The Design and Development of Potent Small Molecules as Anticancer Agents Targeting EGFR TK and Tubulin Polymerization



Abstract
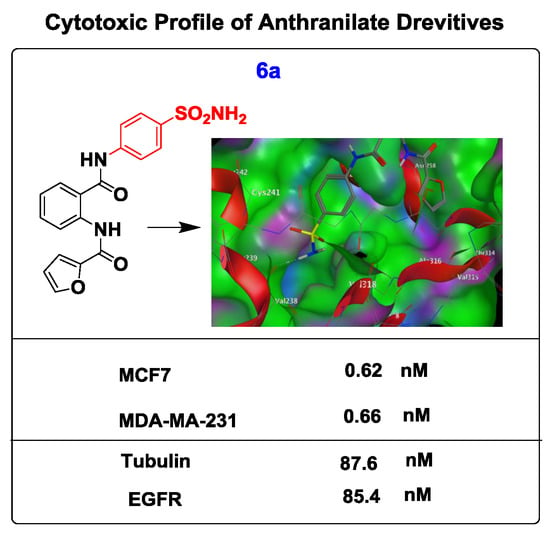
:
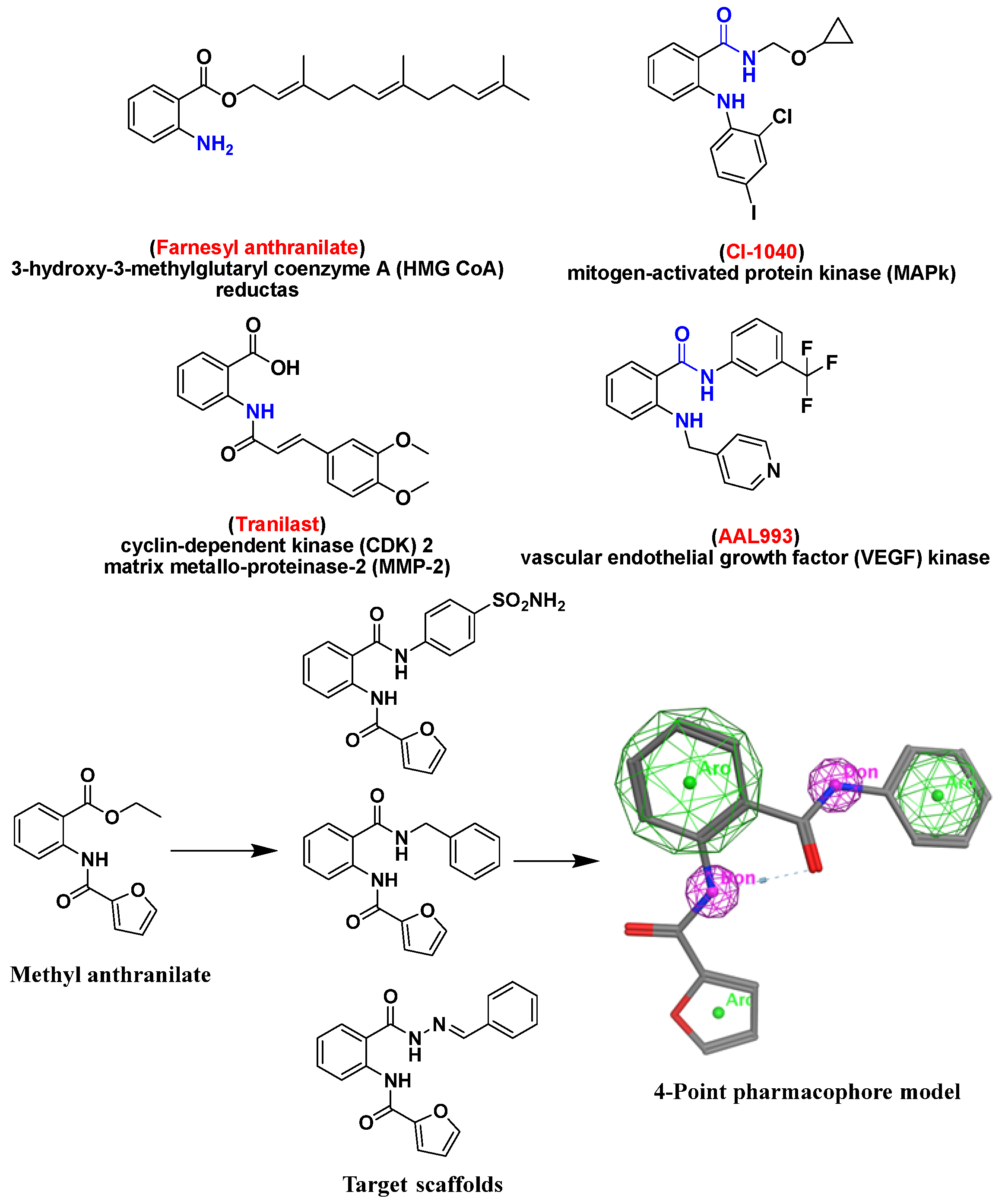
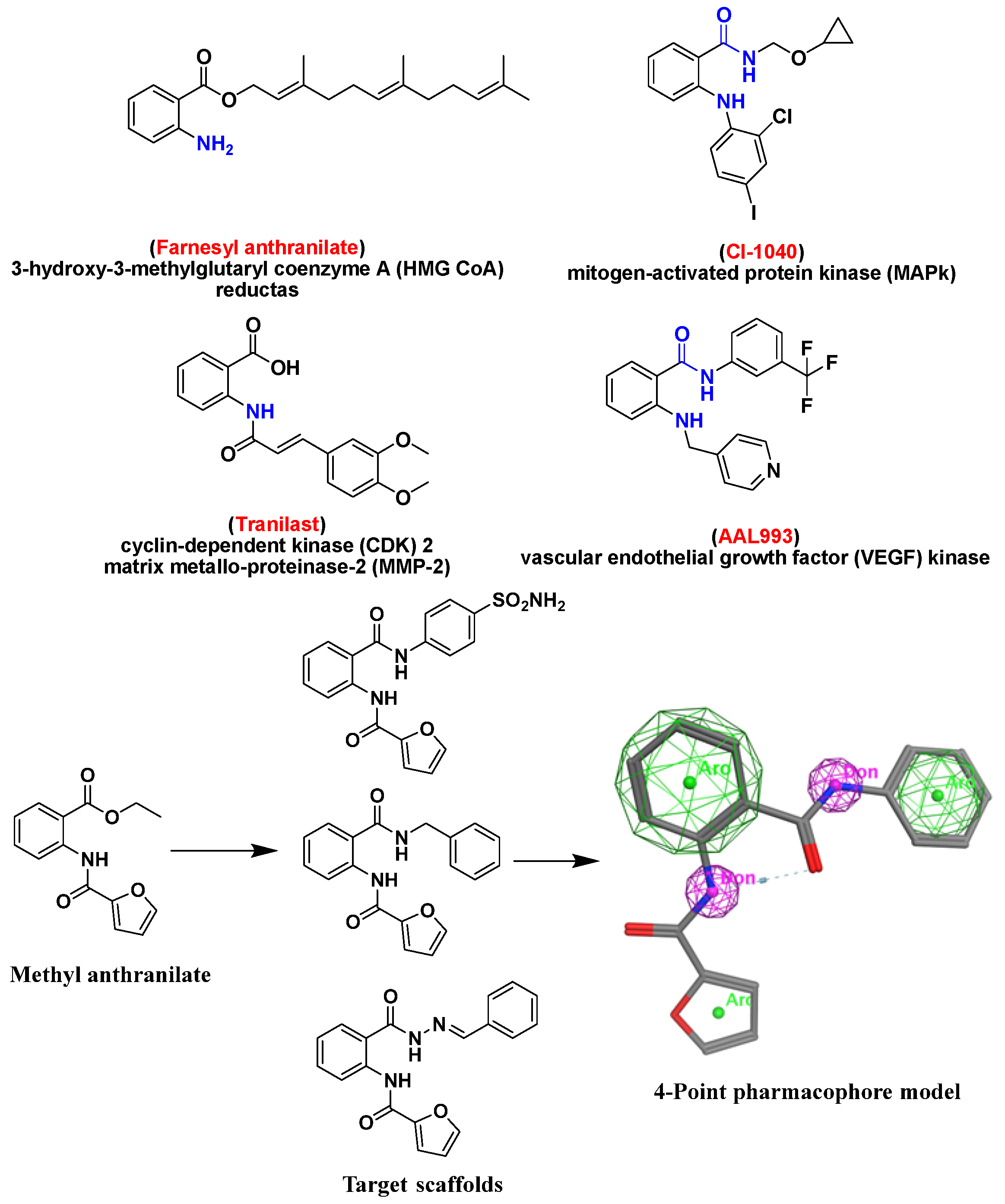
1. Introduction
2. Results and Discussion
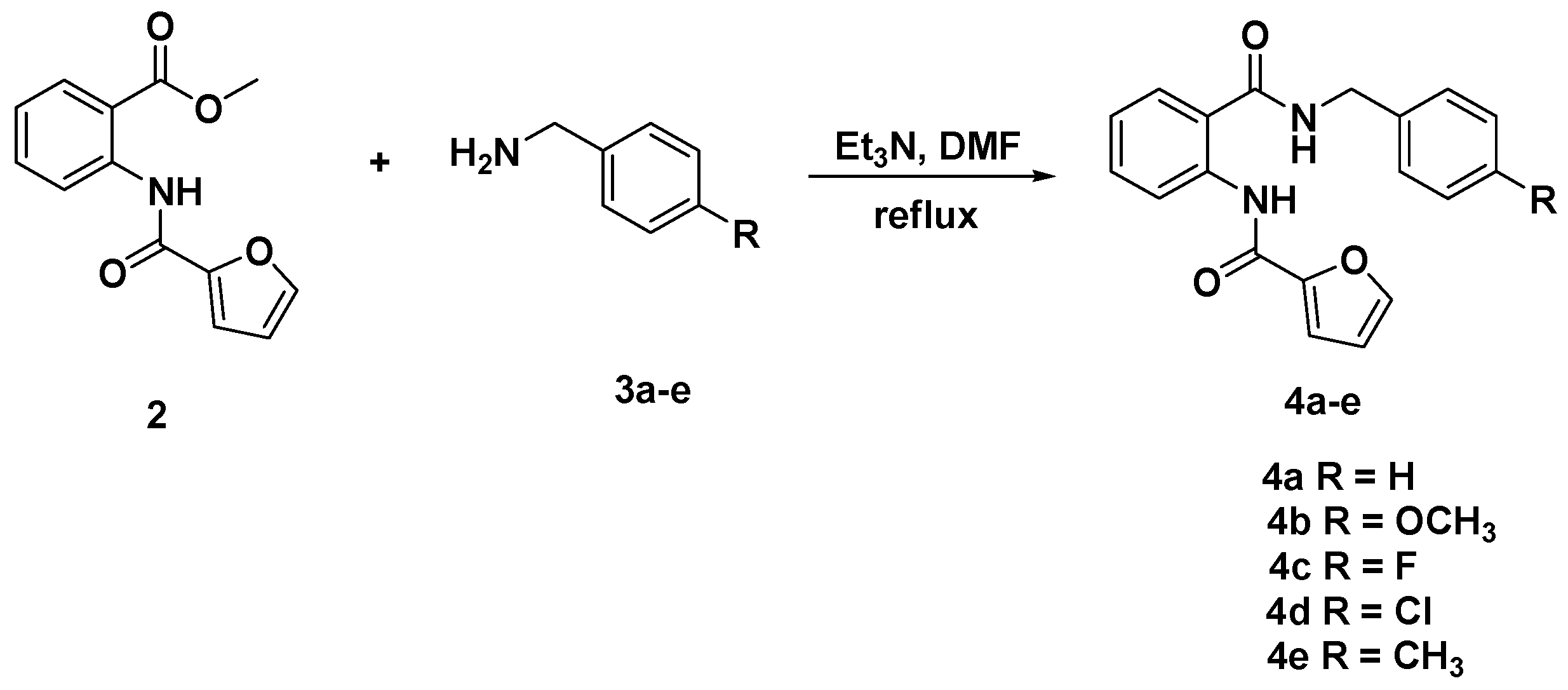
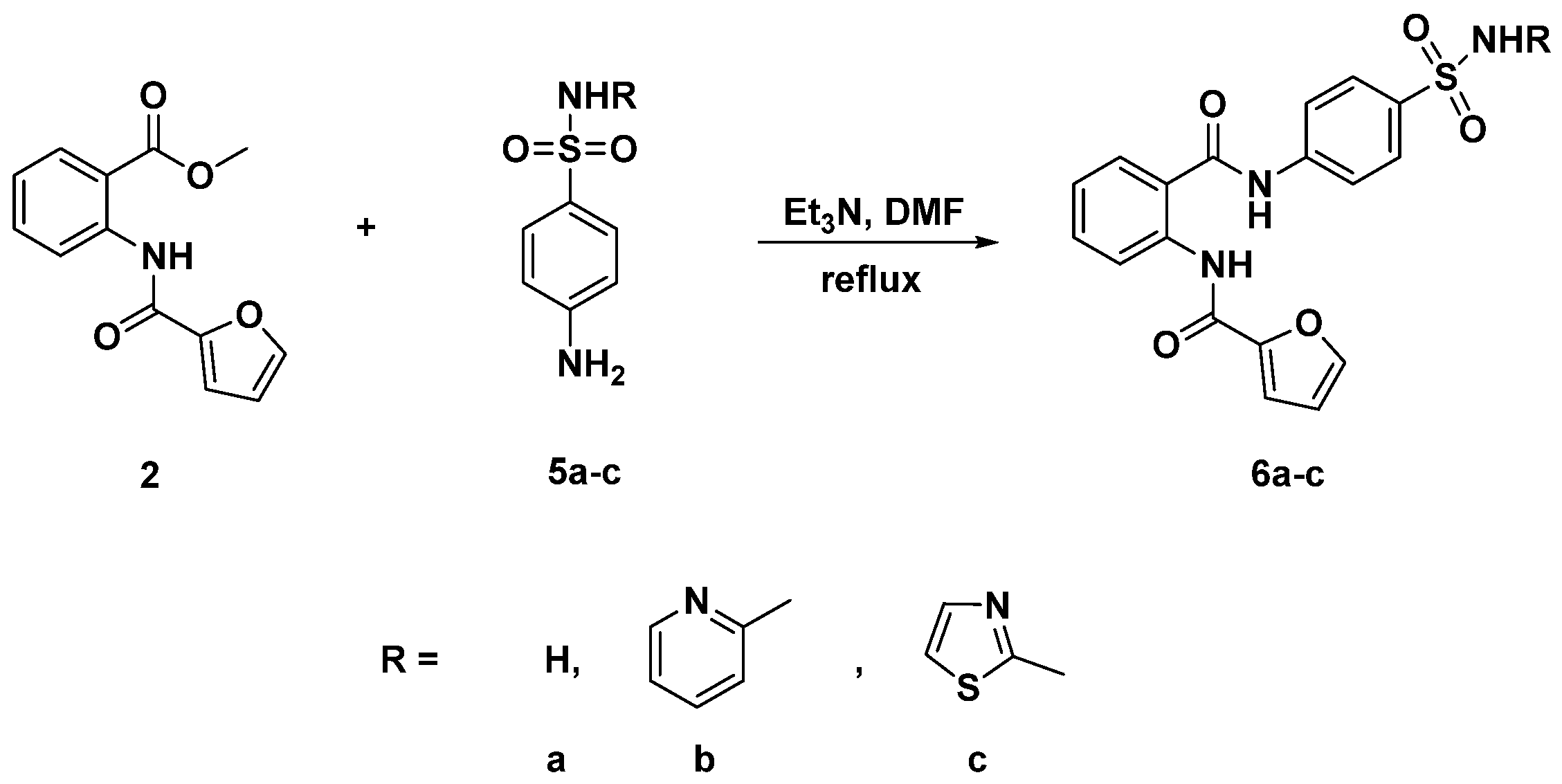
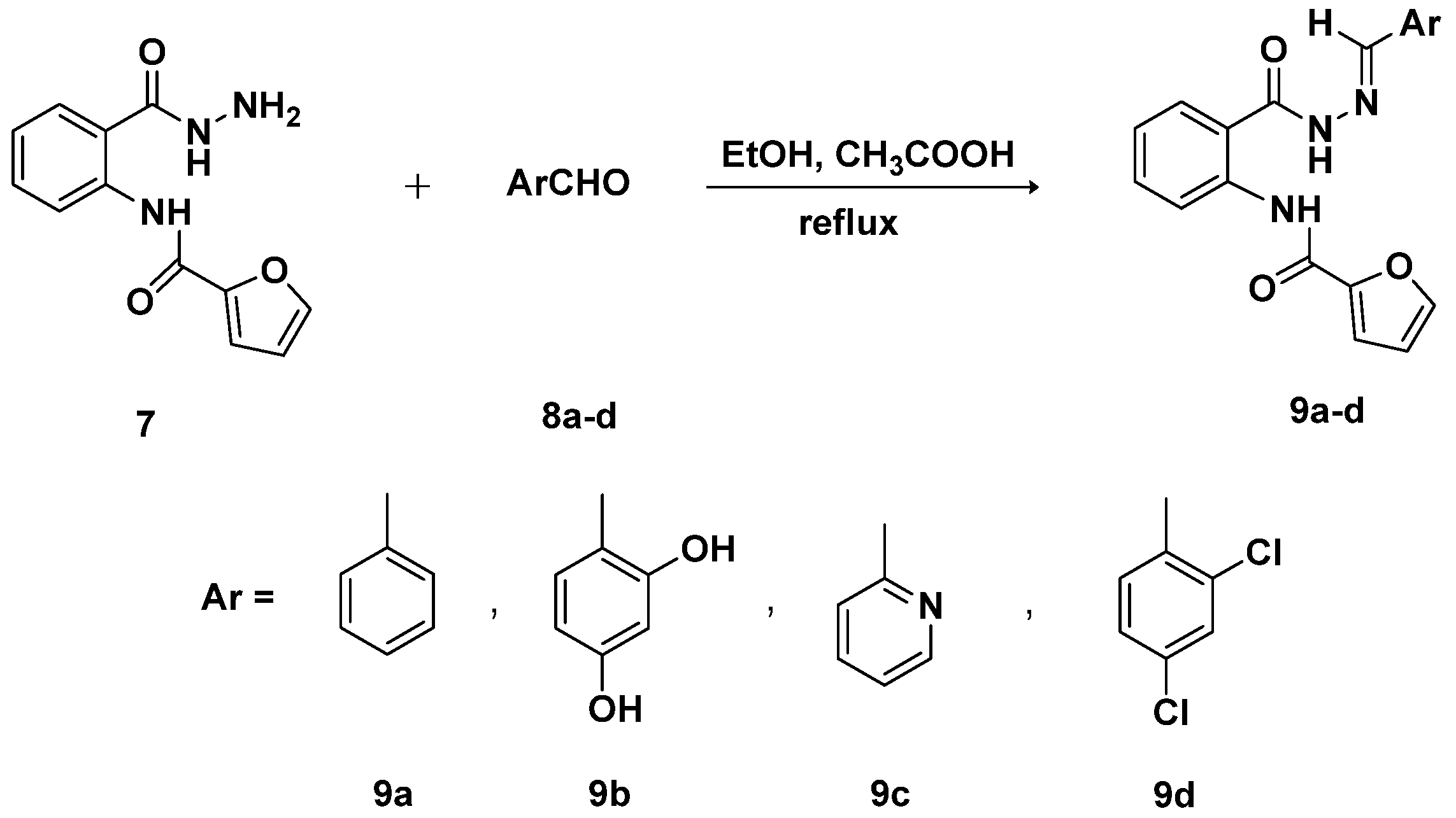
2.1. Chemistry
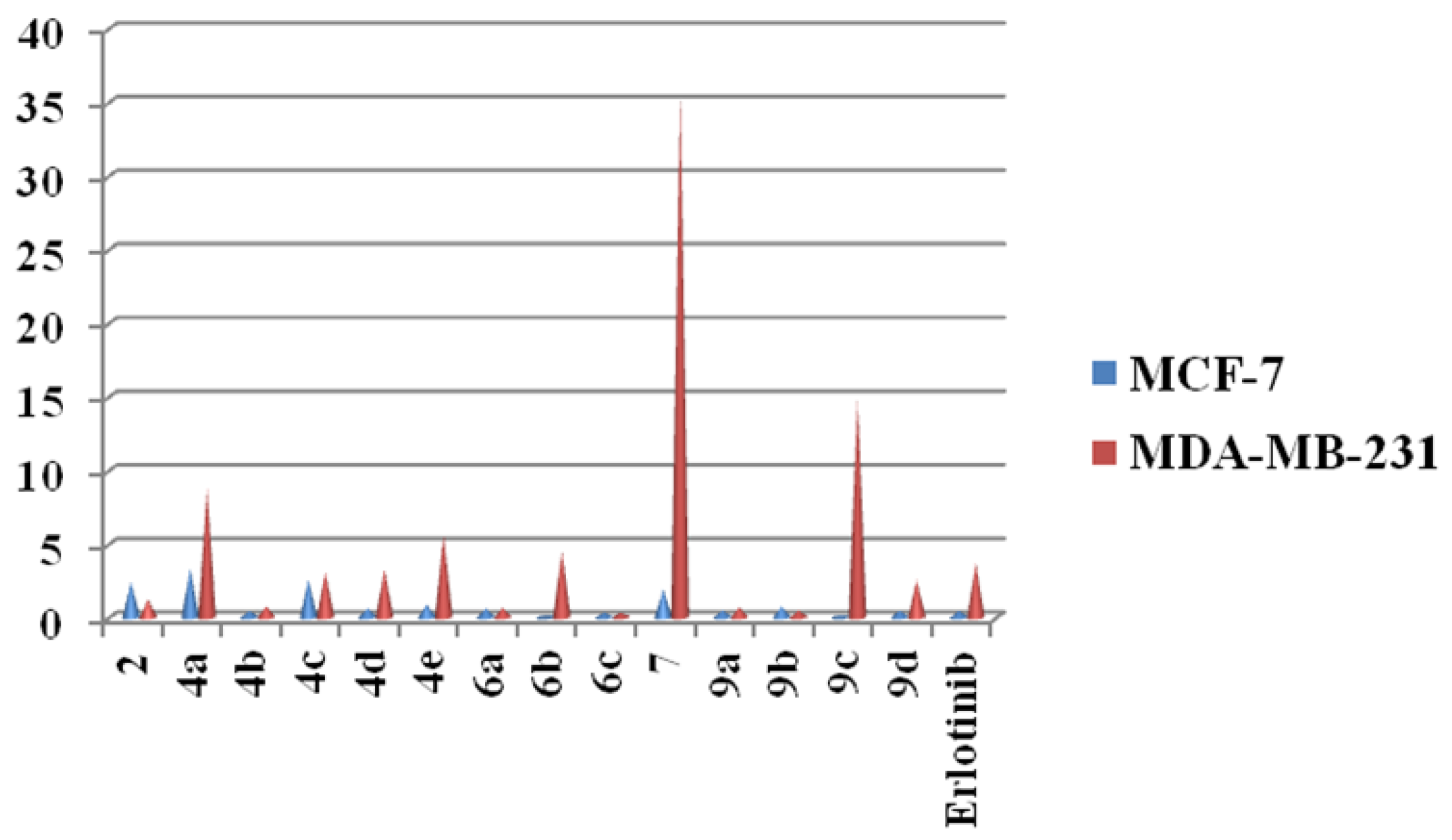
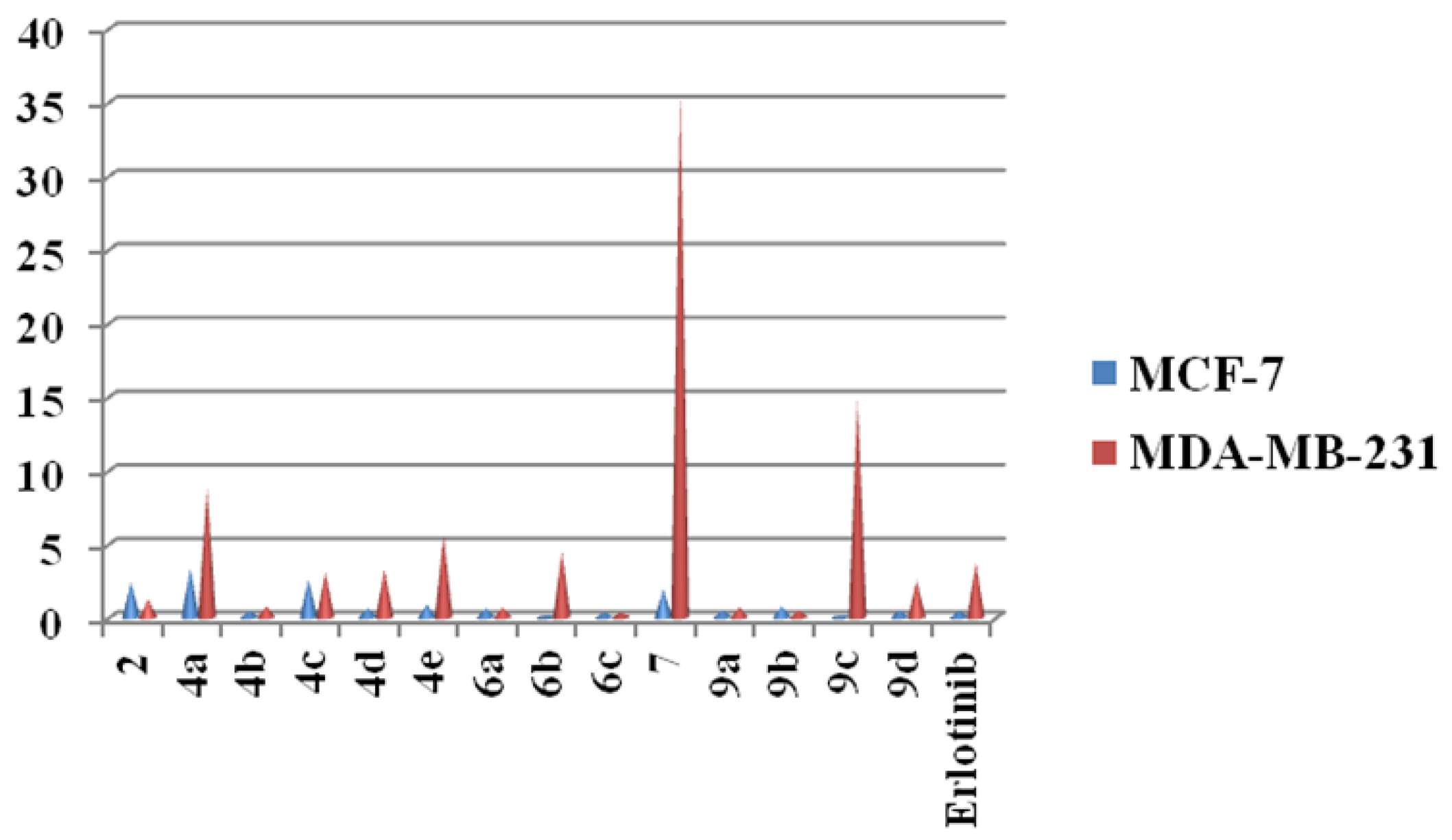
2.2. Cytotoxicity Screening
2.3. Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase (TK) Binding Inhibitory Assay
2.4. Effects on Tubulin Polymerization
2.5. Apoptosis Analysis
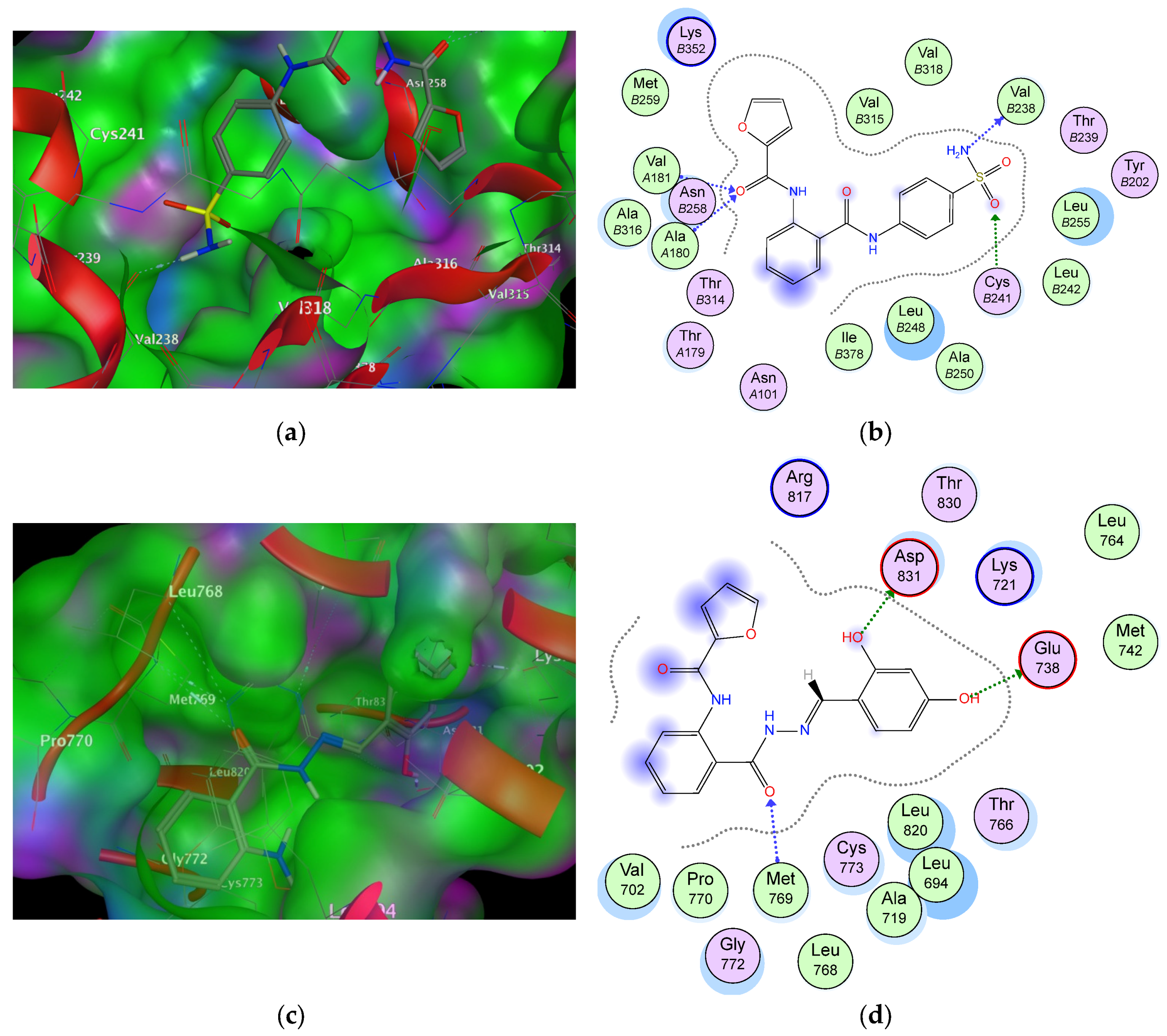
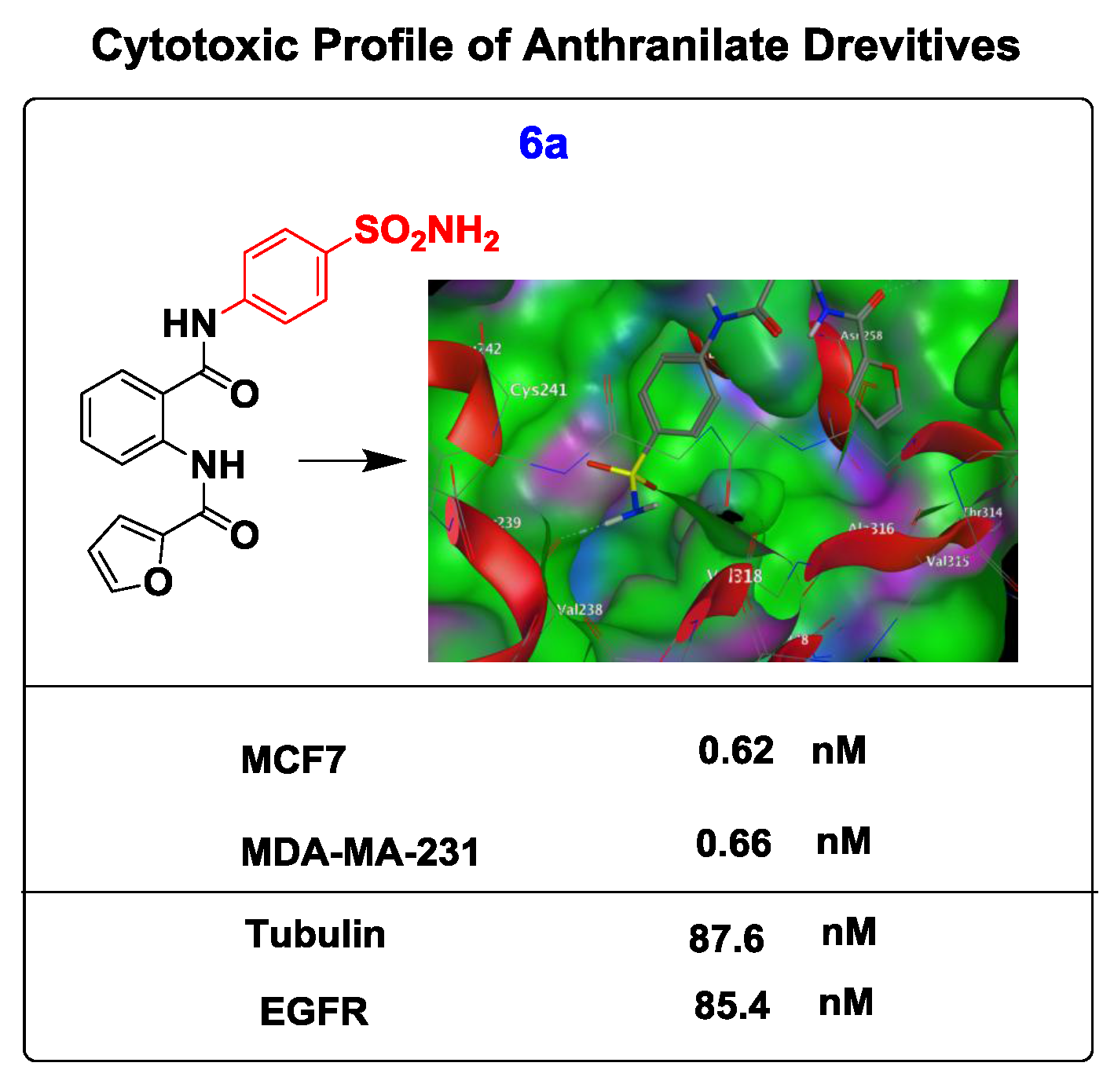
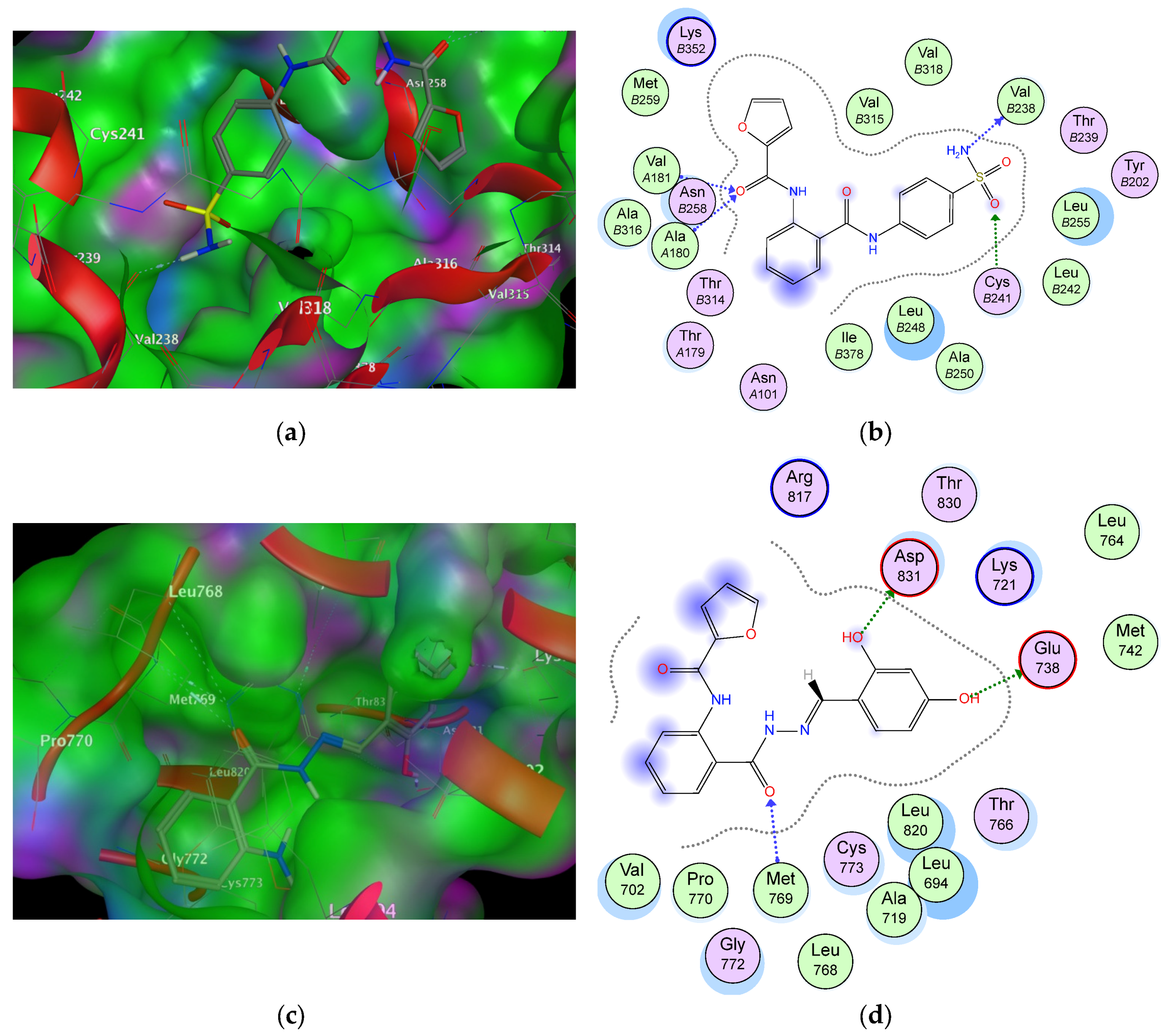
2.6. Molecular Docking Studies and Structure Activity Relationship (SAR) Analyses
3. Materials and Methods
3.1. Chemistry
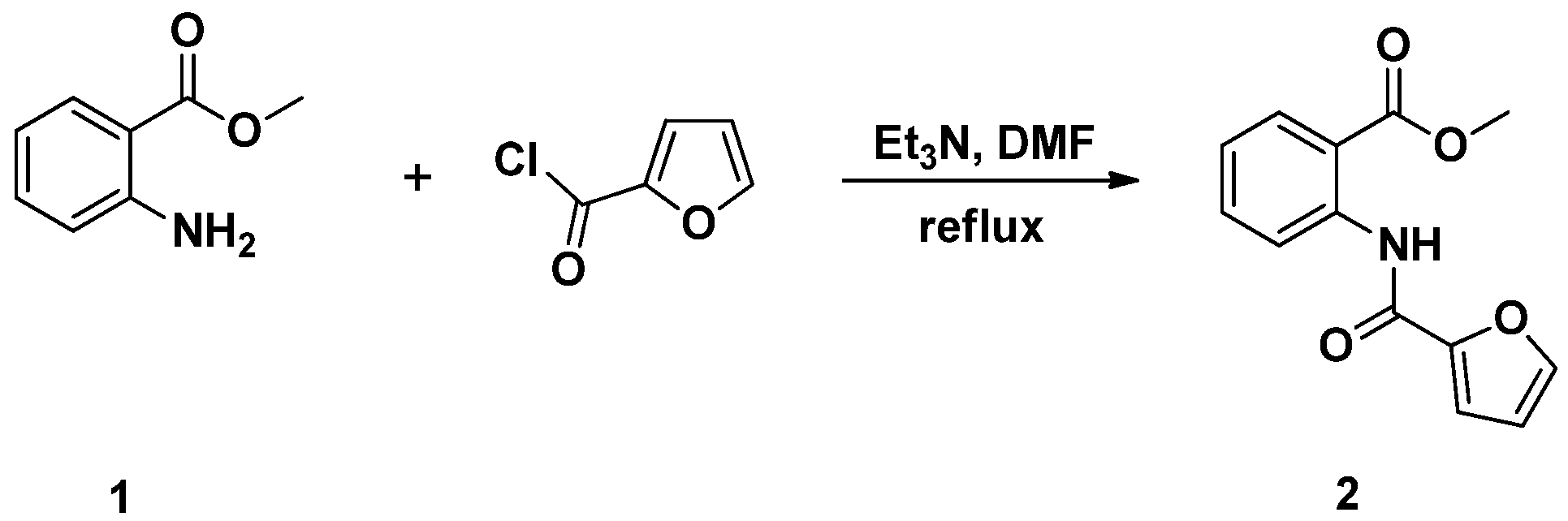
3.2. Synthesis and Characterization of Methyl 2-(Furan-2-carboxamido)benzoate (2)
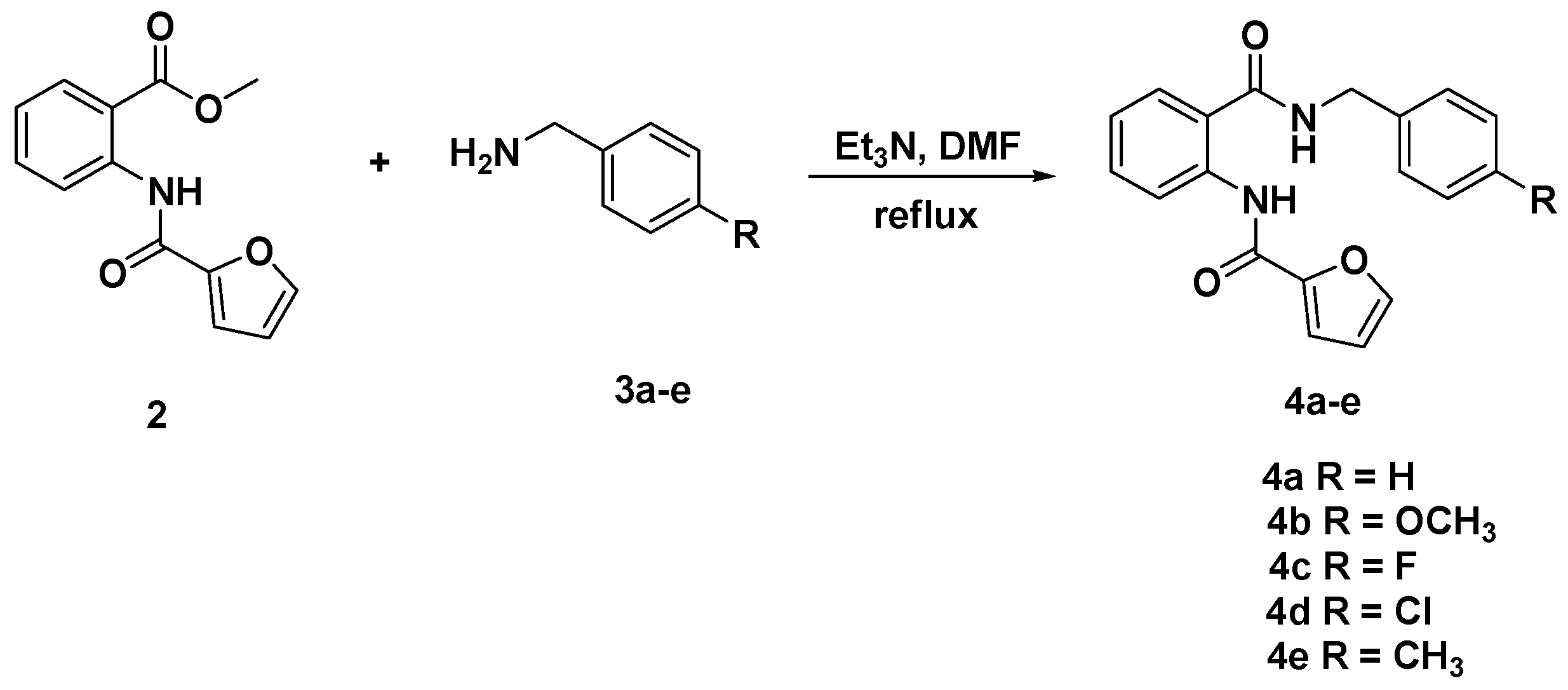
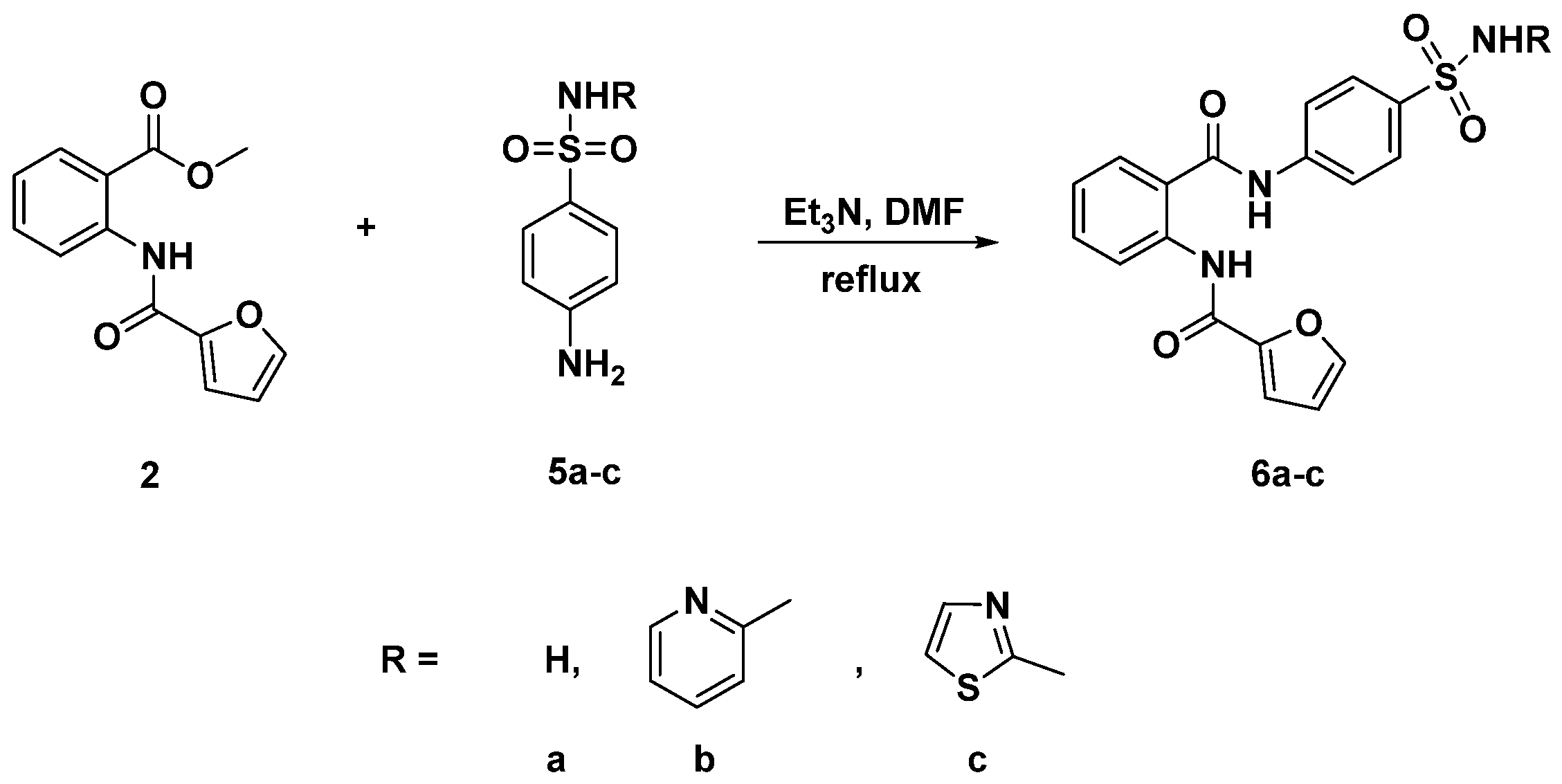
3.3. General Procedure for the Synthesis of Diamides 4a–e and 6a–c
3.3.1. N-[(2-(Benzylcarbamoyl)phenyl]furan-2-carboxamide (4a)
3.3.2. N-{2-[(4-Methoxybenzyl)carbamoyl]phenyl}furan-2-carboxamide (4b)
3.3.3. N-{2-[(4-Fluorobenzyl)carbamoyl]phenyl}furan-2-carboxamide (4c)
3.3.4. N-{2-[(4-Chlorobenzyl)carbamoyl]phenyl}furan-2-carboxamide (4d)
3.3.5. N-{2-[(4-Methylbenzyl)carbamoyl]phenyl}furan-2-carboxamide (4e)
3.3.6. N-{2-[(4-Sulfamoylphenyl)carbamoyl]phenyl}furan-2-carboxamide (6a)
3.3.7. N-{2-[(4-(N-(Pyridin-2-Yl)sulfamoyl]phenyl)carbamoyl)phenyl}furan-2-carboxamide (6b)
3.3.8. N-{2-[(4-(N-(Thiazol-2-Yl)sulfamoyl)phenyl)carbamoyl]phenyl}furan-2-carboxamide (6c)
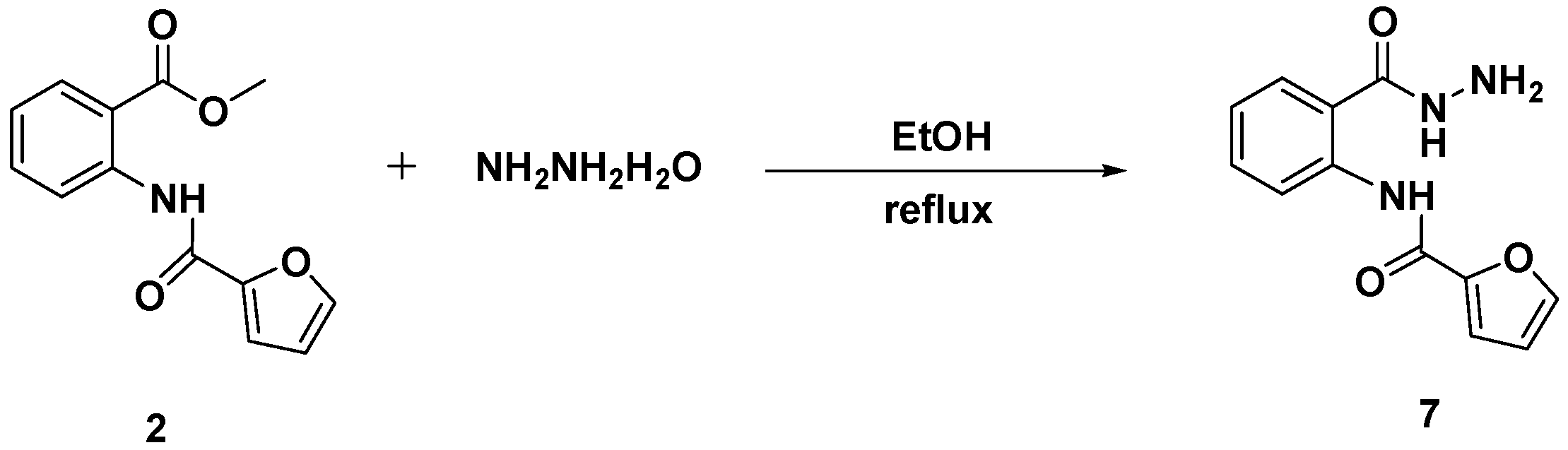
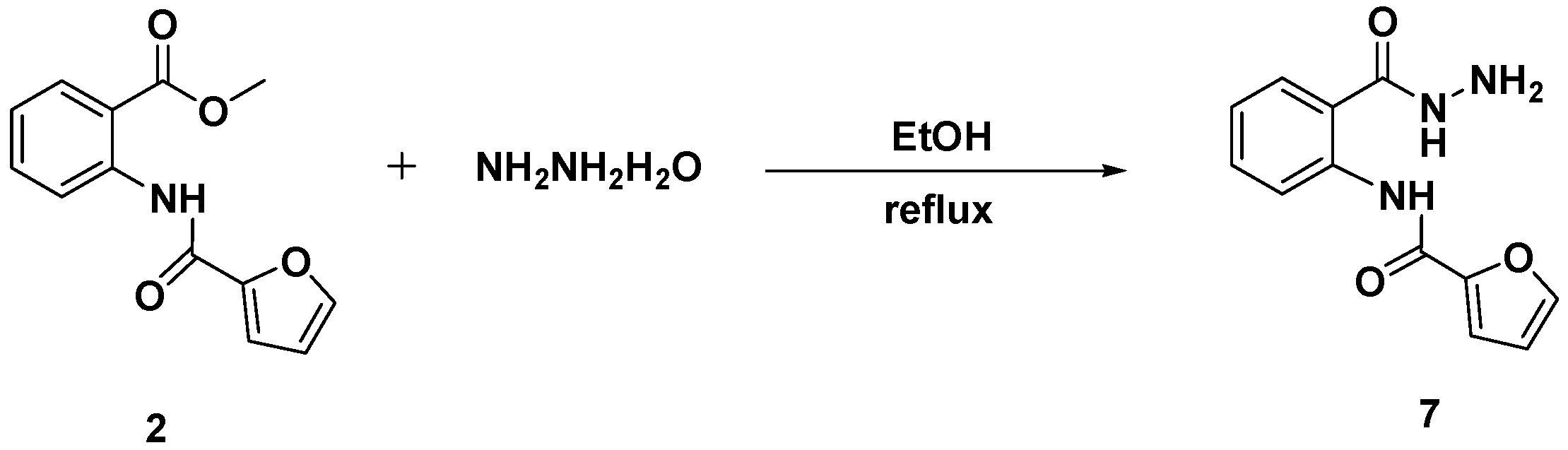
3.4. Synthesis and Characterization of N-[2-(Hydrazinecarbonyl)phenyl]furan-2-carboxamide (7)
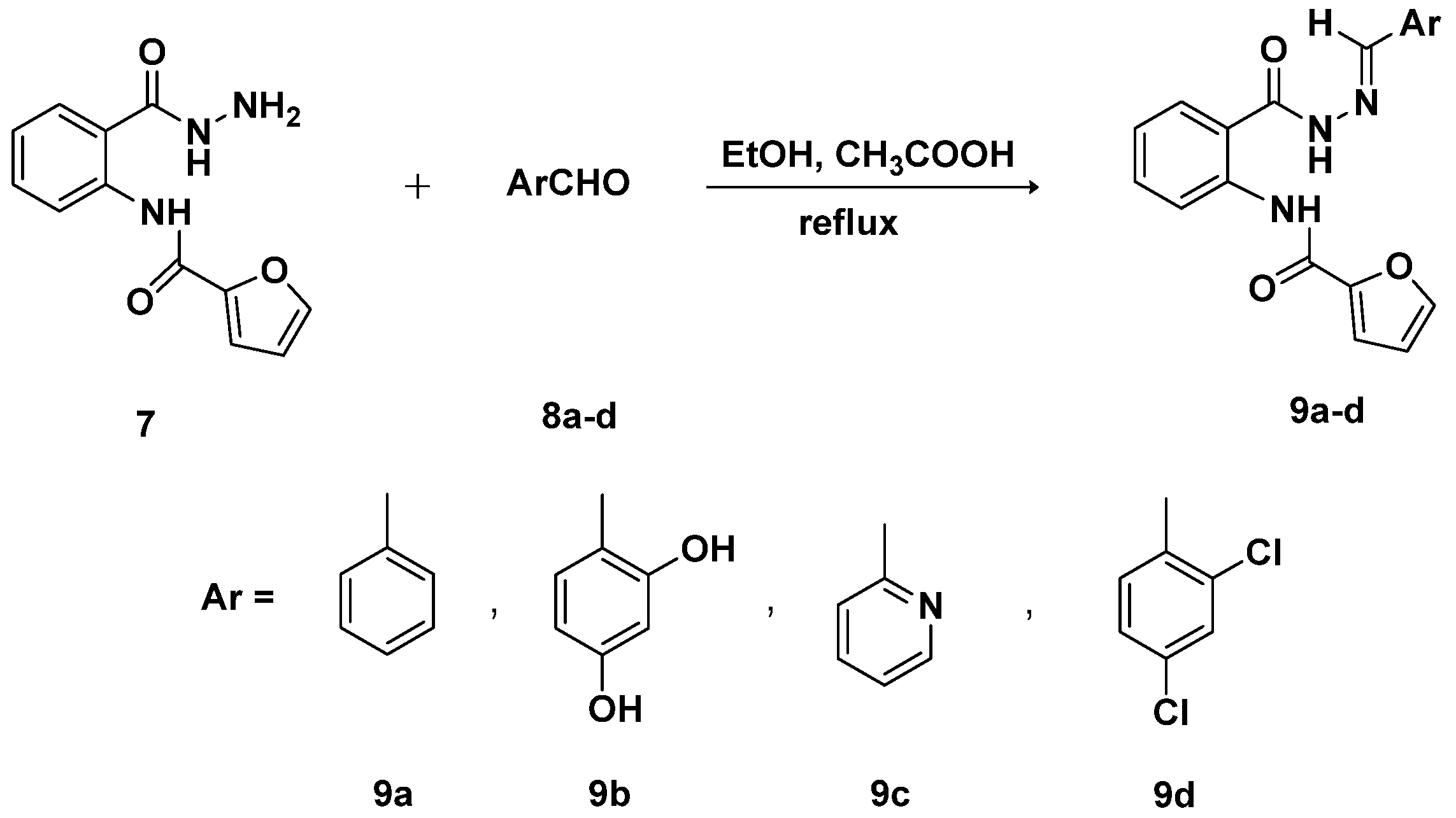
3.5. General Procedure for the Synthesis of Schiff Bases-Type Hydrazones 9a–d
3.5.1. (E)-N-{2-[(2-Benzylidenehydrazine)-1-carbonyl]phenyl}furan-2-carboxamide (9a)
3.5.2. (E)-N-{2-[2-(2,4-Dihydroxybenzylidene)hydrazine-1-carbonyl]phenyl}furan-2-carboxamide (9b)
3.5.3. (E)-N-{2-[2-(Pyridin-2-ylmethylene)hydrazine-1-carbonyl]phenyl}furan-2-carboxamide (9c)
3.5.4. (E)-N-{2-[2-(2,4-Dichlorobenzylidene)hydrazine-1-carbonyl]phenyl}furan-2-carboxamide (9d)
3.6. Cancer Cell Antiproliferative Assay
3.7. EGFR Inhibition Assay
3.8. Enzyme-Linked Immunosorbent Assay for Tubulin Beta (Tubb)
3.9. MDA-MB-231 Cell Line Annexin V-FITC Apoptosis Analysis
3.10. Molecular Docking
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Klener, P.J.; Otahal, P.; Lateckova, L.; Klener, P. Immunotherapy approaches in cancer treatment. Curr. Pharm. Biotechnol. 2015, 16, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Ali, N.A.; O’Brien, J.M., Jr.; Blum, W.; Byrd, J.C.; Klisovic, R.B.; Marcucci, G.; Phillips, G.; Marsh, C.B.; Lemeshow, S.; Grever, M.R. Hyperglycemia in patients with acute myeloid leukemia is associated with increased hospital mortality. Cancer 2007, 110, 96–102. [Google Scholar] [CrossRef] [PubMed]
- Galanski, M.; Arion, V.B.; Jakupec, M.A.; Keppler, B.K. Recent developments in the field of tumor-inhibiting metal complexes. Curr. Pharm. Des. 2003, 9, 2078–2089. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gu, W.; Shan, Y.; Liu, F.; Xu, X.; Yang, Y.; Zhang, Q.; Zhang, Y.; Kuang, H.; Wang, Z.; et al. Design, synthesis and anticancer activity of novel nopinone-based thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 2017, 27, 2360–2363. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T. New challenges and inspired answers for anticancer drug discovery and development. Jpn. J. Clin. Oncol. 2013, 43, 945–953. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Blockage of drug resistance in vitro by disulfiram, a drug used to treat alcoholism. J. Natl. Cancer Inst. 2000, 92, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Pick, A.; Klinkhammer, W.; Wiese, M. Specific inhibitors of the breast cancer resistance protein (BCRP). ChemMedChem 2010, 5, 1498–1505. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.; Crespo, A.; Tiwari, A. Is there a case for selectively promiscuous anticancer drugs? Drug Discov. Today 2009, 14, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Das, U.; Pati, H.N.; Panda, A.K.; De Clercq, E.; Balzarini, J.; Molnar, J.; Barath, Z.; Ocsovszki, I.; Kawase, M.; Zhou, L.; et al. 2-(3-aryl-2-propenoyl)-3-methylquinoxaline-1,4-dioxides: A novel cluster of tumor-specific cytotoxins which reverse multidrug resistance. Bioorg. Med. Chem. 2009, 17, 3909–3915. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Li, C.M.; Wang, Z.; Ross, C.R., II; Chen, J.; Dalton, J.T.; Li, W.; Miller, D.D. Discovery of 4-substituted methoxybenzoyl-aryl-thiazole as novel anticancer agents: Synthesis, biological evaluation, and structure-activity relationships. J. Med. Chem. 2009, 52, 1701–1711. [Google Scholar] [CrossRef] [PubMed]
- Per, W.; Jan, B. The chemistry of anthranilic acid. Curr. Org. Synth. 2006, 3, 379–402. [Google Scholar] [CrossRef]
- Zhang, D.; Wang, G.; Zhao, G.; Xu, W.; Huo, L. Synthesis and cytotoxic activity of novel 3-(1H-indol-3-yl)-1H-pyrazole-5-carbohydrazide derivatives. Eur. J. Med. Chem. 2011, 46, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.A.; Banker, P.; Bickett, D.M.; Boucheron, J.A.; Carter, H.L.; Clancy, D.C.; Cooper, J.P.; Dickerson, S.H.; Garrido, D.M.; Nolte, R.T.; et al. Anthranilimide based glycogen phosphorylase inhibitors for the treatment of type 2 diabetes. Part 3: X-ray crystallographic characterization, core and urea optimization and in vivo efficacy. Bioorg. Med. Chem. Lett. 2009, 19, 1177–1182. [Google Scholar] [CrossRef] [PubMed]
- Lassiani, L.; Pavan, M.V.; Berti, F.; Kokotos, G.; Markidis, T.; Mennuni, L.; Makovec, F.; Varnavas, A. Anthranilic acid based CCK1 receptor antagonists: Blocking the receptor with the same ‘words’ of the endogenous ligand. Bioorg. Med. Chem. 2009, 17, 2336–2350. [Google Scholar] [CrossRef] [PubMed]
- Pinkerton, A.B.; Lee, T.T.; Hoffman, T.Z.; Wang, Y.; Kahraman, M.; Cook, T.G.; Severance, D.; Gahman, T.C.; Noble, S.A.; Shiau, A.K.; et al. Synthesis and SAR of thiophene containing kinesin spindle protein (KSP) inhibitors. Bioorg. Med. Chem. Lett. 2007, 17, 3562–3569. [Google Scholar] [CrossRef] [PubMed]
- Al-Obaid, A.M.; Abdel-Hamide, S.G.; El-Kashef, H.A.; Abdel-Aziz, A.A.M.; El-Azab, A.S.; Al-Khamees, H.A.; El-Subbagh, H.I. Substituted quinazolines, part 3. Synthesis, in vitro antitumor activity and molecular modeling study of certain 2-thieno-4(3H)-quinazolinone analogs. Eur. J. Med. Chem. 2009, 44, 2379–2391. [Google Scholar] [CrossRef] [PubMed]
- Congiu, C.; Cocco, M.T.; Lilliu, V.; Onnis, V. New potential anticancer agents based on the anthranilic acid scaffold: Synthesis and evaluation of biological activity. J. Med. Chem. 2005, 48, 8245–8252. [Google Scholar] [CrossRef] [PubMed]
- Matwijczuk, A.; Kluczyk, D.; Górecki, A.; Niewiadomy, A.; Gagoś, M. Solvent effects on molecular aggregation in 4-(5-Heptyl-1,3,4-thiadiazol-2-yl)benzene-1,3-diol and 4-(5-Methyl-1,3,4-thiadiazol-2-yl)benzene-1,3-diol. J. Phys. Chem. 2016, 120, 7958–7969. [Google Scholar] [CrossRef] [PubMed]
- Brancato, G.; Signore, G.; Neyroz, P.; Polli, D.; Cerullo, G.; Abbandonato, G.; Luca Nucara, L.; Barone, V.; Beltram, F.; Bizzarri, R. Dual fluorescence through Kasha’s rule breaking: An unconventional photomechanism for intracellular probe design. J. Phys. Chem. 2015, 119, 6144–6154. [Google Scholar] [CrossRef] [PubMed]
- Matwijczuk, A.; Kamiński, D.; Górecki, A.; Ludwiczuk, A.; Niewiadomy, A.; Maćkowski, S.; Gagoś, M. spectroscopic studies of dual fluorescence in 2-((4-Fluorophenyl)amino)-5-(2,4-dihydroxybenzeno)-1,3,4-thiadiazole. J. Phys. Chem. 2015, 119, 10791–10805. [Google Scholar] [CrossRef] [PubMed]
- Dyrager, C.; Wickstrom, M.; Friden-Saxin, M.; Friberg, A.; Dahlen, K.; Wallen, E.A.; Gullbo, J.; Grotli, M.; Luthman, K. Inhibitors and promoters of tubulin polymerization: Synthesis and biological evaluation of chalcones and related dienones as potential anticancer agents. Bioorg. Med. Chem. 2011, 19, 2659–2665. [Google Scholar] [CrossRef] [PubMed]
- Jordan, A.; Hadfield, J.A.; Lawrence, N.J.; McGown, A.T. Tubulin as a target for anticancer drugs: Agents which interact with the mitotic spindle. Med. Res. Rev. 1998, 18, 259–296. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Bernhard, D.; Schwaiger, W.; Crazzolara, R.; Tinhofer, I.; Kofler, R.; Csordas, A. Enhanced MTT-reducing activity under growth inhibition by resveratrol in CEM-C7H2 lymphocytic leukemia cells. Cancer Lett. 2003, 195, 193–199. [Google Scholar] [CrossRef]
- Pigault, C.; Follenius-Wund, A.; Schmutz, M.; Freyssinet, J.M.; Brisson, A. Formation of two-dimensional arrays of Annexin V on phosphatidylserine-containing liposomes. J. Mol. Biol. 1994, 236, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Grindheim, A.K.; Saraste, J.; Vedeler, A. Protein phosphorylation and its role in the regulation of Annexin A2 function. Biochim. Biophys. Acta 2017, 1861, 2515–2529. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE). Chemical Computing Group: Quebec, QC, Canada, 2012. Available online: http://www.chemcomp.com (accessed on 30 January 2013).
- Tiwari, S.V.; Siddiqui, S.; Seijas, J.A.; Vazquez-Tato, M.P.; Sarkate, A.P.; Lokwani, D.K.; Nikalje, A.P.G. Microwave-Assisted Facile Synthesis, Anticancer Evaluation and Docking Study of N-((5-(Substituted methylene amino)-1,3,4-thiadiazol-2-yl)methyl) Benzamide Derivatives. Molecules 2017, 22, 995. [Google Scholar] [CrossRef] [PubMed]
- Alswah, M.; Bayoumi, A.H.; Elgamal, K.; Ahmed Elmorsy, A.; Ihmaid, S.; Ahmed, H.E.A. Design, synthesis and cytotoxic evaluation of novel chalcone derivatives bearing triazolo[4,3-a]-quinoxaline moieties as potent anticancer agents with dual EGFR kinase and tubulin polymerization inhibitory effects. Molecules 2018, 23, 48. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | IC50 (nM) | |
|---|---|---|
| MCF-7 | MDA-MB-231 | |
| 2 | 2.43 ± 0.20 | 1.21 ± 0.25 |
| 4a | 3.35 ± 0.11 | 8.82 ± 0.14 |
| 4b | 0.45 ± 0.12 | 0.76 ± 0.11 |
| 4c | 2.59 ± 0.32 | 3.11 ± 0.50 |
| 4d | 0.64 ± 0.25 | 3.29 ± 0.14 |
| 4e | 0.88 ± 0.25 | 5.59 ± 0.20 |
| 6a | 0.62 ± 0.12 | 0.66 ± 0.11 |
| 6b | 0.065 ± 0.32 | 4.45 ± 0.30 |
| 6c | 0.34 ± 0.20 | 0.32 ± 0.21 |
| 7 | 1.91 ± 0.11 | 35.85 ± 0.12 |
| 9a | 0.51 ± 0.16 | 0.68 ± 0.02 |
| 9b | 0.78 ± 0.11 | 0.48 ± 0.01 |
| 9c | 0.15 ± 0.15 | 14.8 ± 0.20 |
| 9d | 0.48 ± 0.14 | 2.53 ± 0.12 |
| Erlotinib | 0.45 ± 0.08 | 3.74 ± 0.01 |
| Compounds | IC50 (nM) | |
|---|---|---|
| EGFR Inhibition | Tubulin Assay | |
| 4b | 30.5 ± 0.25 | 140.8 ± 0.15 |
| 6a | 85.4 ± 0.32 | 87.6 ± 0.13 |
| 6c | 110 ± 0.14 | 155 ± 0.25 |
| 9a | 59.3 ± 0.11 | 410.6 ± 0.12 |
| 9b | 31.2 ± 0.12 | 108.4 ± 0.14 |
| Staurosporine | 59.08 ± 0.2 | - |
| Colchicine | - | 112 ± 0.08 |
| Cop. | Fragment | Target Residues (Distance, Å) | Interaction | Binding Energy (dG) |
|---|---|---|---|---|
| 6a (1SA0) | –NH2 | Val238 (2.5) | H-bonding | −16.3 |
| S=O | Cys241 (2.00) | H-bonding | ||
| C=O–NH | Val181 (1.85), Ala180 (1.91) | H-bonding | ||
| Phenyl (sulphonamide) | Val315, Leu248, Val318 | Hydrophobic | ||
| Furoyl | Met259 | Hydrophobic | ||
| Phenyl (anthranilate) | Thr514, Ile378 | Aromatic stacking | ||
| 9b (1M17) | –OH | Asp831 (2.1) | H-bonding | −15.4 |
| –OH | Glu738 (1.84) | H-bonding | ||
| C=O (anthranilate) | Met769 (2.1) | Hydrophobic | ||
| Phenyl (sulphonamide) | Leu820 | Hydrophobic | ||
| Furoyl | Arg817 | Hydrophobic | ||
| Phenyl (anthranilate) | Val702, Pro770 | Hydrophobic |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ihmaid, S.; Ahmed, H.E.A.; Zayed, M.F. The Design and Development of Potent Small Molecules as Anticancer Agents Targeting EGFR TK and Tubulin Polymerization. Int. J. Mol. Sci. 2018, 19, 408. https://doi.org/10.3390/ijms19020408
Ihmaid S, Ahmed HEA, Zayed MF. The Design and Development of Potent Small Molecules as Anticancer Agents Targeting EGFR TK and Tubulin Polymerization. International Journal of Molecular Sciences. 2018; 19(2):408. https://doi.org/10.3390/ijms19020408
Chicago/Turabian StyleIhmaid, Saleh, Hany E. A. Ahmed, and Mohamed F. Zayed. 2018. "The Design and Development of Potent Small Molecules as Anticancer Agents Targeting EGFR TK and Tubulin Polymerization" International Journal of Molecular Sciences 19, no. 2: 408. https://doi.org/10.3390/ijms19020408
APA StyleIhmaid, S., Ahmed, H. E. A., & Zayed, M. F. (2018). The Design and Development of Potent Small Molecules as Anticancer Agents Targeting EGFR TK and Tubulin Polymerization. International Journal of Molecular Sciences, 19(2), 408. https://doi.org/10.3390/ijms19020408