Novel Bacterial Topoisomerase Inhibitors Exploit Asp83 and the Intrinsic Flexibility of the DNA Gyrase Binding Site

 and
and
Abstract
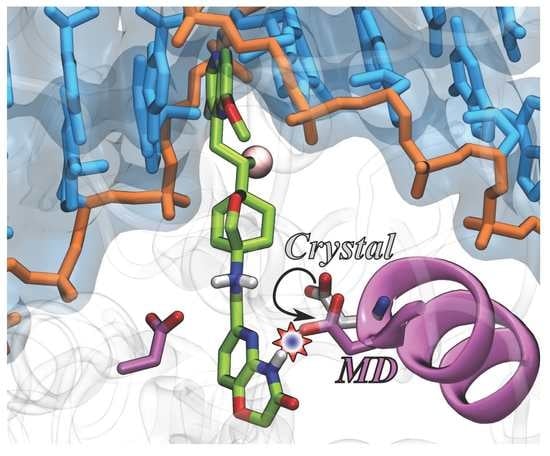
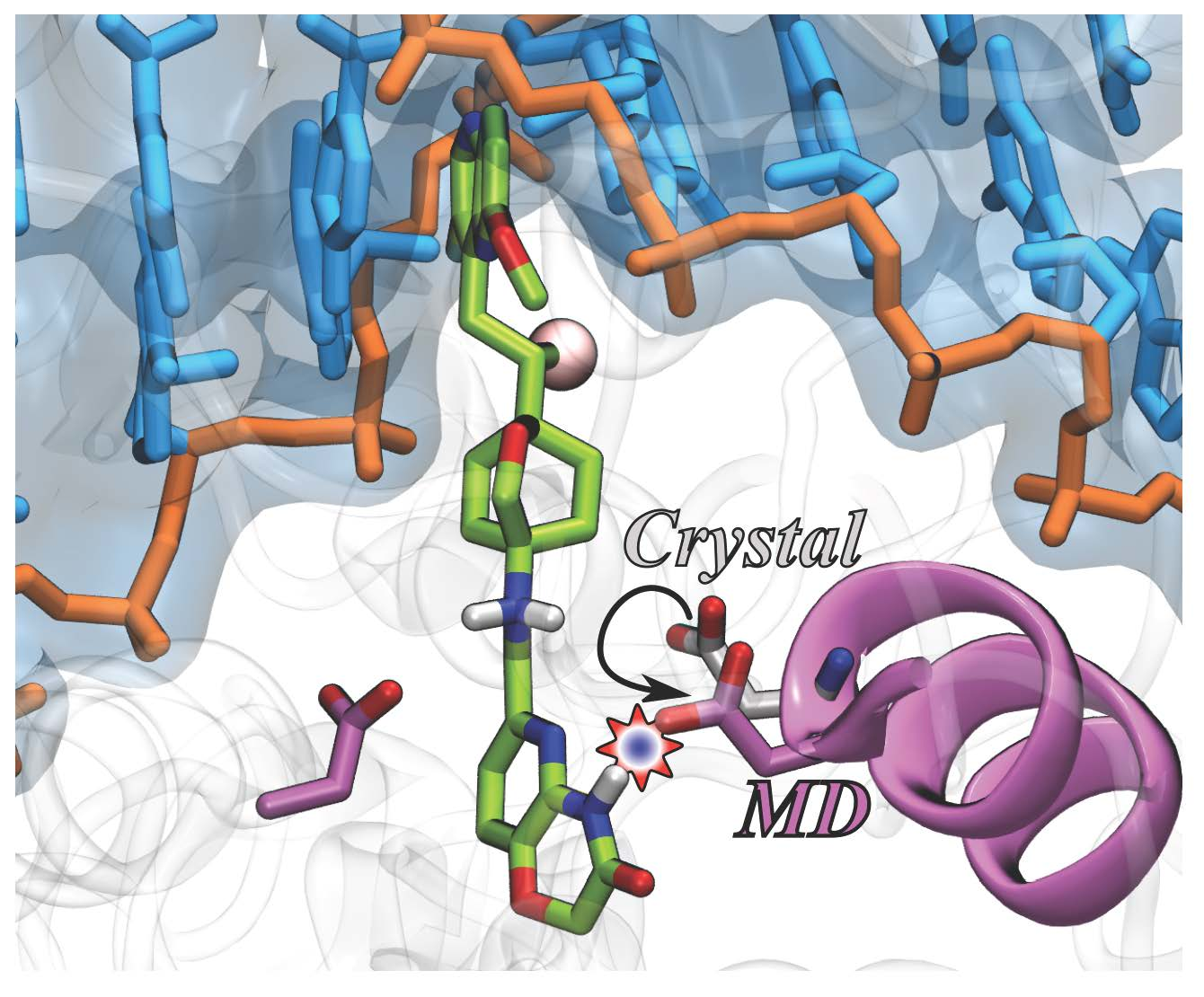
:
{kind=link}
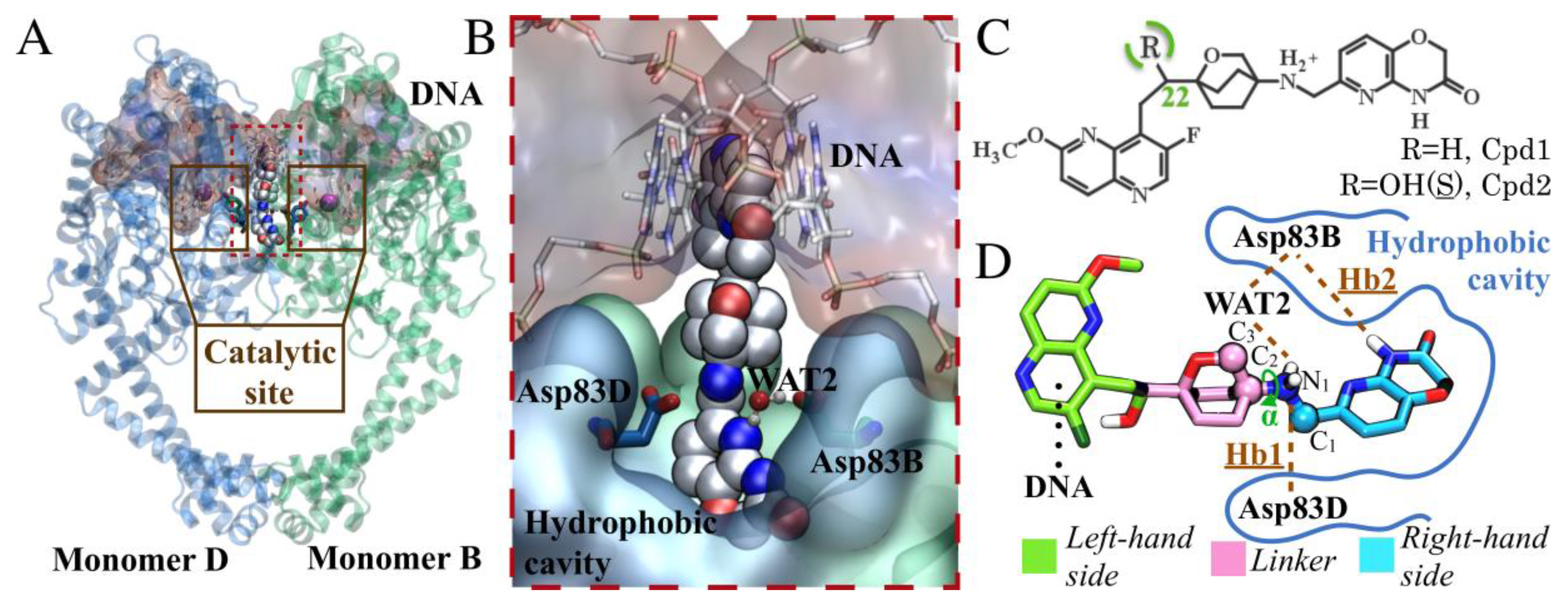{kind=link}
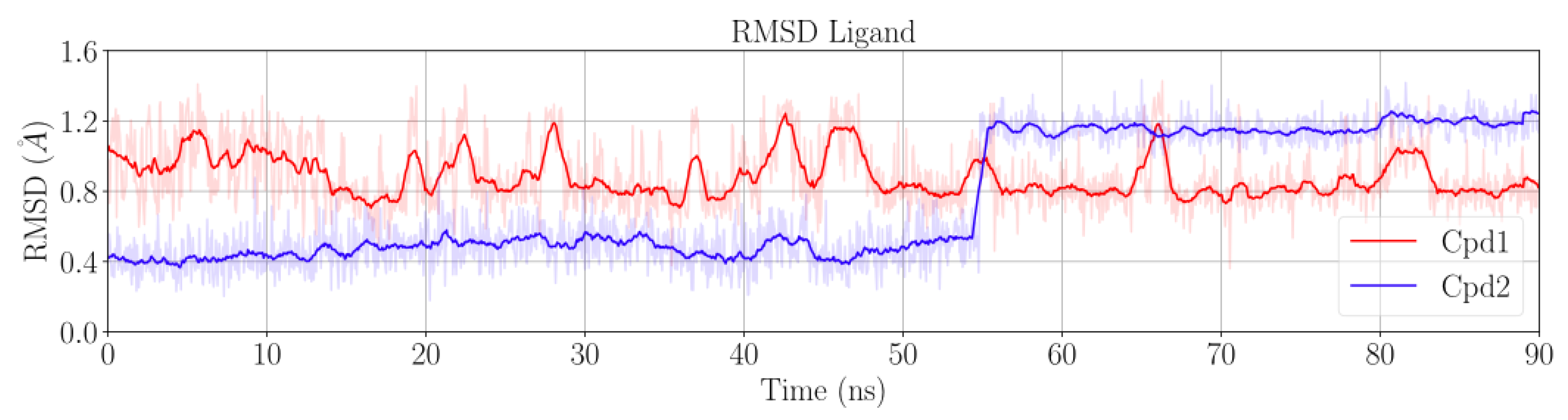{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
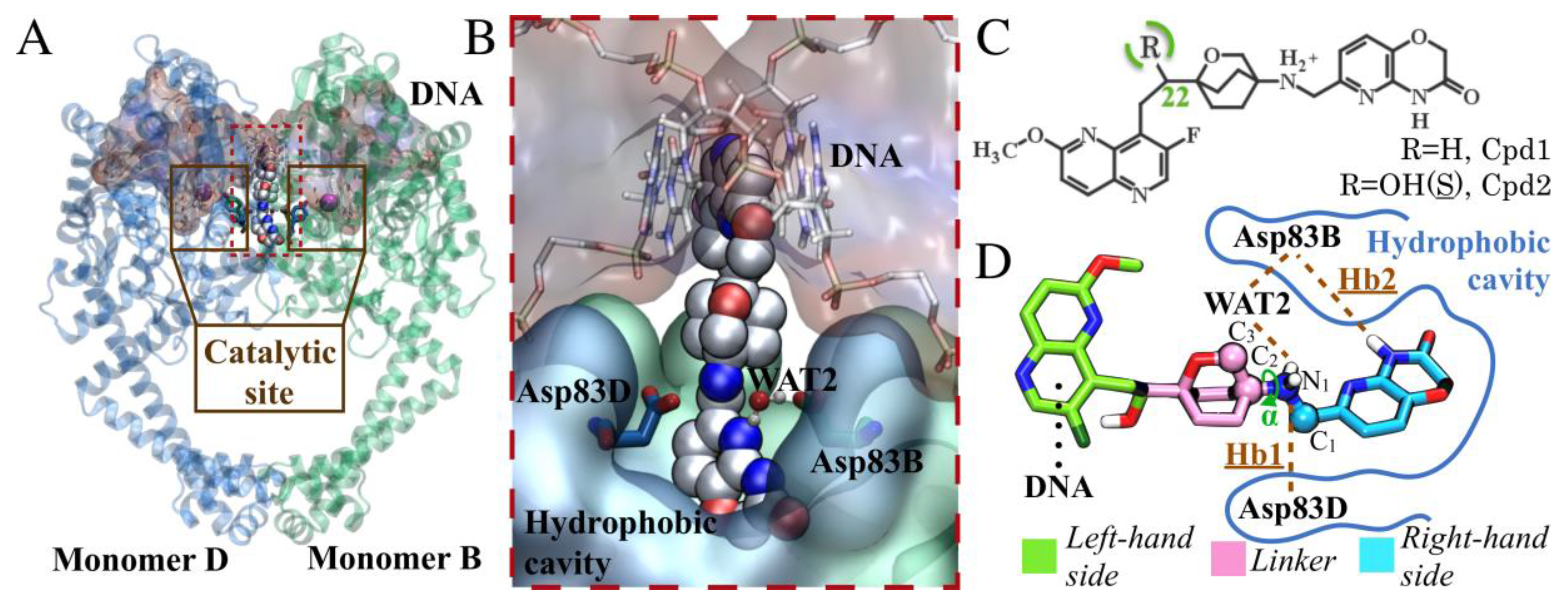
2. Results and Discussion
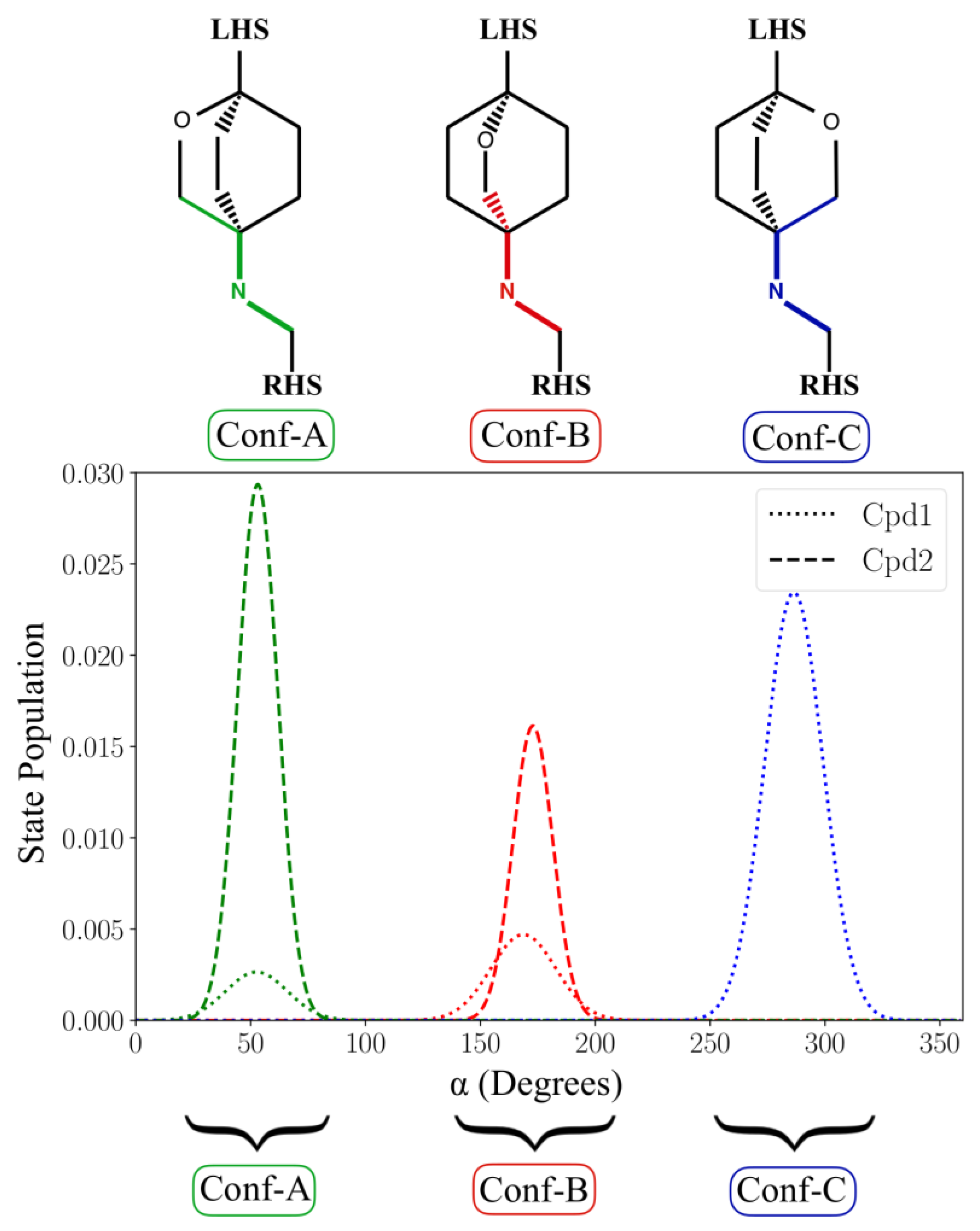
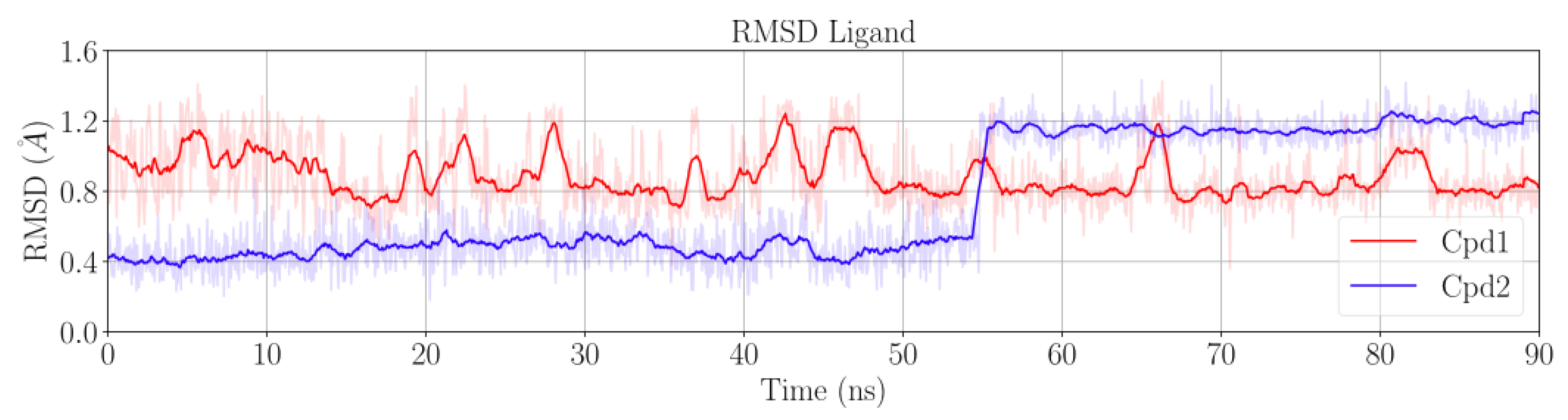
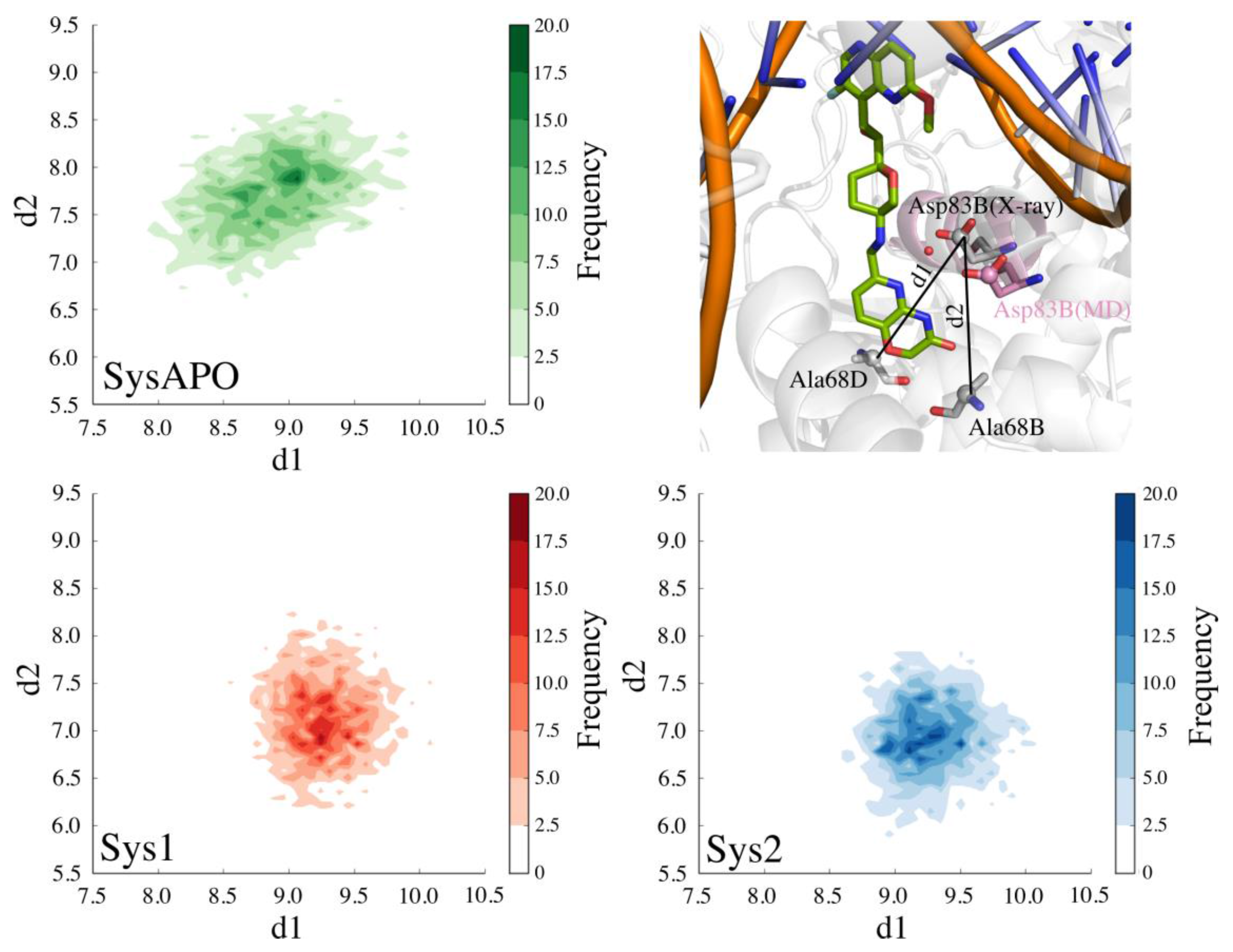
2.1. MD Simulations for NBTI Binding
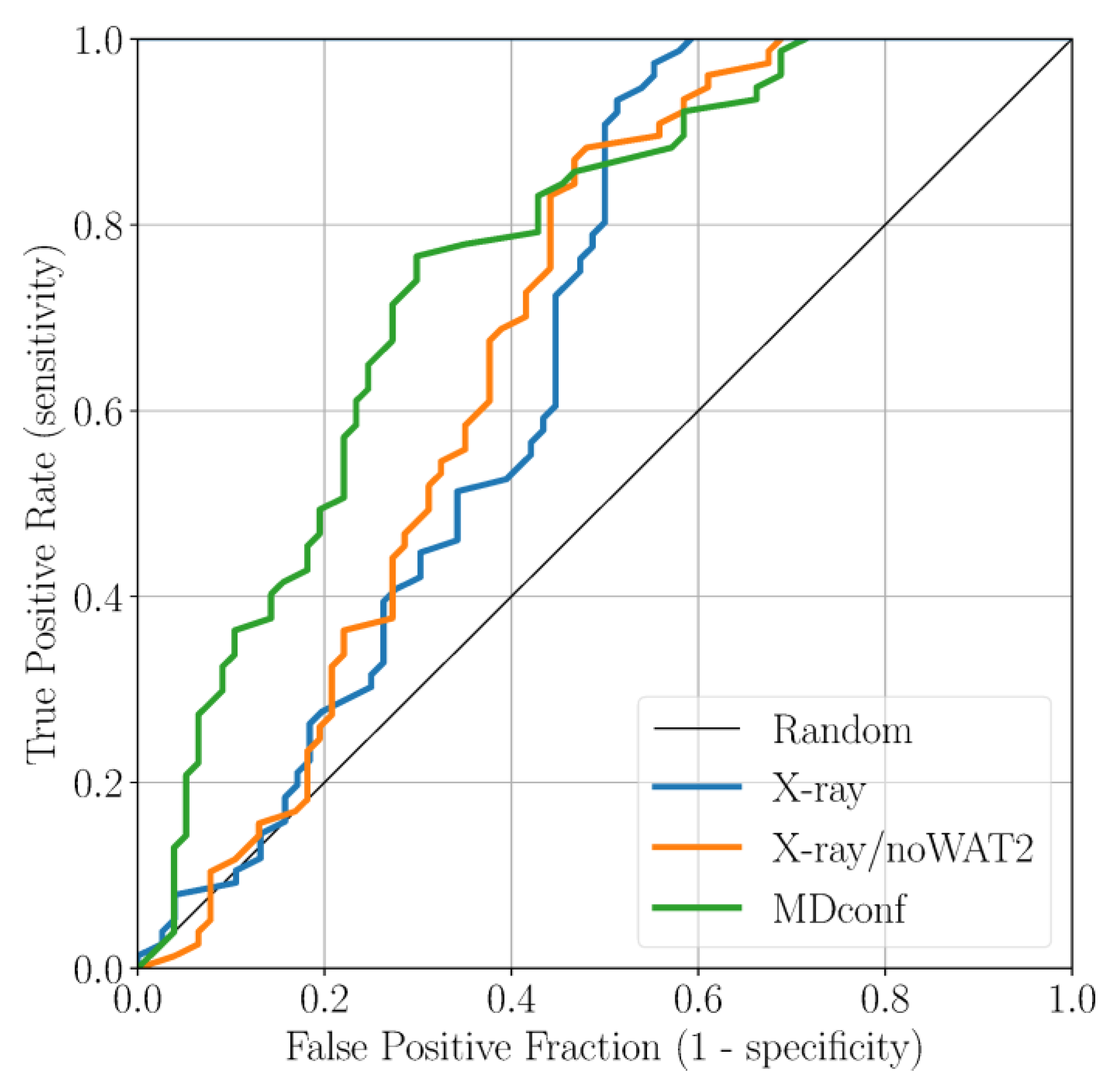
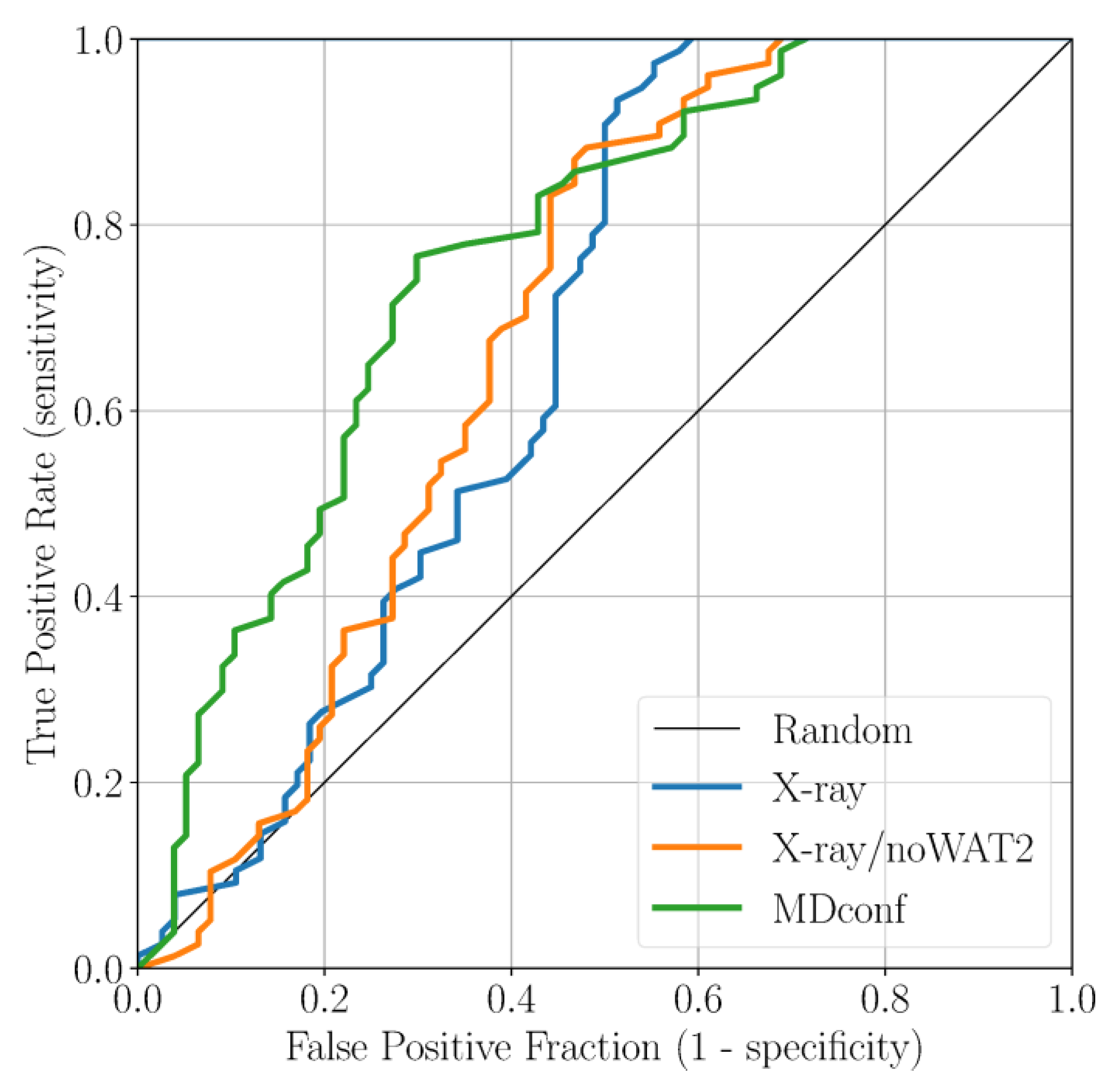
2.2. Accounting for Asp83B Flexibility and H-Bond Formation in Docking Calculations
3. Materials and Methods
3.1. Systems Preparation and MD Simulations
3.2. Analysis of the MD Simulations
3.3. Protein Preparation and Building of the Training Set
3.4. Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Minniti, E.; Greco, M.L.; Riccardi, L.; Simoni, E.; Convertino, M.; Marchetti, C.; Rosini, M.; Sissi, C.; Minarini, A.; et al. An optimized polyamine moiety boosts the potency of human type II topoisomerase poisons as quantified by comparative analysis centered on the clinical candidate F14512. Chem. Commun. 2015, 51, 14310–14313. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, M.; Unniraman, S.; Mahadevan, S.; Nagaraja, V. Effect of different classes of inhibitors on DNA gyrase from Mycobacterium smegmatis. J. Antimicrob. Chemother. 2001, 48, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Rajendram, M.; Hurley, K.A.; Foss, M.H.; Thornton, K.M.; Moore, J.T.; Shaw, J.T.; Weibel, D.B. Gyramides prevent bacterial growth by inhibiting DNA gyrase and altering chromosome topology. ACS Chem. Biol. 2014, 9, 1312–1319. [Google Scholar] [CrossRef] [PubMed]
- Basarab, G.S.; Hill, P.J.; Garner, C.E.; Hull, K.; Green, O.; Sherer, B.A.; Dangel, P.B.; Manchester, J.I.; Bist, S.; Hauck, S.; et al. Optimization of pyrrolamide topoisomerase II inhibitors toward identification of an antibacterial clinical candidate (AZD5099). J. Med. Chem. 2014, 57, 6060–6082. [Google Scholar] [CrossRef] [PubMed]
- Minniti, E.; Byl, J.A.W.; Riccardi, L.; Sissi, C.; Rosini, M.; de Vivo, M.; Minarini, A.; Osheroff, N. Novel xanthone-polyamine conjugates as catalytic inhibitors of human topoisomerase IIα. Bioorg. Med. Chem. Lett. 2017, 27, 4687–4693. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.A.; Riccardi, L.; Minniti, E.; Borgogno, M.; Arencibia, J.M.; Greco, M.L.; Minarini, A.; Sissi, C.; de Vivo, M. Pharmacophore hybridization to discover novel Topoisomerase II poisons with promising antiproliferative activity. J. Med. Chem. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fisher, L.M.; Pan, X.S. Methods to assay inhibitors of DNA gyrase and topoisomerase IV activities. Methods Mol. Med. 2008, 142, 11–23. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, V.; Godbole, A.A.; Henderson, S.R.; Maxwell, A. DNA topoisomerase I and DNA gyrase as targets for TB therapy. Drug Discov. Today 2017, 22, 510–518. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Srikannathasan, V.; Huang, J.; Cui, H.; Fosberry, A.P.; Gu, M.; Hann, M.M.; Hibbs, M.; Homes, P.; Ingraham, K.; et al. Structural basis of DNA gyrase inhibition by antibacterial QPT-1, anticancer drug etoposide and moxifloxacin. Nat. Commun. 2015, 6, 10048. [Google Scholar] [CrossRef] [PubMed]
- Heeb, S.; Fletcher, M.P.; Chhabra, S.R.; Diggle, S.P.; Williams, P.; Cámara, M. Quinolones: From antibiotics to autoinducers. FEMS Microbiol. Rev. 2011, 35, 247–274. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Kerns, R.J.; Osheroff, N. Mechanism of quinolone action and resistance. Biochemistry 2014, 53, 1565–1574. [Google Scholar] [CrossRef] [PubMed]
- Aldred, K.J.; Schwanz, H.A.; Li, G.; McPherson, S.A.; Turnbough, C.L.; Kerns, R.J.; Osheroff, N. Overcoming target-mediated quinolone resistance in topoisomerase IV by introducing metal-ion-independent drug-enzyme interactions. ACS Chem. Biol. 2013, 8, 2660–2668. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.; Janin, Y.L. Non-quinolone inhibitors of bacterial type IIA topoisomerases: A feat of bioisosterism. Chem. Rev. 2014, 114, 2313–2342. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B. Confronting the challenges of discovery of novel antibacterial agents. Bioorg. Med. Chem. Lett. 2014, 24, 3683–3689. [Google Scholar] [CrossRef] [PubMed]
- Fournier, B.; Hooper, D.C. Mutations in topoisomerase IV and DNA gyrase of Staphylococcus aureus: Novel pleiotropic effects on quinolone and coumarin activity. Antimicrob. Agents Chemother. 1998, 42, 121–128. [Google Scholar] [PubMed]
- Basarab, G.S.; Kern, G.H.; McNulty, J.; Mueller, J.P.; Lawrence, K.; Vishwanathan, K.; Alm, R.A.; Barvian, K.; Doig, P.; Galullo, V.; et al. Responding to the challenge of untreatable gonorrhea: ETX0914, a first-in-class agent with a distinct mechanism-of-action against bacterial type II topoisomerases. Sci. Rep. 2015, 5, 11827. [Google Scholar] [CrossRef] [PubMed]
- Collin, F.; Karkare, S.; Maxwell, A. Exploiting bacterial DNA gyrase as a drug target: Current state and perspectives. Appl. Microbiol. Biotechnol. 2011, 92, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Charrier, C.; Salisbury, A.-M.; Savage, V.J.; Duffy, T.; Moyo, E.; Chaffer-Malam, N.; Ooi, N.; Newman, R.; Cheung, J.; Metzger, R.; et al. Novel bacterial topoisomerase inhibitors with potent broad-spectrum activity against drug-resistant bacteria. Antimicrob. Agents Chemother. 2017, 61, e02100-16. [Google Scholar] [CrossRef] [PubMed]
- Kolaric, A.; Minovski, N. Structure-based design of novel combinatorially generated NBTIs as potential DNA gyrase inhibitors against various Staphylococcus aureus mutant strains. Mol. Biosyst. 2017, 13, 1406–1420. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; Bradley, P.; Lu, J.; et al. Oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad spectrum antibacterial agents. ACS Med. Chem. Lett. 2014, 5, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Coates, W.J.; Gwynn, M.N.; Hatton, I.K.; Masters, P.J.; Pearson, N.D.; Rahman, S.S.; Slocombe, B.; Warrack, J.D. Quinolone Derivatives as Antibacterials. Patent EP1051413 (A1), 15 November 2000. [Google Scholar]
- Bax, B.D.; Chan, P.F.; Eggleston, D.S.; Fosberry, A.P.; Gentry, D.R.; Gorrec, F.; Giordano, I.; Hann, M.M.; Hennessy, A.J.; Hibbs, M.; et al. Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nature 2010, 466, 935–940. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; Wei, C.; Liao, Y.; et al. C1-C2-linker substituted 1,5-naphthyridine analogues of oxabicyclooctane-linked NBTIs as broad-spectrum antibacterial agents (part 7). Med. Chem. Commun. 2015, 6, 1773–1780. [Google Scholar] [CrossRef]
- Tan, C.M.; Gill, C.J.; Wu, J.; Toussaint, N.; Yin, J.; Tsuchiya, T.; Garlisi, C.G.; Kaelin, D.E.; Meinke, P.T.; Miesel, L.; et al. In vitro and in vivo characterization of the novel oxabicyclooctane-linked bacterial topoisomerase inhibitor AM-8722, a selective, potent inhibitor of bacterial DNA gyrase. Antimicrob. Agents Chemother. 2016, 60, 4830–4839. [Google Scholar] [CrossRef] [PubMed]
- Hameed, S.P.; Raichurkar, A.; Madhavapeddi, P.; Menasinakai, S.; Sharma, S.; Kaur, P.; Nandishaiah, R.; Panduga, V.; Reddy, J.; Sambandamurthy, V.K.; et al. Benzimidazoles: Novel mycobacterial gyrase inhibitors from scaffold morphing. ACS Med. Chem. Lett. 2014, 5, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.J.; Hennessy, A.J.; Bax, B.D.; Brooks, G.; Brown, B.S.; Brown, P.; Cailleau, N.; Chen, D.; Dabbs, S.; Davies, D.T.; et al. Novel hydroxyl tricyclics (e.g., GSK966587) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2013, 23, 5437–5441. [Google Scholar] [CrossRef] [PubMed]
- Miles, T.J.; Hennessy, A.J.; Bax, B.D.; Brooks, G.; Brown, B.S.; Brown, P.; Cailleau, N.; Chen, D.; Dabbs, S.; Davies, D.T.; et al. Novel tricyclics (e.g., GSK945237) as potent inhibitors of bacterial type IIA topoisomerases. Bioorg. Med. Chem. Lett. 2016, 26, 2464–2469. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.F.; Germe, T.; Bax, B.D.; Huang, J.; Thalji, R.K.; Bacqué, E.; Checchia, A.; Chen, D.; Cui, H.; Ding, X.; et al. Thiophene antibacterials that allosterically stabilize DNA-cleavage complexes with DNA gyrase. Proc. Natl. Acad. Sci. USA 2017, 114, E4492–E4500. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Black, T.; Nargund, R.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; et al. Tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of left-hand-side moiety (Part-2). Bioorg. Med. Chem. Lett. 2015, 25, 1831–1835. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Meinke, P.T.; Wu, J.; Miesel, L.; Tan, C.M.; Olsen, D.B.; Lagrutta, A.; Fukuda, H.; Kishii, R.; et al. Structure activity relationship of C-2 ether substituted 1,5-naphthyridine analogs of oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part-5). Bioorg. Med. Chem. Lett. 2015, 25, 3630–3635. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; Wei, C.; Liao, Y.; et al. Structure activity relationship of pyridoxazinone substituted RHS analogs of oxabicyclooctane-linked 1,5-naphthyridinyl novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part-6). Bioorg. Med. Chem. Lett. 2015, 25, 3636–3643. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Gill, C.J.; Black, T.; Nargund, R.; Meinke, P.T.; Olsen, D.B.; et al. Hydroxy tricyclic 1,5-naphthyridinone oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents-SAR of RHS moiety (Part-3). Bioorg. Med. Chem. Lett. 2015, 25, 2473–2478. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.B.; Kaelin, D.E.; Wu, J.; Miesel, L.; Tan, C.M.; Meinke, P.T.; Olsen, D.B.; Lagrutta, A.; Wei, C.; Peng, X.; et al. Structure activity relationship of substituted 1,5-naphthyridine analogs of oxabicyclooctane-linked novel bacterial topoisomerase inhibitors as broad-spectrum antibacterial agents (Part-4). Bioorg. Med. Chem. Lett. 2015, 25, 2409–2415. [Google Scholar] [CrossRef] [PubMed]
- La Sala, G.; Riccardi, L.; Gaspari, R.; Cavalli, A.; Hantschel, O.; De Vivo, M. HRD motif as the central hub of the signaling network for activation loop autophosphorylation in Abl kinase. J. Chem. Theory Comput. 2016, 12, 5563–5574. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Bauer, I.; Campomanes, P.; Cavalli, A.; Armirotti, A.; Girotto, S.; Rothlisberger, U.; de Vivo, M. Keys to lipid selection in fatty acid amide hydrolase catalysis: Structural flexibility, gating residues and multiple binding pockets. PLoS Comput. Biol. 2015, 11, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Rothlisberger, U.; Cavalli, A.; De Vivo, M. Computational insights into function and inhibition of fatty acid amide hydrolase. Eur. J. Med. Chem. 2015, 91, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Veselkov, D.A.; Laponogov, I.; Pan, X.S.; Selvarajah, J.; Skamrova, G.B.; Branstrom, A.; Narasimhan, J.; Prasad, J.V.N.V.; Fisher, L.M.; Sanderson, M.R. Structure of a quinolone-stabilized cleavage complex of topoisomerase IV from Klebsiella pneumoniae and comparison with a related Streptococcus pneumoniae complex. Acta Crystallogr. Sect. D Struct. Biol. 2016, 72, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Piton, J.; Petrella, S.; Delarue, M.; André-Leroux, G.; Jarlier, V.; Aubry, A.; Mayer, C. Structural insights into the quinolone resistance mechanism of Mycobacterium tuberculosis DNA gyrase. PLoS ONE 2010, 5, e12245. [Google Scholar] [CrossRef] [PubMed]
- Lahiri, S.D.; Kutschke, A.; McCormack, K.; Alm, R.A. Insights into the mechanism of inhibition of novel bacterial topoisomerase inhibitors from characterization of resistant mutants of Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 5278–5287. [Google Scholar] [CrossRef] [PubMed]
- De Vivo, M.; Cavalli, A. Recent advances in dynamic docking for drug discovery. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2017, 7, e1320. [Google Scholar] [CrossRef]
- De Vivo, M.; Masetti, M.; Bottegoni, G.; Cavalli, A. Role of molecular dynamics and related methods in drug discovery. J. Med. Chem. 2016, 59, 4035–4061. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.P.; Friesner, R.A.; Xiang, Z.; Honig, B. On the Role of the Crystal Environment in Determining Protein Side-chain Conformations. J. Mol. Biol. 2002, 320, 597–608. [Google Scholar] [CrossRef]
- Jacobson, M.P.; Pincus, D.L.; Rapp, C.S.; Day, T.J.F.; Honig, B.; Shaw, D.E.; Friesner, R.A. A hierarchical approach to all-atom protein loop prediction. Proteins Struct. Funct. Bioinform. 2004, 55, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A.; Murphy, R.B.; Friesner, R.A.; Beard, H.S.; Frye, L.L.; Pollard, W.T.; Banks, J.L. Glide: A new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef] [PubMed]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollmann, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Palermo, G.; Campomanes, P.; Neri, M.; Piomelli, D.; Cavalli, A.; Rothlisberger, U.; de Vivo, M. Wagging the tail: Essential role of substrate flexibility in FAAH catalysis. J. Chem. Theory Comput. 2013, 9, 1202–1213. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, L.; Arencibia, J.M.; Bono, L.; Armirotti, A.; Girotto, S.; de Vivo, M. Lid domain plasticity and lipid flexibility modulate enzyme specificity in human monoacylglycerol lipase. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Sastry, G.M.; Adzhigirey, M.; Day, T.; Annabhimoju, R.; Sherman, W. Protein and ligand preparation: Parameters, protocols, and influence on virtual screening enrichments. J. Comput. Aided Mol. Des. 2013, 27, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.H.M.; Søndergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Banks, J.L.; Beard, H.S.; Cao, Y.; Cho, A.E.; Damm, W.; Farid, R.; Felts, A.K.; Halgren, T.A.; Mainz, D.T.; Maple, J.R.; et al. Integrated modeling program, applied chemical theory (IMPACT). J. Comput. Chem. 2005, 26, 1752–1780. [Google Scholar] [CrossRef] [PubMed]
- Shelley, J.C.; Cholleti, A.; Frye, L.L.; Greenwood, J.R.; Timlin, M.R.; Uchimaya, M. Epik: A software program for pKa prediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 2007, 21, 681–691. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, J.R.; Calkins, D.; Sullivan, A.P.; Shelley, J.C. Towards the comprehensive, rapid, and accurate prediction of the favorable tautomeric states of drug-like molecules in aqueous solution. J. Comput. Aided Mol. Des. 2010, 24, 591–604. [Google Scholar] [CrossRef] [PubMed]
- Truchon, J.F.; Bayly, C.I. Evaluating virtual screening methods: Good and bad metrics for the “early recognition” problem. J. Chem. Inf. Model. 2007, 47, 488–508. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Hevener, K.E.; White, S.W.; Lee, R.E.; Boyett, J.M. A statistical framework to evaluate virtual screening. BMC Bioinform. 2009, 10, 225. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco-Ulloa, S.; La Sala, G.; Miscione, G.P.; De Vivo, M. Novel Bacterial Topoisomerase Inhibitors Exploit Asp83 and the Intrinsic Flexibility of the DNA Gyrase Binding Site. Int. J. Mol. Sci. 2018, 19, 453. https://doi.org/10.3390/ijms19020453
Franco-Ulloa S, La Sala G, Miscione GP, De Vivo M. Novel Bacterial Topoisomerase Inhibitors Exploit Asp83 and the Intrinsic Flexibility of the DNA Gyrase Binding Site. International Journal of Molecular Sciences. 2018; 19(2):453. https://doi.org/10.3390/ijms19020453
Chicago/Turabian StyleFranco-Ulloa, Sebastian, Giuseppina La Sala, Gian Pietro Miscione, and Marco De Vivo. 2018. "Novel Bacterial Topoisomerase Inhibitors Exploit Asp83 and the Intrinsic Flexibility of the DNA Gyrase Binding Site" International Journal of Molecular Sciences 19, no. 2: 453. https://doi.org/10.3390/ijms19020453
APA StyleFranco-Ulloa, S., La Sala, G., Miscione, G. P., & De Vivo, M. (2018). Novel Bacterial Topoisomerase Inhibitors Exploit Asp83 and the Intrinsic Flexibility of the DNA Gyrase Binding Site. International Journal of Molecular Sciences, 19(2), 453. https://doi.org/10.3390/ijms19020453