The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death

 ,
,
Abstract
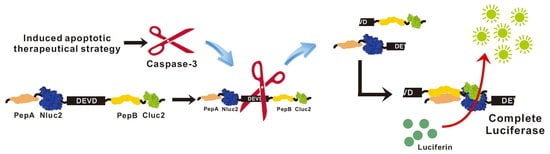
:
1. Introduction
2. Result
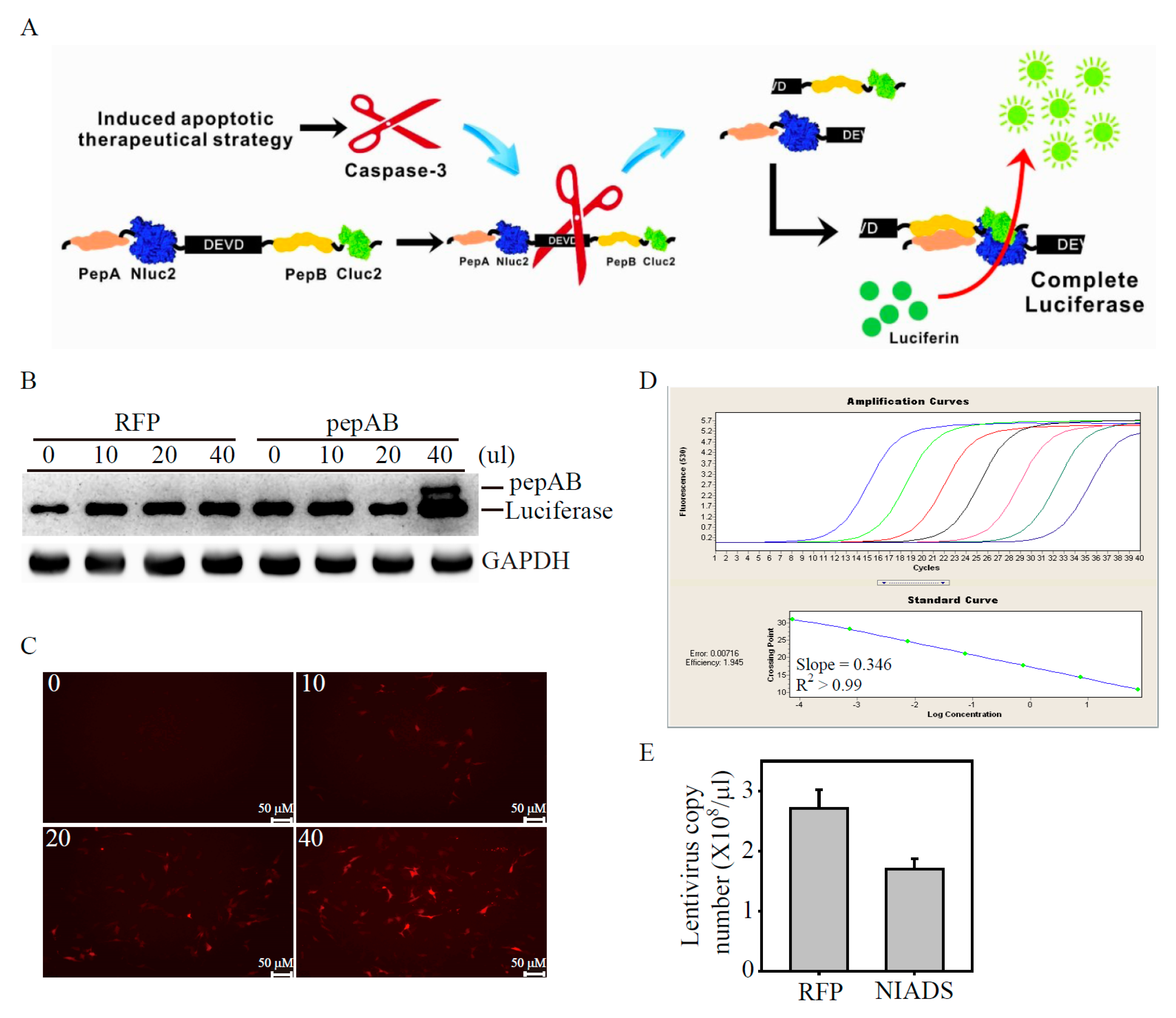
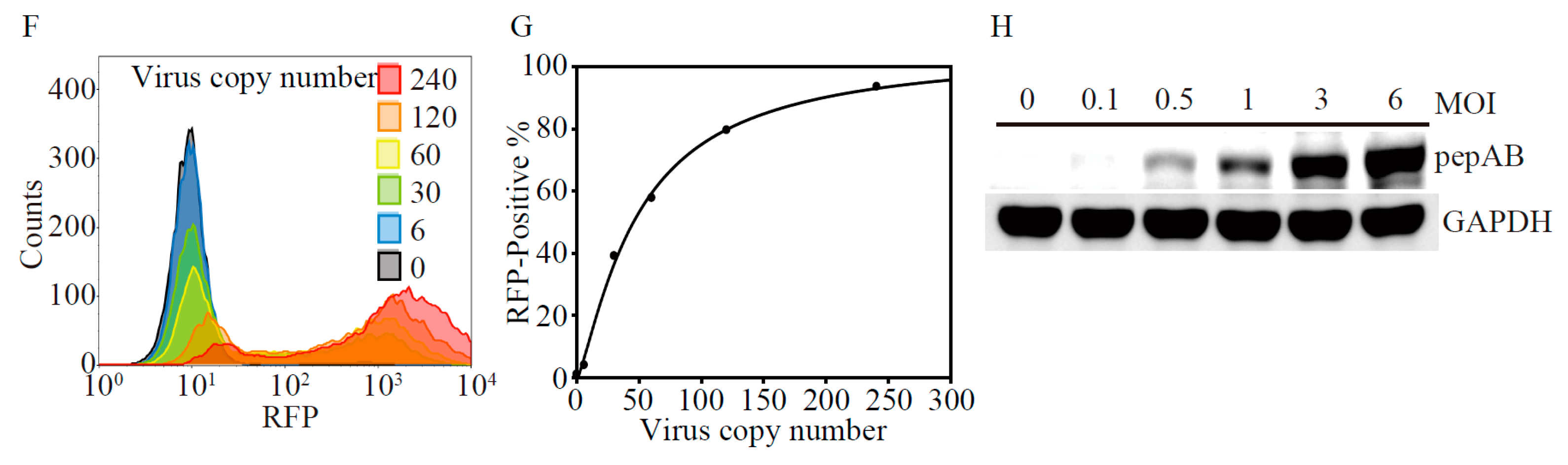
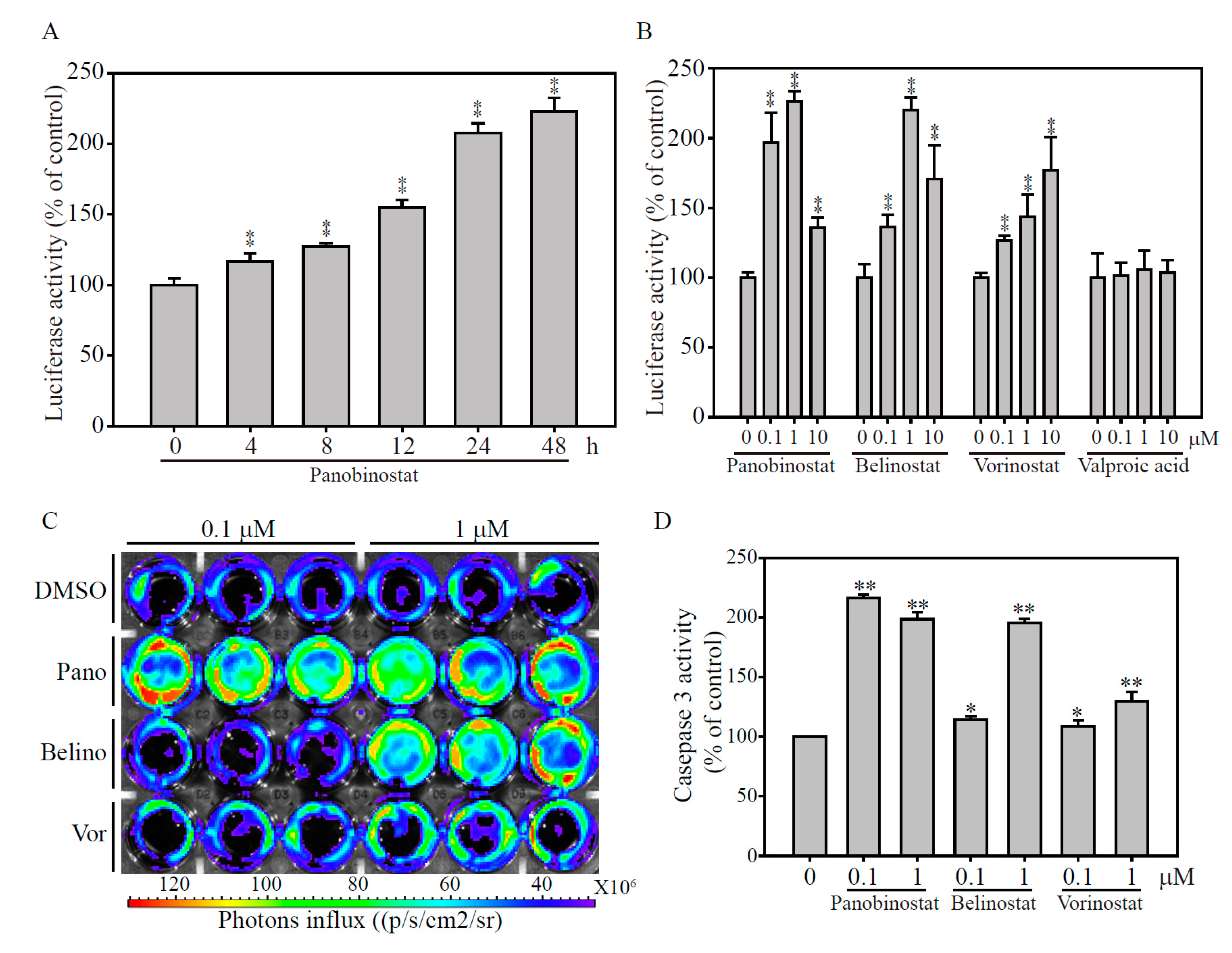
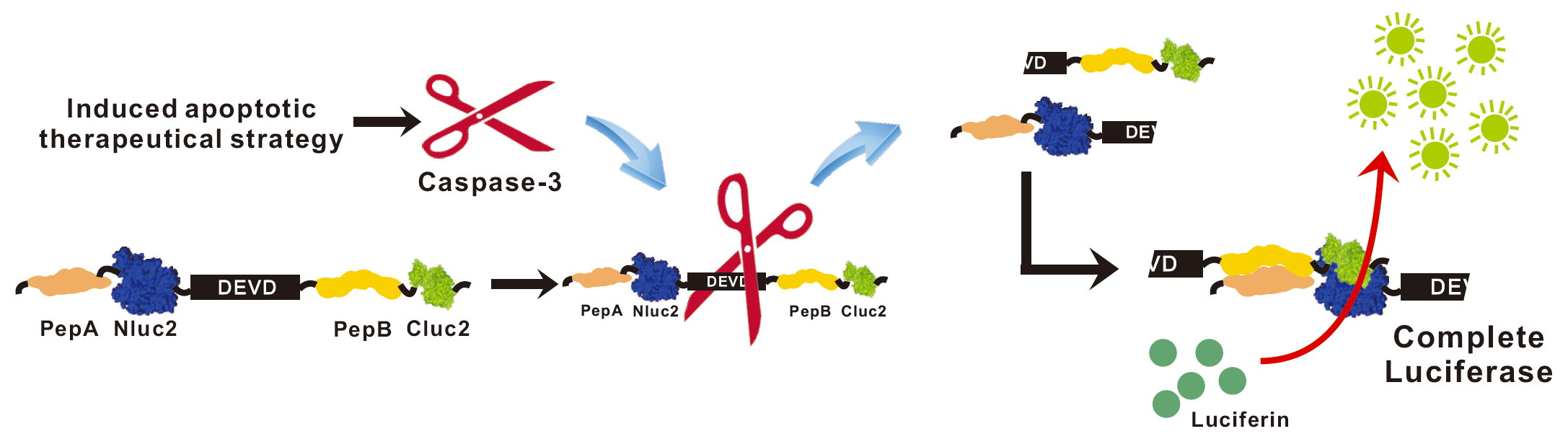
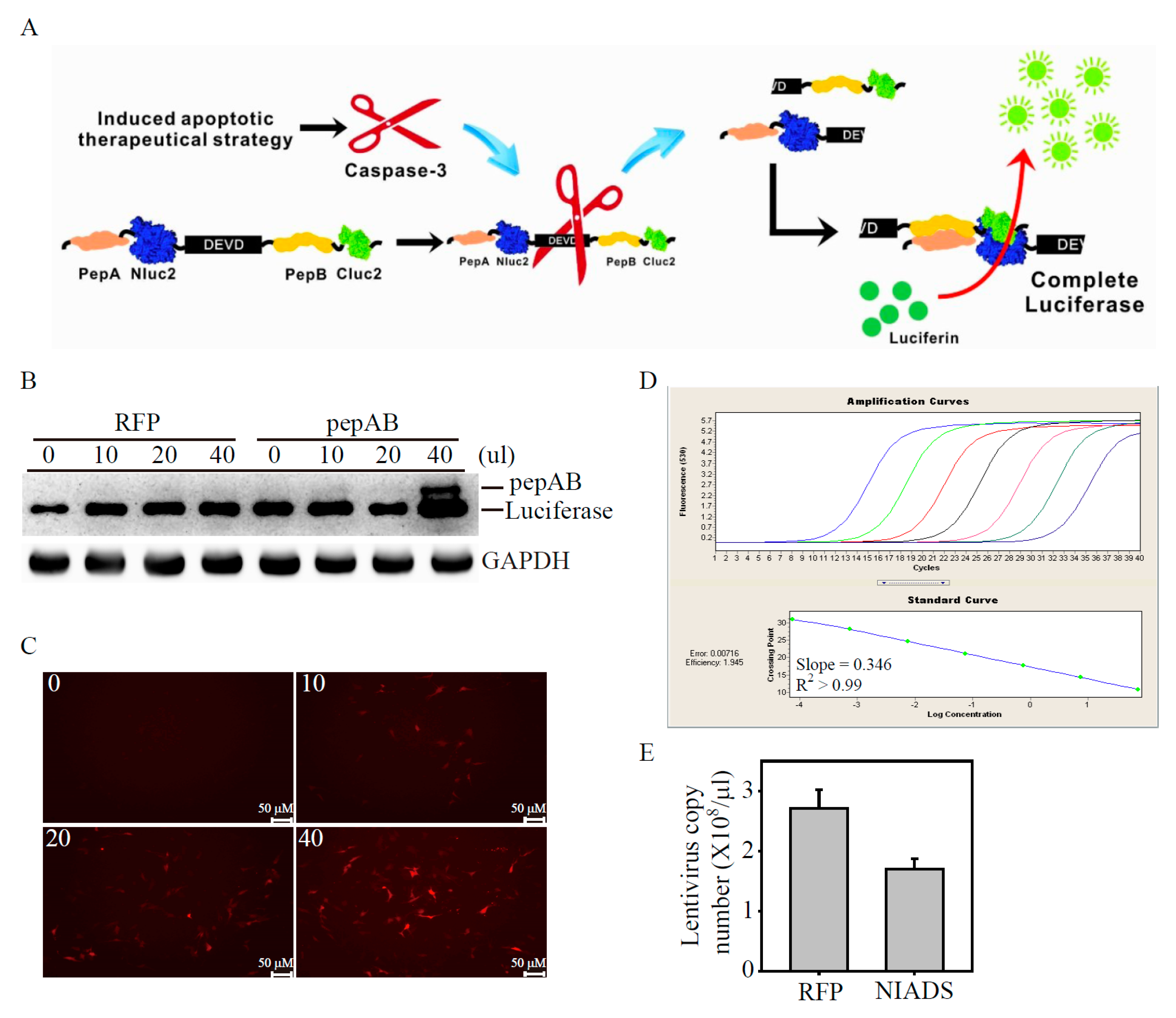
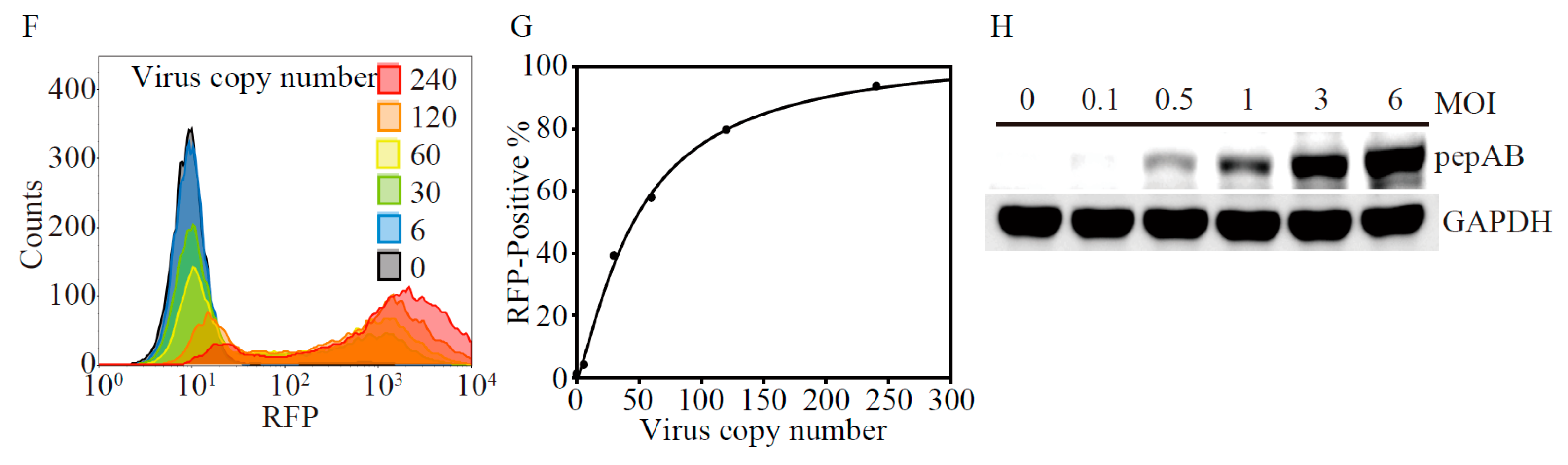
2.1. Development of Lentivirus Mediates Non-Invasive Caspase-3 Reporter Assay
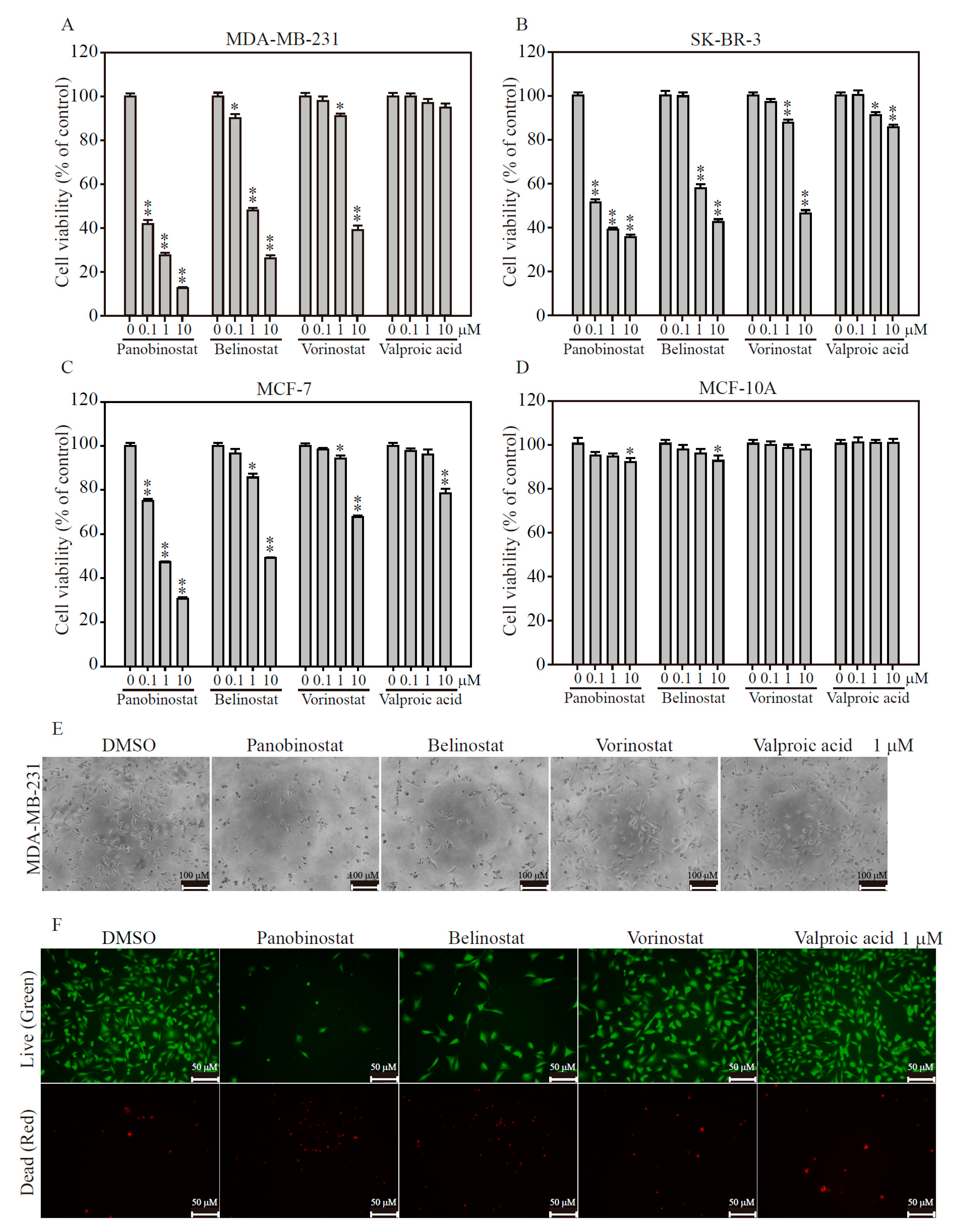
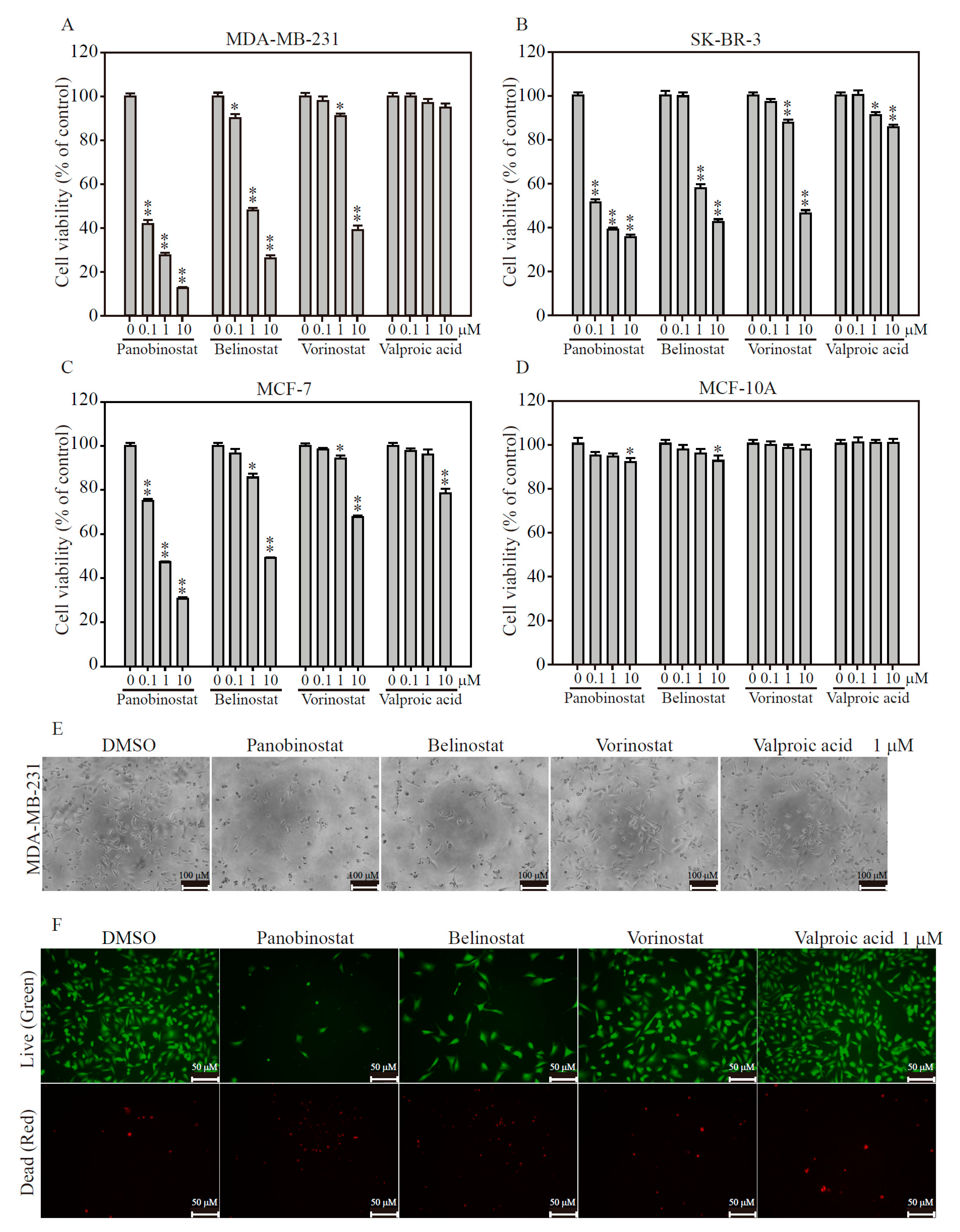
2.2. HDACi Induces Cell Death in TNBC and HER2-Enrich Breast Cancer Subtypes
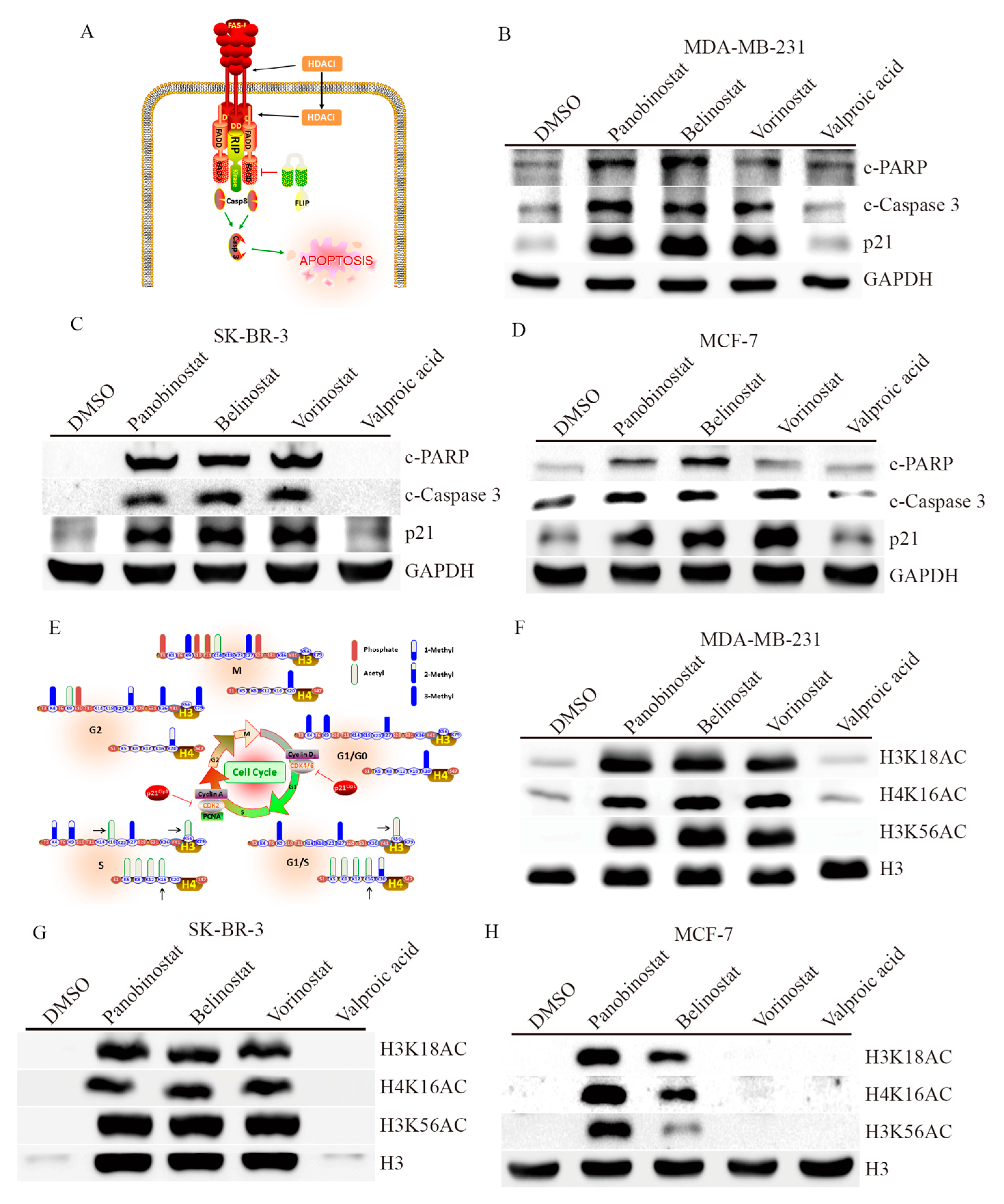
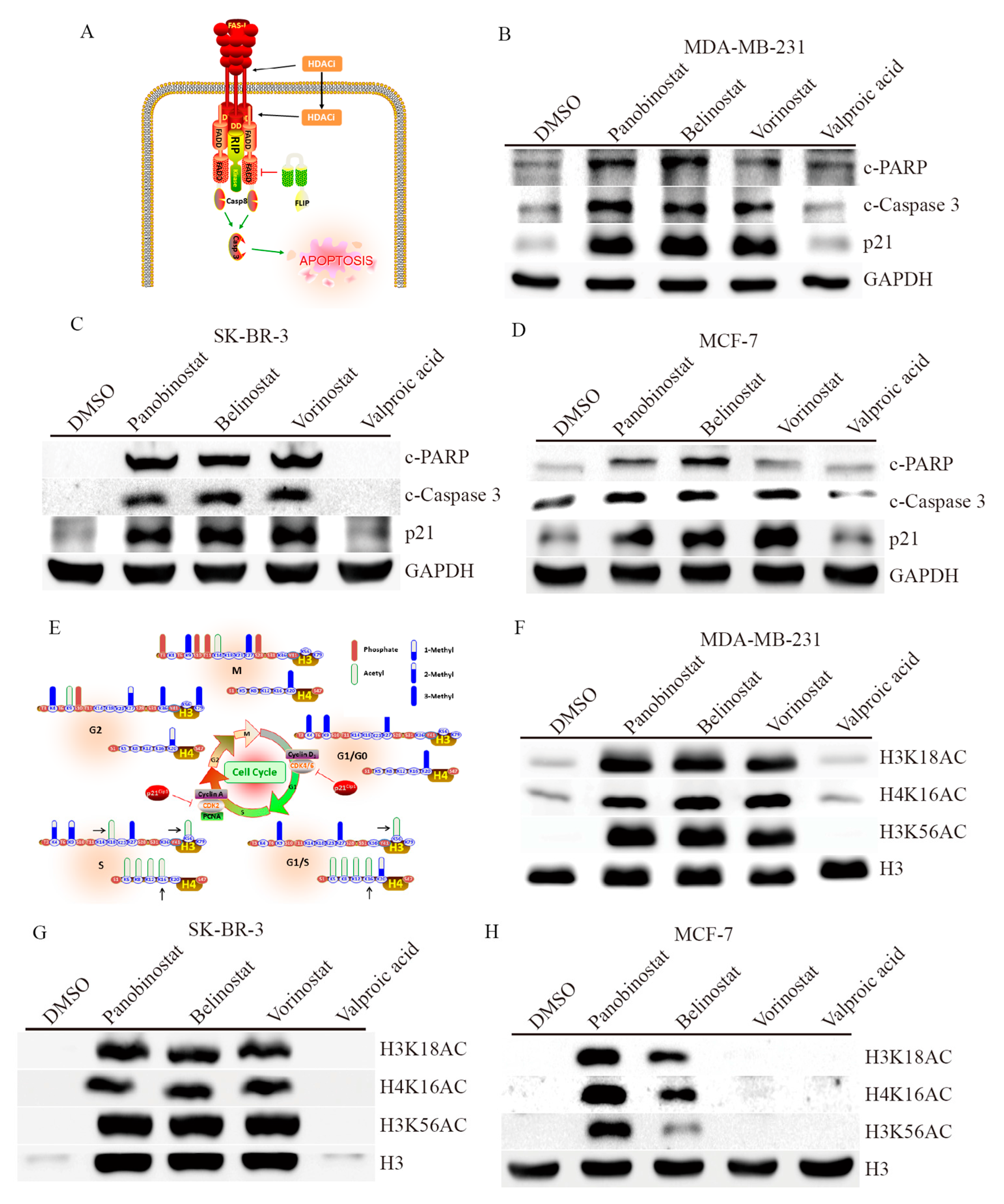
2.3. HDACi Induces Apoptosis and Cell Cycle Regulation through Histone Modification
2.4. Validation of the papAB Bioluminescence Apoptosis Reporter In Vitro
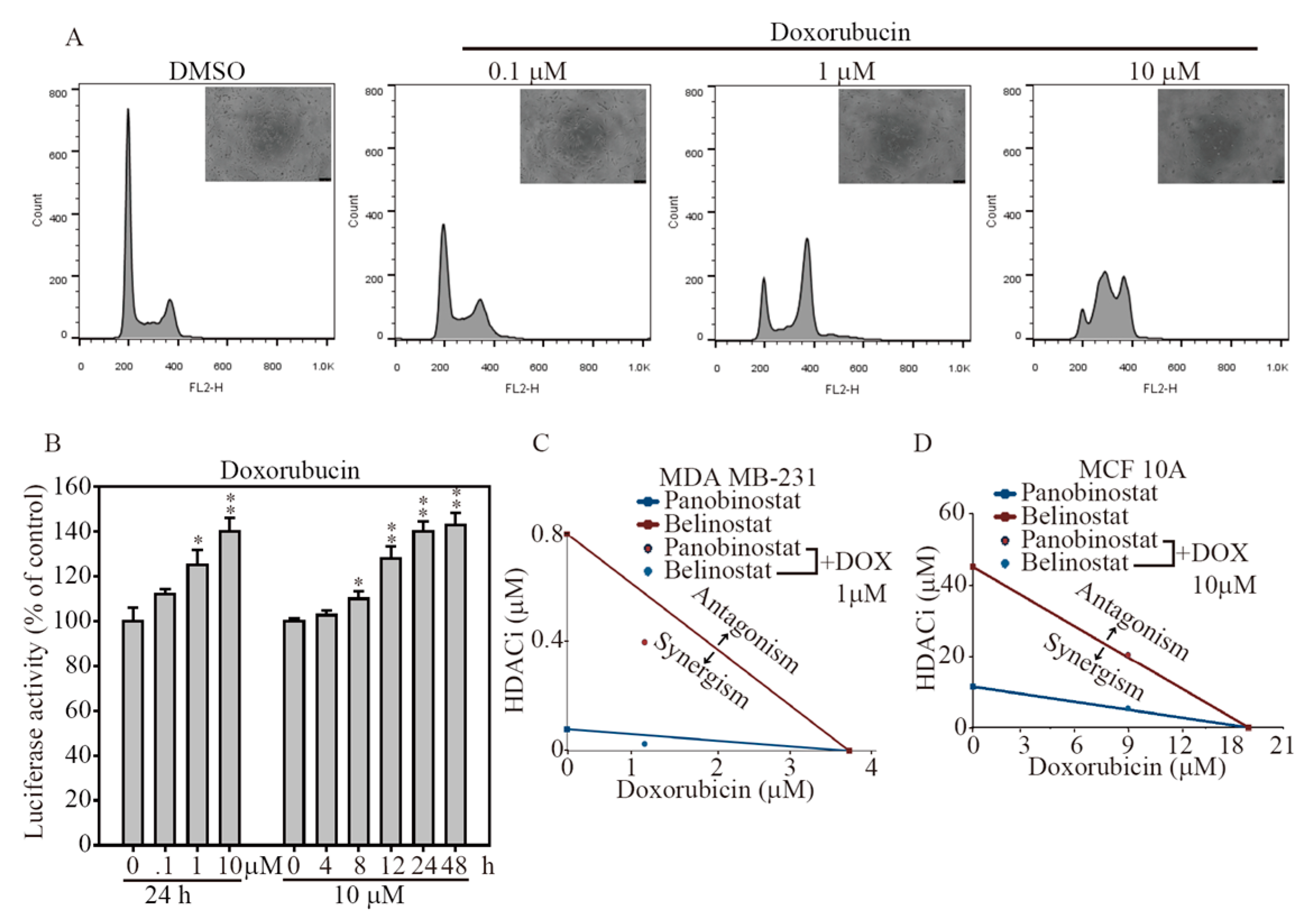
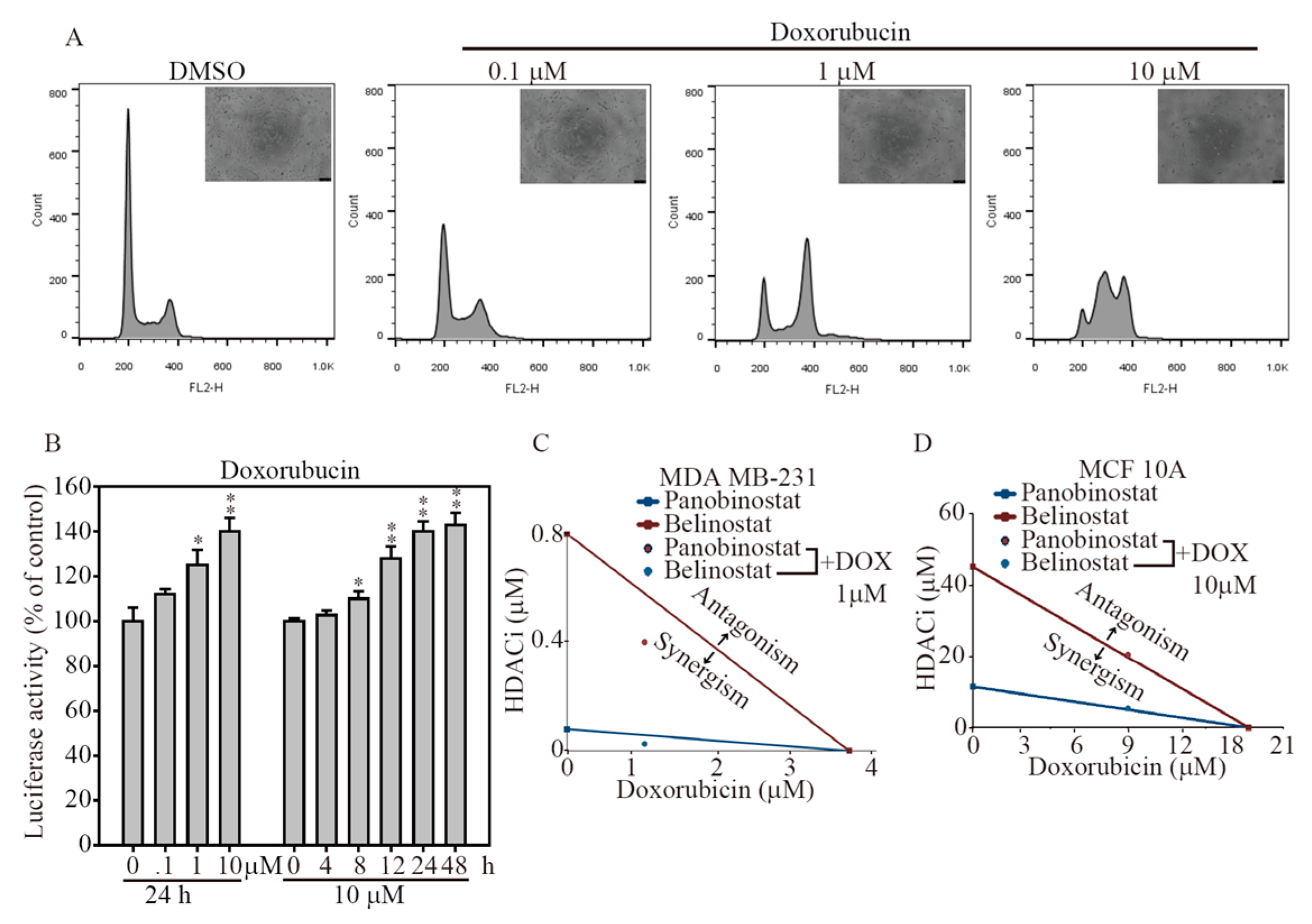
2.5. HDACi and Doxorubicin Combination Synergically-Induced Apoptosis Event
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Culture Conditions
4.2. Lentiviral Production of Non-Invasive Apoptosis Detection Sensor (NIADS) and Cell Transduction
4.3. Transfection and Cell Line Selection
4.4. Real-Time Quantitative Polymerase Chain Reaction (qPCR)
4.5. Absolute qPCR
4.6. Protein Extraction, Western Blotting, and Antibodies
4.7. MTT Viability Assay
4.8. Live/Dead Cell Assay
4.9. Cellular Bioluminescence Assay
4.10. Bioluminescence (IVIS) Imaging
4.11. Flow Cytometry Analysis
4.12. Statistical methods
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 6, a026831. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.Y.; Huang, H.W.; Shu, C.W.; Hou, M.F.; Yuan, S.S.; Wang, H.R.; Chang, Y.T.; Farooqi, A.A.; Tang, J.Y.; Chang, H.W. DNA methylation, histone acetylation and methylation of epigenetic modifications as a therapeutic approach for cancers. Cancer Lett. 2016, 373, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Rekowski, M.; Giannis, A. Histone acetylation modulation by small molecules: A chemical approach. Biochim. Biophys. Acta 2010, 1799, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Liang, G.; Weisenberger, D.J. DNA methylation aberrancies as a guide for surveillance and treatment of human cancers. Epigenetics 2017, 12, 416–432. [Google Scholar] [CrossRef] [PubMed]
- Waldmann, T.; Schneider, R. Targeting histone modifications—Epigenetics in cancer. Curr. Opin. Cell Biol. 2013, 25, 184–189. [Google Scholar] [CrossRef] [PubMed]
- Losson, H.; Schnekenburger, M.; Dicato, M.; Diederich, M. Natural Compound Histone Deacetylase Inhibitors (HDACi): Synergy with Inflammatory Signaling Pathway Modulators and Clinical Applications in Cancer. Molecules 2016, 21, 1608. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Mayer, I.A.; Diamond, J.R.; Moroose, R.L.; Isakoff, S.J.; Starodub, A.N.; Shah, N.C.; O’Shaughnessy, J.; Kalinsky, K.; Guarino, M.; et al. Efficacy and Safety of Anti-Trop-2 Antibody Drug Conjugate Sacituzumab Govitecan (IMMU-132) in Heavily Pretreated Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2017, 35, 2141–2148. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Teijido, P.; Cabal, M.L.; Fernandez, I.P.; Perez, Y.F. Tumor-Infiltrating Lymphocytes in Triple Negative Breast Cancer: The Future of Immune Targeting. Clin. Med. Insights Oncol. 2016, 10 (Suppl. 1), 31–39. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, S.; Lesniak, W.G.; Gabrielson, M.; Lisok, A.; Wharram, B.; Sysa-Shah, P.; Azad, B.B.; Pomper, M.G.; Nimmagadda, S. A humanized antibody for imaging immune checkpoint ligand PD-L1 expression in tumors. Oncotarget 2016, 7, 10215–10227. [Google Scholar] [CrossRef] [PubMed]
- Sellers, W.R.; Fisher, D.E. Apoptosis and cancer drug targeting. J. Clin. Investig. 1999, 104, 1655–1661. [Google Scholar] [CrossRef] [PubMed]
- Lakshmaiah, K.C.; Jacob, L.A.; Aparna, S.; Lokanatha, D.; Saldanha, S.C. Epigenetic therapy of cancer with histone deacetylase inhibitors. J. Cancer Res. Ther. 2014, 10, 469–478. [Google Scholar] [PubMed]
- Zhang, J.; Zhong, Q. Histone deacetylase inhibitors and cell death. Cell. Mol. Life Sci. 2014, 71, 3885–3901. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, B.; Evans, T.R. Developing histone deacetylase inhibitors as anti-cancer therapeutics. Curr. Med. Chem. 2011, 18, 1658–1671. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Harper, N.; Walewska, R.; Dyer, M.J.; Cohen, G.M. Enhanced Fas-associated death domain recruitment by histone deacetylase inhibitors is critical for the sensitization of chronic lymphocytic leukemia cells to TRAIL-induced apoptosis. Mol. Cancer Ther. 2009, 8, 3088–3097. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Kanakousaki, K.; Buttitta, L. How the cell cycle impacts chromatin architecture and influences cell fate. Front. Genet. 2015, 6, 19. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, D.; Avvakumov, N.; Cote, J. Histone phosphorylation: A chromatin modification involved in diverse nuclear events. Epigenetics 2012, 7, 1098–1108. [Google Scholar] [CrossRef] [PubMed]
- Chou, T.-C.; Talalay, P. Quantitative analysis of dose-effect relationships: The combined effects of multiple drugs or enzyme inhibitors. Adv. Enzyme Regul. 1984, 22, 27–55. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Pampfer, S. Apoptosis in rodent peri-implantation embryos: Differential susceptibility of inner cell mass and trophectoderm cell lineages—A review. Placenta 2000, 21 (Suppl. A), S3–S10. [Google Scholar] [CrossRef] [PubMed]
- Fan, F.; Wood, K.V. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007, 5, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, A.; Ji, K.; Sameni, M.; Mazumder, S.H.; Xu, Y.; Moin, K.; Sloane, B.F. Live-Cell Imaging of Protease Activity: Assays to Screen Therapeutic Approaches. Methods Mol. Biol. 2017, 1574, 215–225. [Google Scholar] [PubMed]
- Specht, E.A.; Braselmann, E.; Palmer, A.E. A Critical and Comparative Review of Fluorescent Tools for Live-Cell Imaging. Annu. Rev. Physiol. 2017, 79, 93–117. [Google Scholar] [CrossRef] [PubMed]
- McCann, T. Live cell imaging: An industrial perspective. Methods Mol. Biol. 2010, 591, 47–66. [Google Scholar] [PubMed]
- Joseph, J.; Seervi, M.; Sobhan, P.K.; Retnabai, S.T. High throughput ratio imaging to profile caspase activity: Potential application in multiparameter high content apoptosis analysis and drug screening. PLoS ONE 2011, 6, e20114. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W.; Ali, A.; Thornberry, N.A.; Vaillancourt, J.P.; Ding, C.K.; Gallant, M.; Gareau, Y.; Griffin, P.R.; Labelle, M.; Lazebnik, Y.A.; et al. Identification and inhibition of the ICE/CED-3 protease necessary for mammalian apoptosis. Nature 1995, 376, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Archana, M.; Bastian; Yogesh, T.L.; Kumaraswamy, K.L. Various methods available for detection of apoptotic cells—A review. Indian J. Cancer 2013, 50, 274–283. [Google Scholar] [PubMed]
- Thorburn, A. Apoptosis and autophagy: Regulatory connections between two supposedly different processes. Apoptosis 2008, 13, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Tsapras, P.; Nezis, I.P. Caspase involvement in autophagy. Cell Death Differ. 2017, 24, 1369–1379. [Google Scholar] [CrossRef] [PubMed]
- Gelles, J.D.; Chipuk, J.E. Robust high-throughput kinetic analysis of apoptosis with real-time high-content live-cell imaging. Cell Death Dis. 2016, 7, e2493. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.; Eom, G.H. HDAC and HDAC Inhibitor: From Cancer to Cardiovascular Diseases. Chonnam Med. J. 2016, 52, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Duvic, M.; Vu, J. Vorinostat: A new oral histone deacetylase inhibitor approved for cutaneous T-cell lymphoma. Expert Opin. Investig. Drugs 2007, 16, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Tu, S.H.; Hsieh, Y.C.; Huang, L.C.; Lin, C.Y.; Hsu, K.W.; Hsieh, W.S.; Chi, W.M.; Lee, C.H. A rapid and quantitative method to detect human circulating tumor cells in a preclinical animal model. BMC Cancer 2017, 17, 440. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Panobinostat | Belinostat | Vorinostat | Valproic Acid |
|---|---|---|---|---|
| MDA-MB-231 | 0.024 | 0.9 | 7.87 | >10 |
| SK-BR-3 | 0.117 | 1.41 | 8.94 | >10 |
| MCF-7 | 0.778 | 9.64 | >10 | >10 |
| MCF-10A | >10 | >10 | >10 | >10 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hsu, K.-W.; Huang, C.-Y.; Tam, K.-W.; Lin, C.-Y.; Huang, L.-C.; Lin, C.-L.; Hsieh, W.-S.; Chi, W.-M.; Chang, Y.-J.; Wei, P.-L.; et al. The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death. Int. J. Mol. Sci. 2018, 19, 452. https://doi.org/10.3390/ijms19020452
Hsu K-W, Huang C-Y, Tam K-W, Lin C-Y, Huang L-C, Lin C-L, Hsieh W-S, Chi W-M, Chang Y-J, Wei P-L, et al. The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death. International Journal of Molecular Sciences. 2018; 19(2):452. https://doi.org/10.3390/ijms19020452
Chicago/Turabian StyleHsu, Kai-Wen, Chien-Yu Huang, Ka-Wai Tam, Chun-Yu Lin, Li-Chi Huang, Ching-Ling Lin, Wen-Shyang Hsieh, Wei-Ming Chi, Yu-Jia Chang, Po-Li Wei, and et al. 2018. "The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death" International Journal of Molecular Sciences 19, no. 2: 452. https://doi.org/10.3390/ijms19020452
APA StyleHsu, K.-W., Huang, C.-Y., Tam, K.-W., Lin, C.-Y., Huang, L.-C., Lin, C.-L., Hsieh, W.-S., Chi, W.-M., Chang, Y.-J., Wei, P.-L., Chen, S.-T., & Lee, C.-H. (2018). The Application of Non-Invasive Apoptosis Detection Sensor (NIADS) on Histone Deacetylation Inhibitor (HDACi)-Induced Breast Cancer Cell Death. International Journal of Molecular Sciences, 19(2), 452. https://doi.org/10.3390/ijms19020452