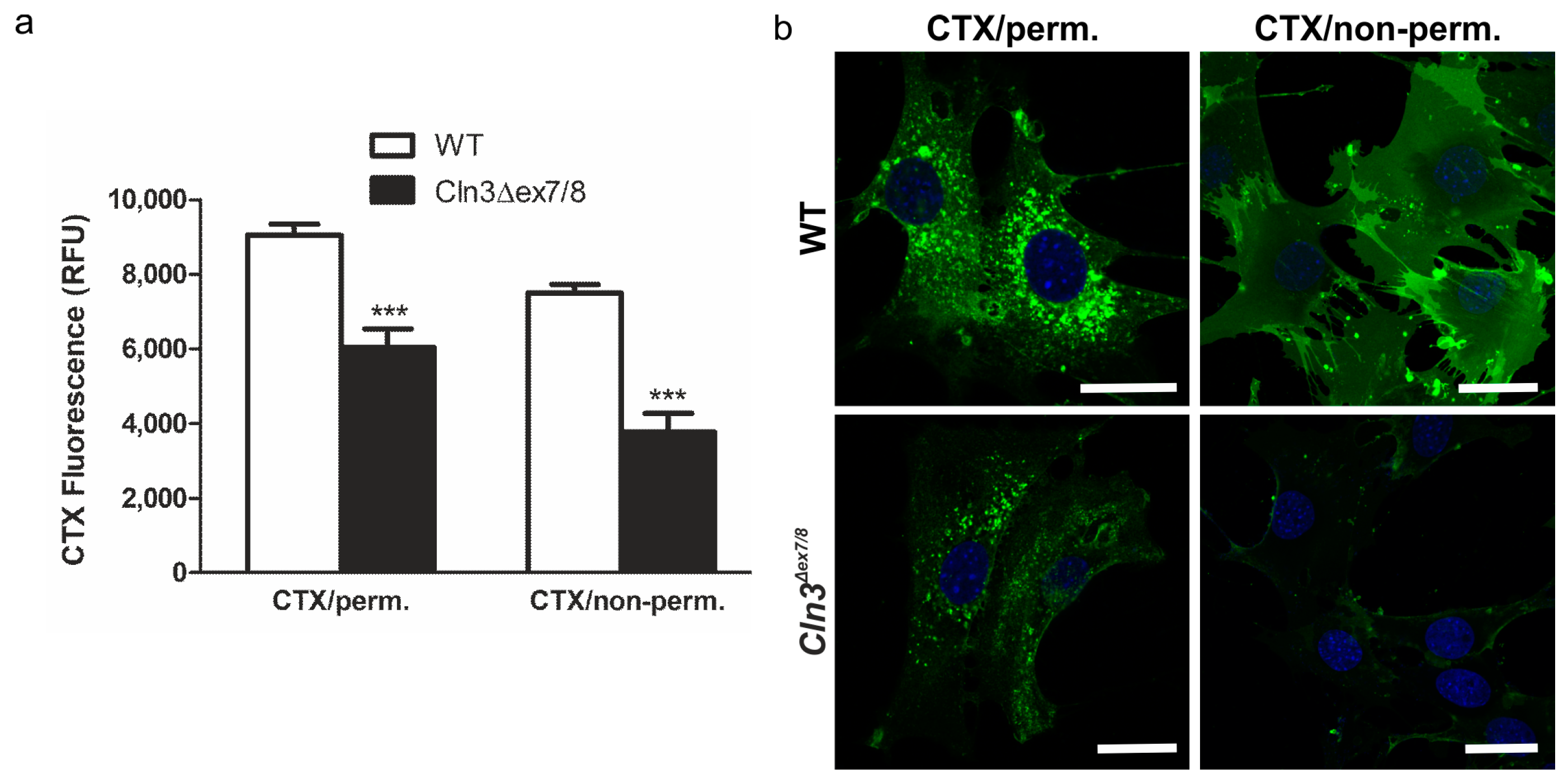
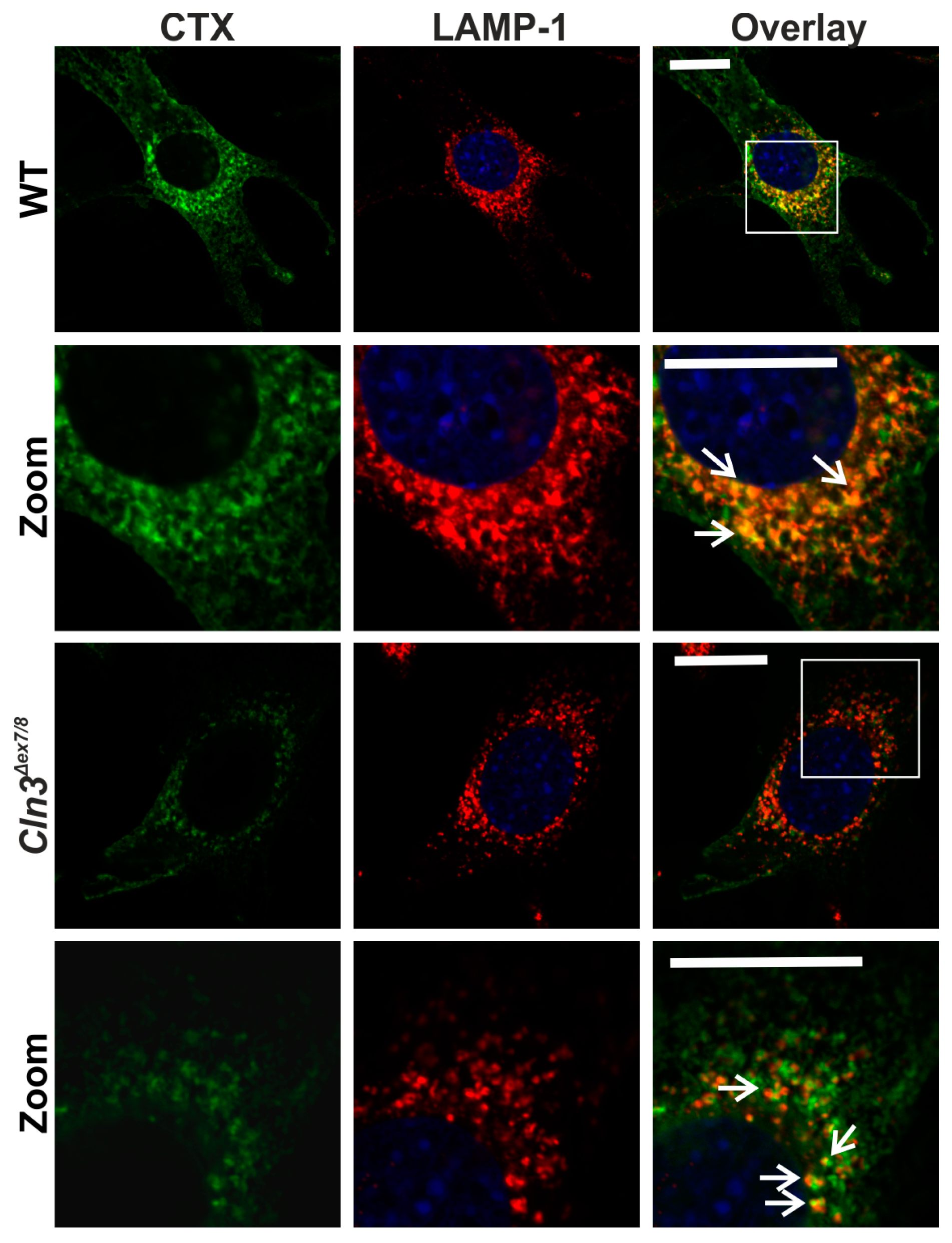
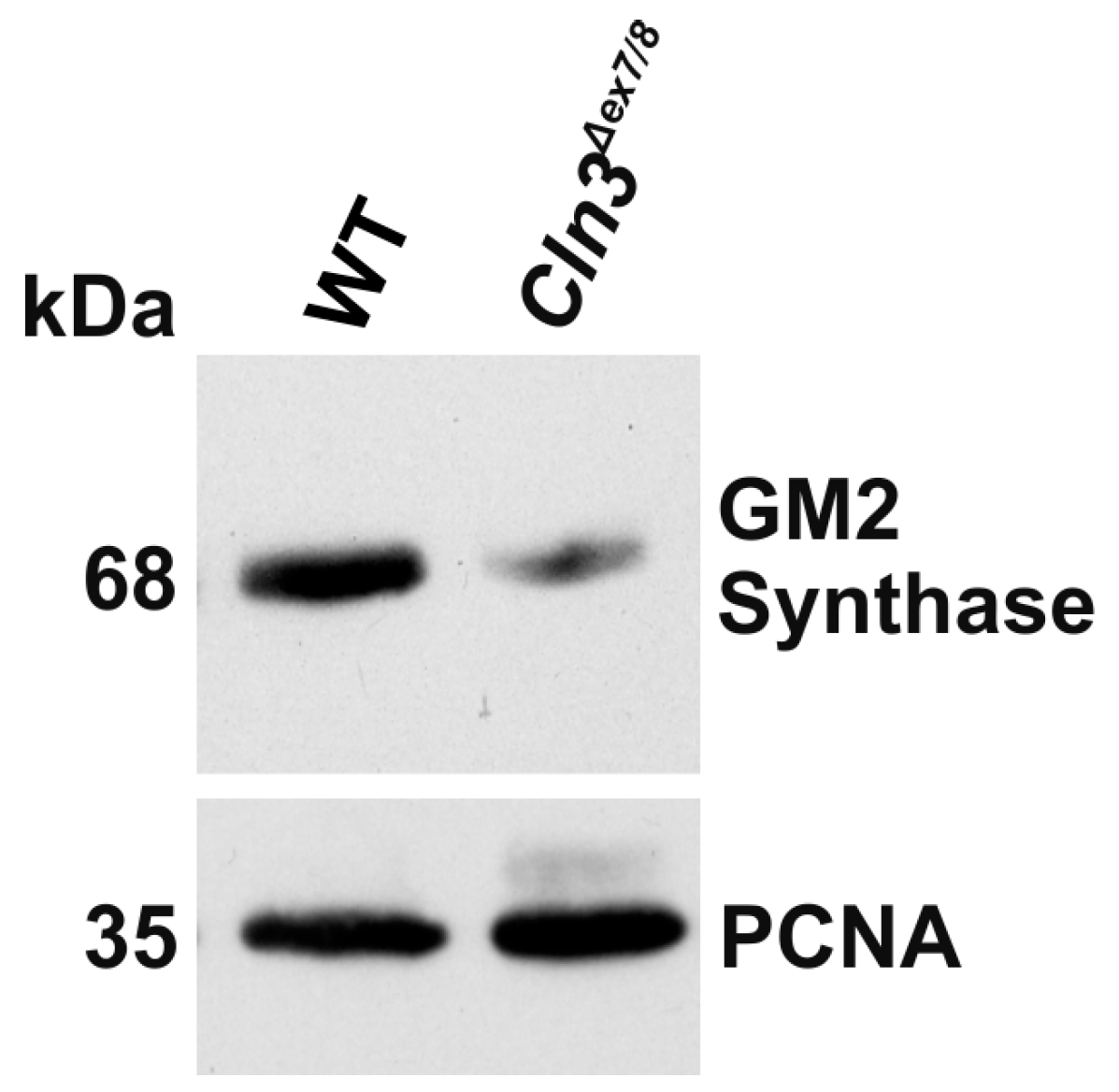
2. Results
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies and Toxins; Immunostaining
4.3. RNA Isolation, cDNA Synthesis and Quantitative Real-Time PCR
4.4. Western Blot and Dot Blot Analysis
4.5. Flow Cytometry and CTX-FITC Fluorometry
4.6. High-Performance Liquid Chromatography of Gangliosides
4.7. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 2AA | Anthranillic acid |
| AD | Alzheimer’s disease |
| Akt | RAC-α serine/threonine-protein kinase |
| BSA | Bovine serum albumin |
| Cb | Cerebellar |
| CLEAR | Coordinated Lysosomal Expression and Regulation |
| CLN3 | Ceroid lipofuscinosis, neuronal |
| CNS | Central nervous system |
| CTX | Cholera toxin subunit B |
| DEPC | Diethylpyrocarbonate |
| ECL | Enhanced chemiluminescence |
| ER | Endoplasmic reticulum |
| FACS | Fluorescence-activated cell sorting |
| FITC | Fluorescein isothiocyanate |
| GalCer | Galactosylceramide |
| GSL | Glycosphingolipid |
| GU | Glucose units |
| HD | Huntington’s disease |
| HPLC | High performance liquid chromatography |
| JNCL | Juvenile neuronal ceroid lipofuscinosis |
| LacCer | Lactosylceramide |
| LAMP-1 | Lysosomal-associated membrane protein 1 |
| MAPK | Mitogen-activated protein kinase |
| NP-HPLC | Normal phase high-performance liquid chromatography |
| PBS | Phosphate buffered saline |
| PD | Parkinson’s disease |
| qRT-PCR | quantitative real-time polymerase chain reaction |
| SDS | Sodium dodecyl sulfate |
| SPE | Solid Phase Extraction |
| TFEB | Transcription Factor EB |
| TrkA | Tropomyosin receptor kinase A |
References
- Cotman, S.L.; Vrbanac, V.; Lebel, L.A.; Lee, R.L.; Johnson, K.A.; Donahue, L.R.; Teed, A.M.; Antonellis, K.; Bronson, R.T.; Lerner, T.J.; et al. Cln3(Deltaex7/8) knock-in mice with the common JNCL mutation exhibit progressive neurologic disease that begins before birth. Hum. Mol. Genet. 2002, 11, 2709–2721. [Google Scholar] [CrossRef] [PubMed]
- Fossale, E.; Wolf, P.; Espinola, J.A.; Lubicz-Nawrocka, T.; Teed, A.M.; Gao, H.; Rigamonti, D.; Cattaneo, E.; MacDonald, M.E.; Cotman, S.L. Membrane trafficking and mitochondrial abnormalities precede subunit c deposition in a cerebellar cell model of juvenile neuronal ceroid lipofuscinosis. BMC Neurosci. 2004, 5, 57. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Espinola, J.A.; Fossale, E.; Massey, A.C.; Cuervo, A.M.; MacDonald, M.E.; Cotman, S.L. Autophagy is disrupted in a knock-in mouse model of juvenile neuronal ceroid lipofuscinosis. J. Biol. Chem. 2006, 281, 20483–20493. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Staropoli, J.F.; Biswas, S.; Espinola, J.A.; MacDonald, M.E.; Lee, J.M.; Cotman, S.L. Distinct early molecular responses to mutations causing vLINCL and JNCL presage ATP synthase subunit C accumulation in cerebellar cells. PLoS ONE 2011, 6, e17118. [Google Scholar] [CrossRef] [PubMed]
- Chandrachud, U.; Walker, M.W.; Simas, A.M.; Heetveld, S.; Petcherski, A.; Klein, M.; Oh, H.; Wolf, P.; Zhao, W.N.; Norton, S.; et al. Unbiased Cell-based Screening in a Neuronal Cell Model of Batten Disease Highlights an Interaction between Ca2+ Homeostasis, Autophagy, and CLN3 Protein Function. J. Biol. Chem. 2015, 290, 14361–14380. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, A.D.; Pearce, D.A. Finding the most appropriate mouse model of juvenile CLN3 (Batten) disease for therapeutic studies: The importance of genetic background and gender. Dis. Model. Mech. 2015, 8, 351–361. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, E.; Petcherski, A.; Ramos-Moreno, J.M.; Ruonala, M.O. FRET-Assisted Determination of CLN3 Membrane Topology. PLoS ONE 2014, 9, e102593. [Google Scholar] [CrossRef] [PubMed]
- Cotman, S.L.; Staropoli, J.F. The juvenile Batten disease protein, CLN3, and its role in regulating anterograde and retrograde post-Golgi trafficking. Clin. Lipidol. 2012, 7, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Ramirez-Montealegre, D.; Pearce, D.A. Defective lysosomal arginine transport in juvenile Batten disease. Hum. Mol. Genet. 2005, 14, 3759–3773. [Google Scholar] [CrossRef] [PubMed]
- Vidal-Donet, J.M.; Carcel-Trullols, J.; Casanova, B.; Aguado, C.; Knecht, E. Alterations in ROS activity and lysosomal pH account for distinct patterns of macroautophagy in LINCL and JNCL fibroblasts. PLoS ONE 2013, 8, e55526. [Google Scholar] [CrossRef] [PubMed]
- Kolter, T.; Sandhoff, K. Sphingolipid metabolism diseases. Biochim. Biophys. Acta 2006, 1758, 2057–2079. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, G. Ganglioside/glycosphingolipid turnover: New concepts. Glycoconj. J. 2004, 20, 301–317. [Google Scholar] [CrossRef] [PubMed]
- Daniotti, J.L.; Iglesias-Bartolome, R. Metabolic pathways and intracellular trafficking of gangliosides. IUBMB Life 2011, 63, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, H.J.F.; Daniotti, J.L.; Martina, J.A. Organization of ganglioside synthesis in the Golgi apparatus. Biochim. Biophys. Acta 1999, 1437, 101–118. [Google Scholar] [CrossRef]
- Yu, R.K.; Bieberich, E.; Xia, T.; Zeng, G. Regulation of ganglioside biosynthesis in the nervous system. J. Lipid Res. 2004, 45, 783–793. [Google Scholar] [CrossRef] [PubMed]
- Schnaar, R.L. Brain gangliosides in axon-myelin stability and axon regeneration. FEBS Lett. 2010, 584, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.K.; Nakatani, Y.; Yanagisawa, M. The role of glycosphingolipid metabolism in the developing brain. J. Lipid Res. 2009, 50, S440–S445. [Google Scholar] [CrossRef] [PubMed]
- Harlalka, G.V.; Lehman, A.; Chioza, B.; Baple, E.L.; Maroofian, R.; Cross, H.; Sreekantan-Nair, A.; Priestman, D.A.; Al-Turki, S.; McEntagart, M.E.; et al. Mutations in B4GALNT1 (GM2 synthase) underlie a new disorder of ganglioside biosynthesis. Brain 2013, 136, 3618–3624. [Google Scholar] [CrossRef] [PubMed]
- Simpson, M.A.; Cross, H.; Proukakis, C.; Priestman, D.A.; Neville, D.C.A.; Reinkensmeier, G.; Wang, H.; Wiznitzer, M.; Gurtz, K.; Verganelaki, A.; et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat. Genet. 2004, 36, 1225–1229. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Yoo, Y.; Lim, B.C.; Kim, K.J.; Song, J.; Choi, M.; Chae, J.H. GM3 synthase deficiency due to ST3GAL5 variants in two Korean female siblings: Masquerading as Rett syndrome-like phenotype. Am. J. Med. Genet. A 2016, 170, 2200–2205. [Google Scholar] [CrossRef] [PubMed]
- Puranam, K.L.; Guo, W.-X.; Qian, W.-H.; Nikbakht, K.; Boustany, R.-M.N. CLN3 Defines a Novel Antiapoptotic Pathway Operative in Neurodegeneration and Mediated by Ceramide. Mol. Gen. Metab. 1999, 66, 294–308. [Google Scholar] [CrossRef] [PubMed]
- Persaud-Sawin, D.-A.; McNamara, J.O.; Rylova, S.N.; Vandongen, A.; Boustany, R.-M.N. A Galactosylceramide Binding Domain Is Involved in Trafficking of CLN3 from Golgi to Rafts via Recycling Endosomes. Pediatr. Res. 2004, 56, 449–463. [Google Scholar] [CrossRef] [PubMed]
- Rusyn, E.; Mousallem, T.; Persaud-Sawin, D.-A.; Miller, S.; Boustany, R.-M.N. CLN3p impacts galactosylceramide transport, raft morphology, and lipid content. Pediatr. Res. 2008, 63, 625–631. [Google Scholar] [CrossRef] [PubMed]
- Schubert, W.; Bonnekoh, B.; Pommer, A.J.; Philipsen, L.; Bockelmann, R.; Malykh, Y.; Gollnick, H.; Friedenberger, M.; Bode, M.; Dress, A.W. Analyzing proteome topology and function by automated multidimensional fluorescence microscopy. Nat. Biotechnol. 2006, 24, 1270–1278. [Google Scholar] [CrossRef] [PubMed]
- Ruonala, M.O. (Image Computing & Information Technologies, Frankfurt am Main, Germany). Personal communication, 2018.
- Nelson, T.; Pearce, D.A.; Kovacs, A.D. Lack of specificity of antibodies raised against CLN3, the lysosomal/endosomal transmembrane protein mutated in juvenile Batten disease. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Haskell, R.E.; Derksen, T.A.; Davidson, B.L. Intracellular Trafficking of the JNCL Protein CLN3. Mol. Genet. Metab. 1999, 66, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Oetjen, S.; Kuhl, D.; Hermey, G. Revisiting the neuronal localization and trafficking of CLN3 in juvenile neuronal ceroid lipofuscinosis. J. Neurochem. 2016, 139, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.; Heo, T.-H.; Kim, S.-J. Altered levels of α-synuclein and sphingolipids in Batten disease lymphoblast cells. Gene 2014, 539, 181–185. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Pal, R.; Nelvagal, H.R.; Lotfi, P.; Stinnett, G.R.; Seymour, M.L.; Chaudhury, A.; Bajaj, L.; Bondar, V.V.; Bremner, L.; et al. mTORC1-independent TFEB activation via Akt inhibition promotes cellular clearance in neurodegenerative storage diseases. Nat. Commun. 2017, 8, 14338. [Google Scholar] [CrossRef] [PubMed]
- Maccioni, H.J. Glycosylation of glycolipids in the Golgi complex. J. Neurochem. 2007, 103 (Suppl. 1), 81–90. [Google Scholar] [CrossRef] [PubMed]
- Fishman, P.H.; Max, S.R.; Tallman, J.F.; Brady, R.O.; Maclaren, N.K.; Cornblath, M. Deficient Ganglioside Biosynthesis: A novel human sphingolipidosis. Science 1975, 187, 68–70. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, K.A.; Sun, J.; Liu, Y.; Kawai, H.; Crawford, T.O.; Proia, R.L.; Griffin, J.W.; Schnaar, R.L. Mice lacking complex gangliosides develop Wallerian degeneration and myelination defects. Proc. Natl. Acad. Sci. USA 1999, 96, 7532–7537. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, K.; Yamamoto, A.; Furukawa, K.; Yamashiro, S.; Shin, M.; Okada, M.; Fukumoto, S.; Haraguchi, M.; Takeda, N.; Fujimura, K.; et al. Mice with disrupted GM2/GD2 synthase gene lack complex gangliosides but exhibit only subtle defects in their nervous system. Proc. Natl. Acad. Sci. USA 1996, 93, 10662–10667. [Google Scholar] [CrossRef] [PubMed]
- Kawai, H.; Allende, M.L.; Wada, R.; Kono, M.; Sango, K.; Deng, C.; Miyakawa, T.; Crawley, J.N.; Werth, N.; Bierfreund, U.; et al. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J. Biol. Chem. 2001, 276, 6885–6888. [Google Scholar] [CrossRef] [PubMed]
- Ngamukote, S.; Yanagisawa, M.; Ariga, T.; Ando, S.; Yu, R.K. Developmental changes of glycosphingolipids and expression of glycogenes in mouse brains. J. Neurochem. 2007, 103, 2327–2341. [Google Scholar] [CrossRef] [PubMed]
- Schauer, R. Sialic acids as regulators of molecular and cellular interactions. Curr. Opin. Struct. Biol. 2009, 19, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Tettamanti, G.; Bonali, F.; Marchesini, S.; Zambotti, V. A new procedure for the extraction, purification and fractionation of brain gangliosides. Biochim. Biophys. Acta 1973, 296, 160–170. [Google Scholar] [CrossRef]
- Schnaar, R.L.; Gerardy-Schahn, R.; Hildebrandt, H. Sialic Acids in the Brain: Gangliosides and Polysialic Acid in Nervous System Development, Stability, Disease, and Regeneration. Phys. Rev. 2014, 94, 461–518. [Google Scholar] [CrossRef] [PubMed]
- Susuki, K.; Baba, H.; Tohyama, K.; Kanai, K.; Kuwabara, S.; Hirata, K.; Furukawa, K.; Furukawa, K.; Rasband, M.N.; Yuki, N. Gangliosides contribute to stability of paranodal junctions and ion channel clusters in myelinated nerve fibers. Glia 2007, 55, 746–757. [Google Scholar] [CrossRef] [PubMed]
- Manev, H.; Favaron, M.; Vicini, S.; Guidotti, A.; Costa, E. Glutamate-induced neuronal death in primary cultures of cerebellar granule cells: Protection by synthetic derivatives of endogenous sphingolipids. J. Pharmacol. Exp. Ther. 1990, 252, 419–427. [Google Scholar] [PubMed]
- Ichikawa, N.; Iwabuchi, K.; Kurihara, H.; Ishii, K.; Kobayashi, T.; Sasaki, T.; Hattori, N.; Mizuno, Y.; Hozumi, K.; Yamada, Y.; et al. Binding of laminin-1 to monosialoganglioside GM1 in lipid rafts is crucial for neurite outgrowth. J. Cell Sci. 2009, 122, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Prioni, S.; Mauri, L.; Loberto, N.; Casellato, R.; Chigorno, V.; Karagogeos, D.; Prinetti, A.; Sonnino, S. Interactions between gangliosides and proteins in the exoplasmic leaflet of neuronal plasma membranes: A study performed with a tritium-labeled GM1 derivative containing a photoactivable group linked to the oligosaccharide chain. Glycoconj. J. 2004, 21, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Traka, M.; Goutebroze, L.; Denisenko, N.; Bessa, M.; Nifli, A.; Havaki, S.; Iwakura, Y.; Fukamauchi, F.; Watanabe, K.; Soliven, B.; et al. Association of TAG-1 with Caspr2 is essential for the molecular organization of juxtaparanodal regions of myelinated fibers. J. Cell Biol. 2003, 162, 1161–1172. [Google Scholar] [CrossRef] [PubMed]
- Golabek, A.A.; Kida, E.; Walus, M.; Kaczmarski, W.; Michalewski, M.P.; Wisniewski, K.E. CLN3 Protein Regulates Lysosomal pH and Alters Intracellular Processing of Alzheimer’s Amyloid-β Protein Precursor and Cathepsin D in Human Cells. Mol. Genet. Metab. 2000, 70, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Di Pardo, A.; Maglione, V.; Alpaugh, M.; Horkey, M.; Atwal, R.S.; Sassone, J.; Ciammola, A.; Steffan, J.S.; Fouad, K.; Truant, R.; et al. Ganglioside GM1 induces phosphorylation of mutant huntingtin and restores normal motor behavior in Huntington disease mice. Proc. Natl. Acad. Sci. USA 2012, 109, 3528–3533. [Google Scholar] [CrossRef] [PubMed]
- Maglione, V.; Marchi, P.; di Pardo, A.; Lingrell, S.; Horkey, M.; Tidmarsh, E.; Sipione, S. Impaired Ganglioside Metabolism in Huntington’s Disease and Neuroprotective Role of GM1. J. Neurosci. 2010, 30, 4072–4080. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Gollomp, S.M.; Sendek, S.; Colcher, A.; Cambi, F.; Du, W. A randomized, controlled, delayed start trial of GM1 ganglioside in treated Parkinson’s disease patients. J. Neurol. Sci. 2013, 324, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.S.; Sendek, S.; Daskalakis, C.; Cambi, F. GM1 ganglioside in Parkinson’s disease: Results of a five year open study. J. Neurol. Sci. 2010, 292, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Trbojevic-Cepe, M.; Kracun, I.; Jusic, A.; Pavlicek, I. Gangliosides of human cerebrospinal fluid in various neurologic diseases. J. Neurol. Sci. 1991, 105, 192–199. [Google Scholar] [CrossRef]
- Kracun, I.; Rosner, H.; Drnovsek, V.; Heffer-Lauc, M.; Cosovic, C.; Lauc, G. Human brain gangliosides in development, aging and disease. Int. J. Dev. Biol. 1991, 35, 289–295. [Google Scholar] [PubMed]
- Kalanj, S.; Kracun, I.; Rosner, H.; Cosovic, C. Regional distribution of brain gangliosides in Alzheimer’s disease. Neurol. Croat. 1991, 40, 269–281. [Google Scholar] [PubMed]
- Clark, L.N.; Chan, R.; Cheng, R.; Liu, X.; Park, N.; Parmalee, N.; Kisselev, S.; Cortes, E.; Torres, P.A.; Pastores, G.M.; et al. Gene-wise association of variants in four lysosomal storage disorder genes in neuropathologically confirmed Lewy body disease. PLoS ONE 2015, 10, e0125204. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.-S.; Chen, L.-Y.; Wang, S.S.; Chang, Y.; Chen, W.-Y. Examining the levels of ganglioside and cholesterol in cell membrane on attenuation the cytotoxicity of beta-amyloid peptide. Colloids Surf. B Biointerfaces 2008, 65, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, A.; Harrison, F.E.; McCord, M.; Zhao, J.; Bruchey, A.; Davies, S.S.; Jackson Roberts, L., 2nd; Mathews, P.M.; Matsuoka, Y.; Ariga, T.; et al. Elimination of GD3 synthase improves memory and reduces amyloid-beta plaque load in transgenic mice. Neurobiol. Aging 2009, 30, 1777–1791. [Google Scholar] [CrossRef] [PubMed]
- Alter, M. GM1 ganglioside for acute ischemic stroke. Trial design issues. Ann. N. Y. Acad. Sci. 1998, 845, 391–401. [Google Scholar] [CrossRef] [PubMed]
- Schengrund, C.-L.; Mummert, C.M. Exogenous gangliosides. How do they cross the blood-brain barrier and how do they inhibit cell proliferation. Ann. N. Y. Acad. Sci. 1998, 845, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.J.; Alexander, N.; Benedict, J.W.; Chattopadhyay, S.; Shemilt, S.J.; Guerin, C.J.; Cooper, J.D.; Pearce, D.A. IgG entry and deposition are components of the neuroimmune response in Batten disease. Neurobiol. Dis. 2007, 25, 239–251. [Google Scholar] [CrossRef] [PubMed]
- Lim, M.J.; Beake, J.; Bible, E.; Curran, T.M.; Ramirez-Montealegre, D.; Pearce, D.A.; Cooper, J.D. Distinct patterns of serum immunoreactivity as evidence for multiple brain-directed autoantibodies in juvenile neuronal ceroid lipofuscinosis. Neuropathol. Appl. Neurobiol. 2006, 32, 469–482. [Google Scholar] [CrossRef] [PubMed]
- Tecedor, L.; Stein, C.S.; Schultz, M.L.; Farwanah, H.; Sandhoff, K.; Davidson, B.L. CLN3 Loss Disturbs Membrane Microdomain Properties and Protein Transport in Brain Endothelial Cells. J. Neurosci. 2013, 33, 18065–18079. [Google Scholar] [CrossRef] [PubMed]
- Neville, D.C.; Coquard, V.; Priestman, D.A.; te Vruchte, D.J.; Sillence, D.J.; Dwek, R.A.; Platt, F.M.; Butters, T.D. Analysis of fluorescently labeled glycosphingolipid-derived oligosaccharides following ceramide glycanase digestion and anthranilic acid labeling. Anal. Biochem. 2004, 331, 275–282. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
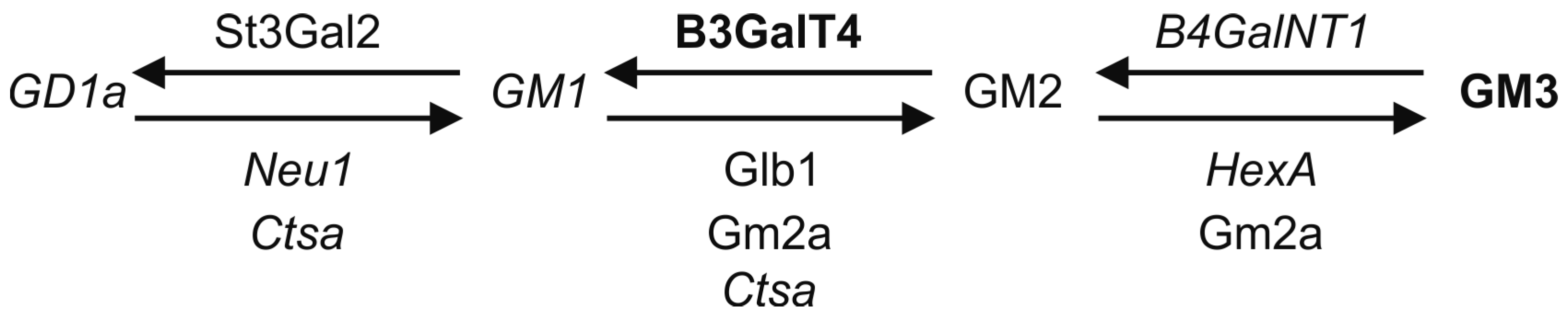
| Gene | Enzyme | Alternative Names | Synthesis Step |
|---|---|---|---|
| ST3Gal5 | Lactosylceramide sialyltransferase | GM3 synthase, Sial-T1 | LacCer to GM3 |
| B4GalNT1 | GM3 N-acetyl-galactosaminyl transferase | GM2 synthase, GalNAc-T, GM3 sialyltransferase | GM3 to GM2 |
| B3GalT-4 | GM2 galactosyltransferase | GM1 synthase, Gal-T2 | GM2 to GM1 |
| ST3Gal2 | GM1 sialyltransferase | GD1a synthase, Sial-T4 | GM1 to GD1a |
| ST3Gal3 | GD1a sialyltransferase | GT1a synthase, Sial-T5 | GD1a to GT1a |
| Gene Symbol | Enzyme/Protein (Function) | Mean Expression Level ± SD | p-Value | |
|---|---|---|---|---|
| WT | Cln3Δex7/8 | |||
| St3gal2 | GM1 sialyltransferase | 1.0 ± 0.01 | 1.02 ± 0.01 | 0.939 |
| B4galNT1 | β-1,4-N-acetyl-galactosaminyl transferase 1 | 1.0 ± 0.03 | 0.12 ± 0.01 | 4 × 10−4 |
| Neu1 | neuraminidase 1 (GD1a to GM1) | 1.0 ± 0.07 | 0.50 ± 0.04 | 0.036 |
| Glb1 | β-galactosidase (GM1 to GM2) | 1.0 ± 0.002 | 0.57 ± 0.006 | 0.060 |
| Gm2a | GM2 activator protein (assists in degradation of GM1 and GM2) | 1.0 ± 0.01 | 0.78 ± 0.01 | 0.474 |
| Ctsa | cathepsin A (assists in degradation of GD1a and GM1a) | 1.0 ± 0.03 | 0.29 ± 0.04 | 0.005 |
| Hexa | hexosaminidase A (GM2 to GM3) | 1.0 ± 0.03 | 0.20 ± 0.04 | 0.007 |
| B3galT4 | β-1,3-galactosyltransferase, polypeptide 4 (GM2 to GM1) | 1.0 ± 0.09 | 4.46 ± 0.52 | 0.056 |
| Target Gene | Primer Name | Sequence 5′-3′ |
|---|---|---|
| St3Gal2 | St3Gal2-fwd | GACGCCAGCACCTCTGAATGGT |
| St3Gal2-rev | TTGTAGCATCATCCACCACCGC | |
| B4GalNT1 | B4GalNT1-fwd | GAGATATACCAGGTGAACCTGAG |
| B4GalNT1-rev | CTGCCGGTTGAGTTTATCCA | |
| Neu1 | Neu1-fwd | CACCCTGAGTTCCGAGTGAACC |
| Neu1-rev | GCTGCTTCTTTCCATCCGTGCT | |
| Glb1 | Glb1-fwd | CGGTCGACCTCCAATTCTTCGG |
| Glb1-rev | TACTTGACCCTTGGACCACCCA | |
| Gm2a | Gm2a-fwd | AGCCTCACGATCCAACCTGACC |
| Gm2a-rev | GAGCTCCACCTTCTGAGGAGCA | |
| Ctsa | Ctsa-fwd | TCACCATCAAGGGTGCCGGA |
| Ctsa-rev | GGATTTCCATGGGTTGCAGCGG | |
| Hexa | Hexa-fwd | CTACATCCAGACGCTGCTGGAC |
| Hexa-rev | TACTGGCATTTCTTCCCGCCAC | |
| B3GalT4 | B3GalT4-fwd | GGTTAAAGCCGTCCTCCCACCT |
| B3GalT4-rev | ACCAGTAGTAGGACCGCCAGGA | |
| RPL13a | RPL13a-fwd | GTTCGGCTGAAGCCTACCAG |
| RPL13a-rev | TTCCGTAACCTCAAGATCTGCT | |
| B2M | B2M-fwd | CCTGGCTCACACTGAATTCACC |
| B2M-rev | ATGTCTCGATCCCAGTAGACGG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Somogyi, A.; Petcherski, A.; Beckert, B.; Huebecker, M.; Priestman, D.A.; Banning, A.; Cotman, S.L.; Platt, F.M.; Ruonala, M.O.; Tikkanen, R. Altered Expression of Ganglioside Metabolizing Enzymes Results in GM3 Ganglioside Accumulation in Cerebellar Cells of a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. Int. J. Mol. Sci. 2018, 19, 625. https://doi.org/10.3390/ijms19020625
Somogyi A, Petcherski A, Beckert B, Huebecker M, Priestman DA, Banning A, Cotman SL, Platt FM, Ruonala MO, Tikkanen R. Altered Expression of Ganglioside Metabolizing Enzymes Results in GM3 Ganglioside Accumulation in Cerebellar Cells of a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. International Journal of Molecular Sciences. 2018; 19(2):625. https://doi.org/10.3390/ijms19020625
Chicago/Turabian StyleSomogyi, Aleksandra, Anton Petcherski, Benedikt Beckert, Mylene Huebecker, David A. Priestman, Antje Banning, Susan L. Cotman, Frances M. Platt, Mika O. Ruonala, and Ritva Tikkanen. 2018. "Altered Expression of Ganglioside Metabolizing Enzymes Results in GM3 Ganglioside Accumulation in Cerebellar Cells of a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis" International Journal of Molecular Sciences 19, no. 2: 625. https://doi.org/10.3390/ijms19020625
APA StyleSomogyi, A., Petcherski, A., Beckert, B., Huebecker, M., Priestman, D. A., Banning, A., Cotman, S. L., Platt, F. M., Ruonala, M. O., & Tikkanen, R. (2018). Altered Expression of Ganglioside Metabolizing Enzymes Results in GM3 Ganglioside Accumulation in Cerebellar Cells of a Mouse Model of Juvenile Neuronal Ceroid Lipofuscinosis. International Journal of Molecular Sciences, 19(2), 625. https://doi.org/10.3390/ijms19020625