Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy


Abstract
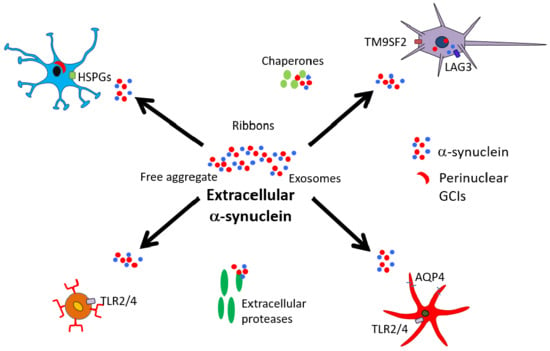
1. Multiple System Atrophy and α-Synuclein
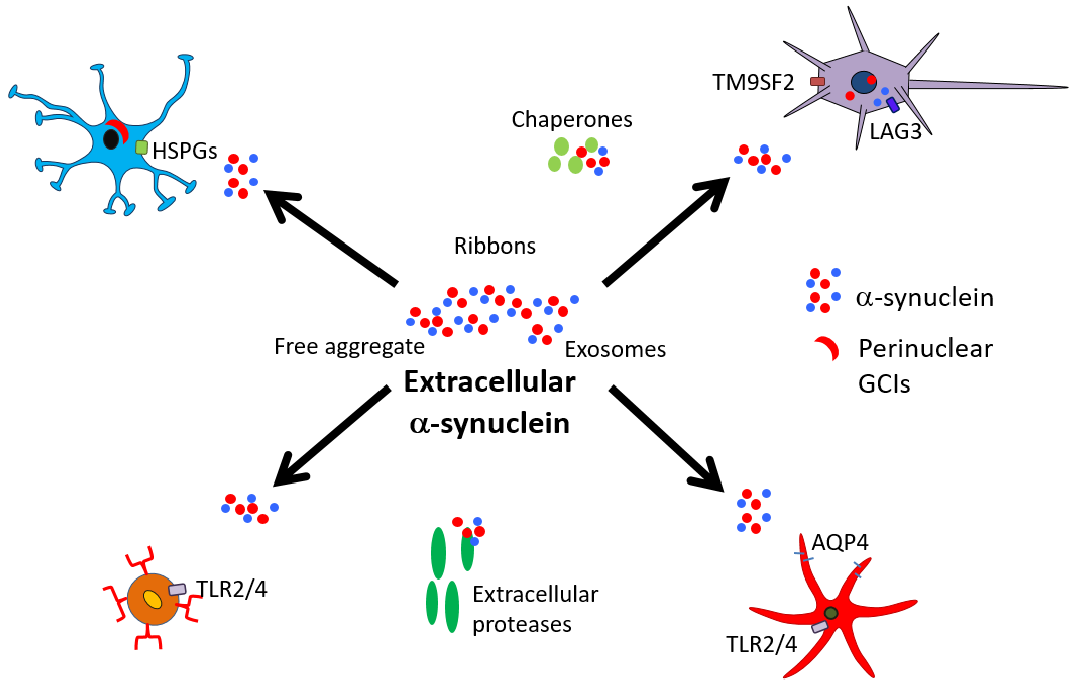
2. Secretion/Release of α-Synuclein
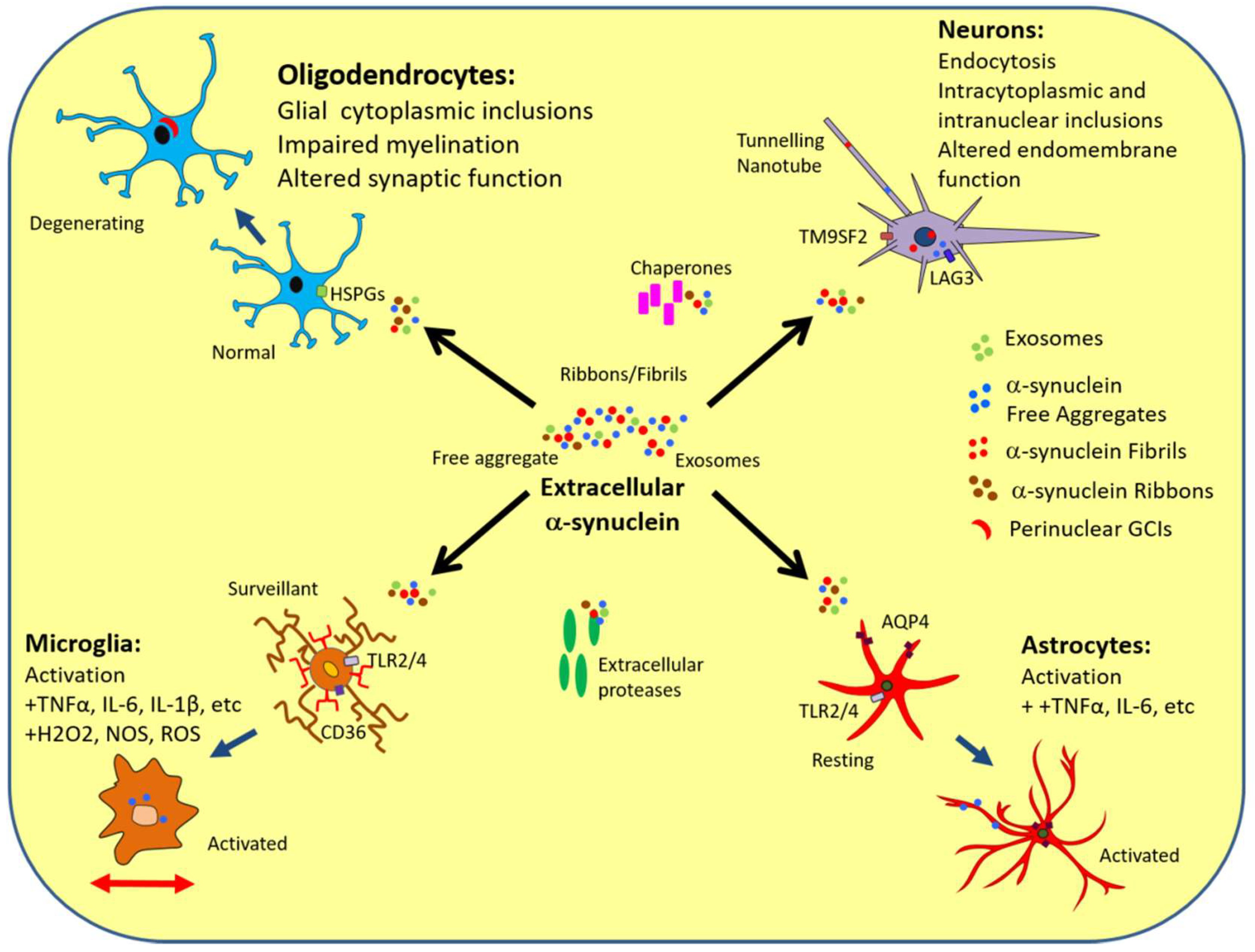
3. α-Synuclein Interactions with Neurons
4. α-Synuclein Interactions with Oligodendrocytes
5. α-Synuclein Interactions with Astrocytes
6. α-Synuclein Interactions with Microglia
7. Interactions outside the CNS
8. α-Synuclein and Extracellular Factors
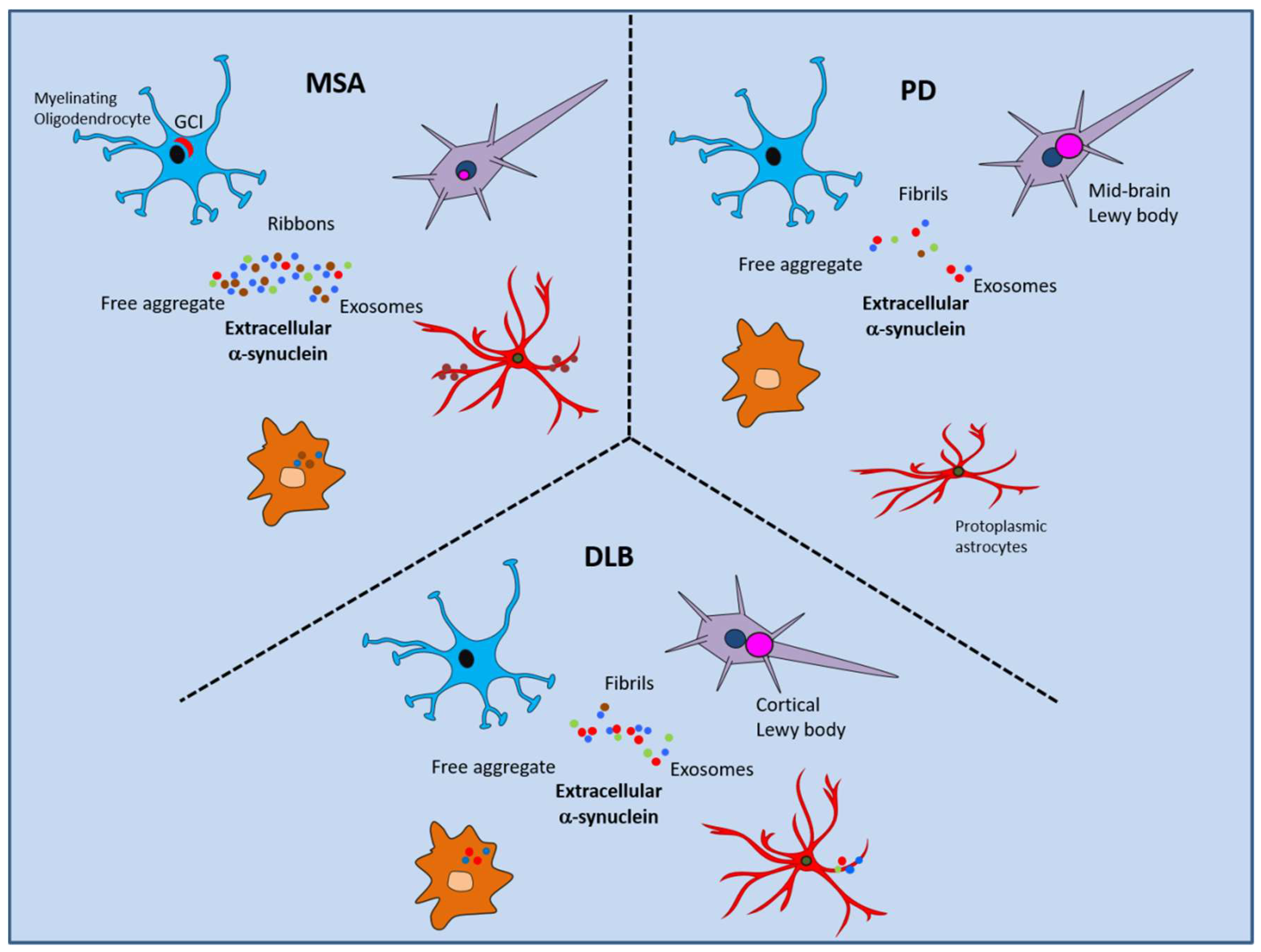
9. Conclusions—Implications of the Extracellular α-Synuclein Interactome
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| α-syn | α-Synuclein |
| CNS | Central Nervous System |
| CSF | Cerebrospinal Fluid |
| DLB | Dementia with Lewy Bodies |
| GCI | Glial Cytoplasmic Inclusions |
| LB | Lewy Body |
| MSA | Multiple System Atrophy |
| MSA-C | Multiple System Atrophy-Cerebellar |
| MSA-P | Multiple System Atrophy-Parkinsonism |
| PD | Parkinson’s disease |
| SNARE | Soluble N-ethylmaleimide-sensitive factor attachment protein receptor |
| TLR | Toll-Like Receptor |
| LAG-3 | Lymphocyte Activating Gene 3 |
References
- Weinreb, P.; Zhen, W.; Poon, A.; Conway, K.; Lansbury, P. NACP, A Protein Implicated in Alzheimer’s Disease and Learning, Is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [Google Scholar] [CrossRef] [PubMed]
- Theillet, F.; Binolfi, A.; Bekei, B.; Martorana, A.; Rose, H.; Stuiver, M.; Verzini, S.; Lorenz, D.; van Rossum, M.; Goldfarb, D.; et al. Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 2016, 530, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Barbour, R.; Kling, K.; Anderson, J.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.; Soriano, F.; Seubert, P.; et al. Red Blood Cells Are the Major Source of A-Synuclein in Blood. Neurodegener. Dis. 2008, 5, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.X.; Campbell, B.C.; McLean, C.A.; Thyagarajan, D.; Gai, W.P.; Kapsa, R.M.; Beyreuther, K.; Masters, C.L.; Culvenor, J.G. Platelet alpha- and gamma-synucleins in Parkinson’s disease and normal control subjects. J. Alzheimer’s Dis. 2002, 4, 309–315. [Google Scholar] [CrossRef] [PubMed]
- Shin, E.C.; Cho, S.E.; Lee, D.K.; Hur, M.W.; Paik, S.R.; Park, J.H.; Kim, J. Expression patterns of alpha-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol. Cells 2000, 10, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of α-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef] [PubMed]
- Burré, J. The Synaptic Function of α-Synuclein. J. Parkinsons. Dis. 2015, 5, 699–713. [Google Scholar] [CrossRef]
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.; Sudhof, T. α-Synuclein Promotes SNARE-Complex Assembly in Vivo and in Vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.; Waymire, J.; Lin, E.; Liu, J.; Guo, F.; Zigmond, M. A role for α-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef]
- Larsen, K.; Schmitz, Y.; Troyer, M.; Mosharov, E.; Dietrich, P.; Quazi, A.; Savalle, M.; Nemani, V.; Chaudhry, F.; Edwards, R.; et al. α-Synuclein Overexpression in PC12 and Chromaffin Cells Impairs Catecholamine Release by Interfering with a Late Step in Exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Jäkälä, P.; Tanila, H. Abnormal compartmentalization of norepinephrine in mouse dentate gyrus in α-synuclein knockout and A30P transgenic mice. J. Neurochem. 2006, 99, 724–732. [Google Scholar] [CrossRef] [PubMed]
- Yavich, L.; Tanilla, H.; Vepsalainen, S.; Jakala, P. Role of α-Synuclein in Presynaptic Dopamine Recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef] [PubMed]
- Wersinger, C.; Sidhu, A. Disruption of the interaction of alpha-synuclein with microtubules enhances cell surface recruitment of the dopamine transporter. Biochemistry 2005, 44, 13612–13624. [Google Scholar] [CrossRef] [PubMed]
- Bartels, T.; Choi, J.; Selkoe, D. α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110. [Google Scholar] [CrossRef] [PubMed]
- Jakes, R.; Spillantini, M.; Goedert, M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994, 345, 27–32. [Google Scholar] [CrossRef]
- Luth, E.; Bartels, T.; Dettmer, U.; Kim, N.; Selkoe, D. Purification of α-Synuclein from Human Brain Reveals an Instability of Endogenous Multimers as the Protein Approaches Purity. Biochemistry 2015, 54, 279–292. [Google Scholar] [CrossRef]
- Dettmer, U.; Newman, A.J.; Soldner, F.; Luth, E.S.; Kim, N.C.; von Saucken, V.E.; Sanderson, J.B.; Jaenisch, R.; Bartels, T.; Selkoe, D. Parkinson-causing α-synuclein missense mutations shift native tetramers to monomers as a mechanism for disease initiation. Nat. Commun. 2015, 6, 7314. [Google Scholar] [CrossRef] [PubMed]
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. α-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364. [Google Scholar] [CrossRef]
- Rcom-H’cheo-Gauthier, A.; Osborne, S.; Meedeniya, A.; Pountney, D. Calcium: α-Synuclein Interactions in α-Synucleinopathies. Front. Neurosci. 2016, 10, 570. [Google Scholar] [CrossRef]
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.; Shen, J.; Takio, K.; Iwatsubo, T. α-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell. Biol. 2002, 4, 160–164. [Google Scholar] [CrossRef]
- El-Agnaf, O.; Salem, S.; Paleologou, K.; Curran, M.; Gibson, M.; Court, J.; Schlossmacher, M.; Allsop, D. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Glanzer, J.; Kadiu, I.; Ricardo-Dukelow, M.; Chaudhuri, A.; Ciborowski, P.; Cerny, R.; Gelman, B.; Thomas, M.; Mosley, R.; et al. Nitrated α-synuclein-activated microglial profiling for Parkinson’s disease. J. Neurochem. 2007, 104, 1504–1525. [Google Scholar] [CrossRef] [PubMed]
- Oueslati, A. Implication of Alpha-Synuclein Phosphorylation at S129 in Synucleinopathies: What Have We Learned in the Last Decade? J. Parkinsons. Dis. 2016, 6, 39–51. [Google Scholar] [CrossRef] [PubMed]
- McCann, H.; Stevens, C.; Cartwright, H.; Halliday, G. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67. [Google Scholar] [CrossRef]
- Asi, Y.; Simpson, J.; Heath, P.; Wharton, S.; Lees, A.; Revesz, T.; Houlden, H.; Holton, J. α-Synuclein mRNA expression in oligodendrocytes in MSA. Glia 2014, 62, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Djelloul, M.; Holmqvist, S.; Boza-Serrano, A.; Azevedo, C.; Yeung, M.; Goldwurm, S.; Frisén, J.; Deierborg, T.; Roybon, L. α-Synuclein Expression in the Oligodendrocyte Lineage: An In Vitro and In Vivo Study Using Rodent and Human Models. Stem Cell. Rep. 2015, 5, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Reyes, J.; Rey, N.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. α-Synuclein transfers from neurons to oligodendrocytes. Glia 2013, 62, 387–398. [Google Scholar] [CrossRef]
- Longo, D.; Fanciulli, A.; Wenning, G. Multiple-System Atrophy. N. Engl. J. Med. 2015, 372, 249–263. [Google Scholar]
- Ubhi, K.; Low, P.; Masliah, E. Multiple system atrophy: A clinical and neuropathological perspective. Trends Neurosci. 2011, 34, 581–590. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.; Ruf, W.; Putcha, P.; Joyner, D.; Hashimoto, T.; Glabe, C.; Hyman, B.; McLean, P. Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J. 2011, 25, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Fontaine, S.; Zheng, D.; Sabbagh, J.; Martin, M.; Chaput, D.; Darling, A.; Trotter, J.; Stothert, A.; Nordhues, B.; Lussier, A.; et al. DnaJ/Hsc70 chaperone complexes control the extracellular release of neurodegenerative-associated proteins. EMBO J. 2016, 35, 1537–1549. [Google Scholar] [CrossRef] [PubMed]
- Danzer, K.; Kranich, L.; Ruf, W.; Cagsal-Getkin, O.; Winslow, A.; Zhu, L.; Vanderburg, C.; McLean, P. Exosomal cell-to-cell transmission of α synuclein oligomers. Mol. Neurodegener. 2012, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Erviti, L.; Seow, Y.; Schapira, A.; Gardiner, C.; Sargent, I.; Wood, M.; Cooper, J. Lysosomal dysfunction increases exosome-mediated α-synuclein release and transmission. Neurobiol. Dis. 2011, 42, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.; Ntzouni, M.; Margaritis, L.; Stefanis, L.; Vekrellis, K. Cell-Produced-Synuclein Is Secreted in a Calcium-Dependent Manner by Exosomes and Impacts Neuronal Survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [PubMed]
- Peelaerts, W.; Bousset, L.; van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; van den Haute, C.; Melki, R.; Baekelandt, V. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Iwatsubo, T. Extracellular α-synuclein levels are regulated by neuronal activity. Mol. Neurodegener. 2018, 13, 9. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, V.E.; Nikolopoulou, G.; Antoniadou, I.; Karachaliou, A.; Arianoglou, G.; Emmanouilidou, E.; Sardi, S.P.; Stefanis, L.; Vekrellis, K. Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum. Mol. Genet. 2018, 27, 1696–1710. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Eitan, E.; Wu, T.Y.; Mattson, M.P. Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol. Aging 2018, 61, 52–65. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Tredici, K.; Rüb, U.; de Vos, R.; Jansen Steur, E.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Paviour, D.; Price, S.; Lees, A.; Fox, N. MRI derived brain atrophy in PSP and MSA-P. J. Neurol. 2007, 254, 478–481. [Google Scholar] [CrossRef] [PubMed]
- Brettschneider, J.; Irwin, D.; Boluda, S.; Byrne, M.; Fang, L.; Lee, E.; Robinson, J.; Suh, E.; van Deerlin, V.; Toledo, J.; et al. Progression of α-synuclein pathology in multiple system atrophy of the cerebellar type. Neuropathol. Appl. Neurobiol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Luk, K.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.; Lee, V. Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science 2012, 338, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Osterberg, V.; Spinelli, K.; Weston, L.; Luk, K.; Woltjer, R.; Unni, V. Progressive Aggregation of α-Synuclein and Selective Degeneration of Lewy Inclusion-Bearing Neurons in a Mouse Model of Parkinsonism. Cell. Rep. 2015, 10, 1252–1260. [Google Scholar] [CrossRef] [PubMed]
- Paumier, K.; Luk, K.; Manfredsson, F.; Kanaan, N.; Lipton, J.; Collier, T.; Steece-Collier, K.; Kemp, C.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse α-synuclein fibrils into rats triggers α-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Sacino, A.; Brooks, M.; McGarvey, N.; McKinney, A.; Thomas, M.; Levites, Y.; Ran, Y.; Golde, T.; Giasson, B. Induction of CNS α-synuclein pathology by fibrillar and non-amyloidogenic recombinant α-synuclein. Acta Neuropathol. Commun. 2013, 1, 38. [Google Scholar] [CrossRef] [PubMed]
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of α-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778. [Google Scholar] [CrossRef] [PubMed]
- Desplats, P.; Lee, H.; Bae, E.; Patrick, C.; Rockenstein, E.; Crews, L.; Spencer, B.; Masliah, E.; Lee, S. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of α-synuclein. Proc. Natl. Acad. Sci. USA 2009, 106, 13010–13015. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Wang, Y.; Fong, H.; Murray, T.; Zhang, J. Identification of Proteins Involved in Microglial Endocytosis of α-Synuclein. J. Proteome Res. 2007, 6, 3614–3627. [Google Scholar] [CrossRef]
- Lee, H.; Suk, J.; Bae, E.; Lee, J.; Paik, S.; Lee, S. Assembly-dependent endocytosis and clearance of extracellular α-synuclein. Int. J. Biochem. Cell. Biol. 2008, 40, 1835–1849. [Google Scholar] [CrossRef] [PubMed]
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases α-synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38. [Google Scholar] [CrossRef]
- Mao, X.; Ou, M.; Karuppagounder, S.; Kam, T.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.; Brahmachari, S.; Shin, J.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353. [Google Scholar] [CrossRef] [PubMed]
- Schimmöller, F.; Diaz, E.; Mühlbauer, B.; Pfeffer, S. Characterization of a 76 kDa endosomal, multispanning membrane protein that is highly conserved throughout evolution. Gene 1998, 216, 311–318. [Google Scholar] [CrossRef]
- Wadman, M. Rogue protein’s partners offer hope in Parkinson’s disease. Science 2016, 354, 956. [Google Scholar] [CrossRef] [PubMed]
- Usenovic, M.; Tresse, E.; Mazzulli, J.; Taylor, J.; Krainc, D. Deficiency of ATP13A2 Leads to Lysosomal Dysfunction, α-Synuclein Accumulation, and Neurotoxicity. J. Neurosci. 2012, 32, 4240–4246. [Google Scholar] [CrossRef]
- Siebert, M.; Sidransky, E.; Westbroek, W. Glucocerebrosidase is shaking up the synucleinopathies. Brain 2014, 137, 1304–1322. [Google Scholar] [CrossRef] [PubMed]
- Ronzitti, G.; Bucci, G.; Emanuele, M.; Leo, D.; Sotnikova, T.D.; Mus, L.V.; Soubrane, C.H.; Dallas, M.L.; Thalhammer, A.; Cingolani, L.A.; et al. Exogenous α-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 2014. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.; Budka, H. Prion Diseases: From Protein to Cell Pathology. Am. J. Pathol. 2008, 172, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M. Alzheimer’s and Parkinson’s diseases: The prion concept in relation to assembled Aβ, tau, and α-synuclein. Science 2015, 349. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B.; Woerman, A.L.; Mordes, D.A.; Watts, J.C.; Rampersaud, R.; Berry, D.B.; Patel, S.; Oehler, A.; Lowe, J.K.; Kravitz, S.N.; et al. Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc. Natl. Acad. Sci. USA 2015, 112, E5308–E5317. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Englund, E.; Holton, J.; Soulet, D.; Hagell, P.; Lees, A.; Lashley, T.; Quinn, N.; Rehncrona, S.; Björklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503. [Google Scholar] [CrossRef]
- Kordower, J.; Chu, Y.; Hauser, R.; Freeman, T.; Olanow, C. Lewy body–like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506. [Google Scholar] [CrossRef] [PubMed]
- Ahn, T.; Langston, J.; Aachi, V.; Dickson, D. Relationship of neighboring tissue and gliosis to α-synuclein pathology in a fetal transplant for Parkinson’s disease. Am. J. Neurodegener. Dis. 2012, 1, 49–59. [Google Scholar]
- Iwai, A. Properties of NACP/α-synuclein and its role in Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2000, 1502, 95–109. [Google Scholar] [CrossRef]
- Rodriguez, J.; Ivanova, M.; Sawaya, M.; Cascio, D.; Reyes, F.; Shi, D.; Sangwan, S.; Guenther, E.; Johnson, L.; Zhang, M.; et al. Structure of the toxic core of α-synuclein from invisible crystals. Nature 2015, 525, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Cookson, M. α-Synuclein and neuronal cell death. Mol. Neurodegener. 2009, 4, 9. [Google Scholar] [CrossRef]
- Recasens, A.; Dehay, B.; Bové, J.; Carballo-Carbajal, I.; Dovero, S.; Pérez-Villalba, A.; Fernagut, P.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362. [Google Scholar] [CrossRef]
- Peng, C.; Gathagan, R.J.; Covell, D.J.; Medellin, C.; Stieber, A.; Robinson, J.L.; Zhang, B.; Pitkin, R.M.; Olufemi, M.F.; Luk, K.C.; et al. Cellular milieu imparts distinct pathological α-synuclein strains in α-synucleinopathies. Nature 2018, 557, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Cykowski, M.; Coon, E.; Powell, S.; Jenkins, S.; Benarroch, E.; Low, P.; Schmeichel, A.; Parisi, J. Expanding the spectrum of neuronal pathology in multiple system atrophy. Brain 2015, 138, 2293–2309. [Google Scholar] [CrossRef]
- Woerman, A.L.; Kazmi, S.A.; Patel, S.; Aoyagi, A.; Oehler, A.; Widjaja, K.; Mordes, D.A.; Olson, S.H.; Prusiner, S.B. Familial Parkinson’s point mutation abolishes multiple system atrophy prion replication. Proc. Natl. Acad. Sci. USA 2018, 115, 409–414. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Mori, F.; Kon, T.; Tanji, K.; Miki, Y.; Tomiyama, M.; Kurotaki, H.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; et al. Accumulation of phosphorylated α-synuclein in subpial and periventricular astrocytes in multiple system atrophy of long duration. Neuropathology 2016, 36, 157–167. [Google Scholar] [CrossRef]
- Braak, H.; Sastre, M.; Del Tredici, K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 2007, 114, 231–241. [Google Scholar] [CrossRef]
- Piao, Y.; Mori, F.; Hayashi, S.; Tanji, K.; Yoshimoto, M.; Kakita, A.; Wakabayashi, K.; Takahashi, H. α-Synuclein pathology affecting Bergmann glia of the cerebellum in patients with α-synucleinopathies. Acta Neuropathol. 2003, 105, 403–409. [Google Scholar] [PubMed]
- Recasens, A.; Carballo-Carbajal, I.; Parent, A.; Bové, J.; Gelpi, E.; Tolosa, E.; Vila, M. Lack of pathogenic potential of peripheral α-synuclein aggregates from Parkinson’s disease patients. Acta Neuropathol. Commun. 2018, 6, 8. [Google Scholar] [CrossRef] [PubMed]
- Holmes, B.; DeVos, S.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.; Brodsky, F.; Marasa, J.; Bagchi, D.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, E3138–E3147. [Google Scholar] [CrossRef] [PubMed]
- Ihse, E.; Yamakado, H.; Wijk, X.; Lawrence, R.; Esko, J.; Masliah, E. Cellular Internalization of alpha-synuclien aggregagtes by cell surface heparin sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Buchinger, E.; Brueck, D.; Irschick, R.; Wenning, G.K.; Stefanova, N. Limited effects of dysfunctional macroautophagy on the accumulation of extracellularly derived α-synuclein in oligodendroglia: Implications for MSA pathogenesis. BMC Neurosci. 2018, 19, 32. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef]
- De Keyser, J.; Mostert, J.P.; Koch, M.W. Dysfunctional astrocytes as key players in the pathogenesis of central nervous system disorders. J. Neurol. Sci. 2008, 267, 3–16. [Google Scholar] [CrossRef]
- Iglesias, J.; Morales, L.; Barreto, G.E. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol. Neurobiol. 2017, 54, 2518–2538. [Google Scholar] [CrossRef]
- Radford, R.; Rcom-H’cheo-Gauthier, A.; Wong, M.B.; Eaton, E.D.; Quilty, M.; Blizzard, C.; Norazit, A.; Meedeniya, A.; Vickers, J.C.; Gai, W.P.; et al. The degree of astrocyte activation in multiple system atrophy is inversely proportional to the distance to α-synuclein inclusions. Mol. Cell. Neurosci. 2015, 65, 68–81. [Google Scholar] [CrossRef]
- Vieira, B.D.; Radford, R.A.; Chung, R.S.; Guillemin, G.J.; Pountney, D.L. Neuroinflammation in Multiple System Atrophy: Response to and Cause of α-Synuclein Aggregation. Front. Cell. Neurosci. 2015, 9, 437. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Reindl, M.; Neumann, M.; Haass, C.; Poewe, W.; Kahle, P.J.; Wenning, G.K. Oxidative stress in transgenic mice with oligodendroglial alpha-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. Am. J. Pathol. 2005, 166, 869–876. [Google Scholar] [CrossRef]
- Lee, H.J.; Suk, J.E.; Patrick, C.; Bae, E.J.; Cho, J.H.; Rho, S.; Hwang, D.; Masliah, E.; Lee, S.J. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J. Biol. Chem. 2010, 285, 9262–9272. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.; Stefanova, N. Toll-like receptor 4 is required for α-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef] [PubMed]
- Fellner, L.; Stefanova, N. The Role of Glia in A-Synucleinopathies. Mol. Neurobiol. 2012, 47, 575–586. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Li, W.J.; Huang, X.S. Alpha-synuclein levels in patients with multiple system atrophy: A meta-analysis. Int. J. Neurosci. 2018, 128, 477–486. [Google Scholar] [CrossRef] [PubMed]
- Jessen, N.; Munk, A.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Iliff, J.; Wang, M.; Liao, Y.; Plogg, B.; Peng, W.; Gundersen, G.; Benveniste, H.; Vates, G.; Deane, R.; Goldman, S.; et al. A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Sci. Transl. Med. 2012, 4. [Google Scholar] [CrossRef]
- Iliff, J.; Chen, M.; Plog, B.; Zeppenfeld, D.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of Glymphatic Pathway Function Promotes Tau Pathology after Traumatic Brain Injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef]
- Hoshi, A.; Tsunoda, A.; Tada, M.; Nishizawa, M.; Ugawa, Y.; Kakita, A. Expression of aquaporin 1 and aquaporin 4 in the temporal neocortex of patients with Parkinson’s disease. Brain Pathol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Tang, H.; Nie, K.; Wang, L.; Zhao, J.; Gan, R.; Huang, J.; Zhu, R.; Feng, S.; Duan, Z.; et al. Cerebrospinal fluid α-synuclein as a biomarker for Parkinson’s disease diagnosis: A systematic review and meta-analysis. Int. J. Neurosci. 2014, 125, 645–654. [Google Scholar] [CrossRef] [PubMed]
- Lundgaard, I.; Lu, M.; Yang, E.; Peng, W.; Mestre, H.; Hitomi, E.; Deane, R.; Nedergaard, M. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J. Cereb. Blood Flow Metable. 2016. [Google Scholar] [CrossRef] [PubMed]
- Halliday, G.M.; Stevens, C.H. Glia: Initiators and progressors of pathology in Parkinson’s disease. Mov. Disord. 2011, 26, 6–17. [Google Scholar] [CrossRef] [PubMed]
- Prinz, M.; Priller, J.; Sisodia, S.S.; Ransohoff, R.M. Heterogeneity of CNS myeloid cells and their roles in neurodegeneration. Nat. Neurosci. 2011, 14, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 2015. [CrossRef] [PubMed]
- Husemann, J.; Loike, J.D.; Anankov, R.; Febbraio, M.; Silverstein, S.C. Scavenger receptors in neurobiology and neuropathology: Their role on microglia and other cells of the nervous system. Glia 2002, 40, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Scheffel, J.; Regen, T.; VanRossum, D.; Seifert, S.; Ribes, S.; Nau, R. Toll-like receptor activation reveals developmental reorganization and unmasks responder subsets of microglia. Glia 2012, 60, 1930–1943. [Google Scholar] [CrossRef] [PubMed]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Colton, C.A.; Wilcock, D.M. Assessingactivationstates in microglia. CNS Neurol. Disord. Drug Targets 2010, 9, 174–191. [Google Scholar] [CrossRef] [PubMed]
- Welser-Alves, J.V.; Milner, R. Microglia are the major source of TNF-a and TGF-b1 in post natal glial cultures;regulationbycytokines, lipopolysaccharide andvitronectin. Neurochem. Int. 2013, 63, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Yan, Z.F.; Gao, J.H.; Sun, L.; Huang, X.Y.; Liu, Z.; Yu, S.Y.; Cao, C.J.; Zuo, L.J.; Chen, Z.J.; et al. Role and mechanism of microglial activation in iron-induced selective and progressive dopaminergic neurodegeneration. Mol. Neurobiol. 2014, 49, 1153–1165. [Google Scholar] [CrossRef] [PubMed]
- Wenning, G.; Stefanova, N. Recent developments in multiple system atrophy. J. Neurol. 2009, 256, 1791–1808. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, E.; Hall, S.; Surova, Y.; Widner, H.; Hansson, O.; Lindqvist, D. Proinflammatory Cytokines Are Elevated in Serum of Patients with Multiple System Atrophy. PLoS ONE 2013, 8, e62354. [Google Scholar] [CrossRef] [PubMed]
- Koga, S.; Aoki, N.; Uitti, R.; van Gerpen, J.; Cheshire, W.; Josephs, K.; Wszolek, Z.; Langston, J.; Dickson, D. When DLB, PD, and PSP masquerade as MSA. Neurology 2015, 85, 404–412. [Google Scholar] [CrossRef] [PubMed]
- Daniele, S.G.; Béraud, D.; Davenport, C.; Cheng, K.; Yin, H.; Maguire-Zeiss, K.A. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci. Signal. 2015. [Google Scholar] [CrossRef] [PubMed]
- Ejlerskov, P.; Hultberg, J.G.; Wang, J.; Carlsson, R.; Ambjørn, M.; Kuss, M.; Liu, Y.; Porcu, G.; Kolkova, K.; Friis Rundsten, C.; et al. Lack of neuronal IFN-b-IFNAR causes lewy body-and parkinson’s disease-like dementia. Cell 2015, 163, 324–339. [Google Scholar] [CrossRef] [PubMed]
- Tien, L.-T.; Kaizaki, A.; Pang, Y.; Cai, Z.; Bhatt, A.J.; Fan, L.-W. Neonatal exposure tolipopolysaccharide enhances accumulation of a-synuclein aggregation and dopamine transporter proteinexpression in the substantia nigrainresponsestorotenonechallenge inlater life. Toxicology 2013, 308, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Béraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K.A. a-Synuclein alters toll-like receptor expression. Front. Neurosci. 2011, 5, 80. [Google Scholar] [CrossRef] [PubMed]
- Brudek, T.; Winge, K.; Agander, T.K.; Pakkenberg, B. Screening of Toll-like receptors expression inmultiple system atrophy brains. Neurochem. Res. 2013, 38, 1252–1259. [Google Scholar] [CrossRef]
- Fitzner, D.; Schnaars, M.; van Rossum, D.; Krishnamoorthy, G.; Dibaj, P.; Bakhti, M.; Regen, T.; Hanisch, U.; Simons, M. Selective transfer of exosomes from oligodendrocytes to microglia by macropinocytosis. J. Cell Sci. 2011, 124, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Rockenstein, E.; Ostroff, G.; Dikengil, F.; Rus, F.; Mante, M.; Florio, J.; Adame, A.; Trinh, I.; Kim, C.; Overk, C.; et al. Combined Active Humoral and Cellular Immunization Approaches for the Treatment of Synucleinopathies. J. Neurosci. 2018, 38, 1000–1014. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Kim, K.; Lee, S.; Ryu, J.; Chung, K.; Choo, Y.; Jou, I.; Kim, J.; Park, S. On the mechanism of internalization of α-synuclein into microglia: Roles of ganglioside GM1 and lipid raft. J. Neurochem. 2009, 110, 400–411. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Chu, C.; Stewart, T.; Ginghina, C.; Wang, Y.; Nie, H.; Guo, M.; Wilson, B.; Hong, J.; Zhang, J. α-Synuclein, a chemoattractant, directs microglial migration via H2O2-dependent Lyn phosphorylation. Proc. Natl. Acad. Sci. USA 2015, 112, E1926–E1935. [Google Scholar] [CrossRef] [PubMed]
- Su, X.; Maguire-Zeiss, K.; Giuliano, R.; Prifti, L.; Venkatesh, K.; Federoff, H. Synuclein activates microglia in a model of Parkinson’s disease. Neurobiol. Aging 2008, 29, 1690–1701. [Google Scholar] [CrossRef] [PubMed]
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G. Toll-Like Receptor 4 Promotes α-Synuclein Clearance and Survival of Nigral Dopaminergic Neurons. Am. J. Pathol. 2011, 179, 954–963. [Google Scholar] [CrossRef]
- Kim, C.; Ho, D.; Suk, J.; You, S.; Michael, S.; Kang, J.; Joong Lee, S.; Masliah, E.; Hwang, D.; Lee, H.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2013, 4, 1562. [Google Scholar] [CrossRef]
- Roodveldt, C.; Labrador-Garrido, A.; Gonzalez-Rey, E.; Lachaud, C.; Guilliams, T.; Fernandez-Montesinos, R.; Benitez-Rondan, A.; Robledo, G.; Hmadcha, A.; Delgado, M.; et al. Preconditioning of Microglia by α-Synuclein Strongly Affects the Response Induced by Toll-like Receptor (TLR) Stimulation. PLoS ONE 2013, 8, e79160. [Google Scholar] [CrossRef]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584–1593. [Google Scholar] [CrossRef] [PubMed]
- Valdinocci, D.; Radford, R.A.; Siow, S.M.; Chung, R.S.; Pountney, D.L. Potential Modes of Intercellular α-Synuclein Transmission. Int. J. Mol. Sci. 2017, 18, e469. [Google Scholar] [CrossRef]
- Valdinocci, D.; Grant, G.D.; Dickson, T.C.; Pountney, D.L. Epothilone D inhibits microglia-mediated spread of alpha-synuclein aggregates. Mol. Cell. Neurosci. 2018, 89, 80–94. [Google Scholar] [CrossRef]
- Pan-Montojo, F.; Schwarz, M.; Winkler, C.; Arnhold, M.; O’Sullivan, G.A.; Pal, A.; Said, J.; Marsico, G.; Verbavatz, J.M.; Rodrigo-Angulo, M.; Gille, G.; Funk, R.H. Reichmann Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci. Rep. 2012, 2, 898. [Google Scholar] [CrossRef] [PubMed]
- Paillusson, S.; Clairembault, T.; Biraud, M.; Neunlist, M.; Derkinderen, P. Activity-dependent secretion of alpha-synuclein by enteric neurons. J. Neurochem. 2013, 125, 512–517. [Google Scholar] [CrossRef] [PubMed]
- Grathwohl, S.A.; Steiner, J.A.; Britschgi, M.; Brundin, P. Mind the gut: Secretion of α-synuclein by enteric neurons. J. Neurochem. 2013, 125, 487–490. [Google Scholar] [CrossRef] [PubMed]
- Braidy, N.; Gai, W.P.; Xu, Y.H.; Sachdev, P.; Guillemin, G.J.; Jiang, X.M.; Ballard, J.W.; Horan, M.P.; Fang, Z.M.; Chong, B.H.; Chan, D.K. Alpha-Synuclein Transmission and Mitochondrial Toxicity in Primary Human Foetal Enteric Neurons in Vitro. Neurotox. Res. 2014, 25, 170–182. [Google Scholar] [CrossRef] [PubMed]
- Haehner, A.; Hummel, T.; Reichmann, H. Olfactory dysfunction as a diagnostic marker for Parkinson’s disease. Expert Rev. Neurother. 2009, 9, 1773–1779. [Google Scholar] [CrossRef] [PubMed]
- Iranzo, A.; Molinuevo, J.L.; Santamaría, J.; Serradell, M.; Martí, M.J.; Valldeoriola, F.; Tolosa, E. Rapid-eye-movement sleep behaviour disorder as an early marker for an eurodegenerative disorder: A descriptive study. Lancet Neurol. 2006, 5, 572–577. [Google Scholar] [CrossRef]
- Cersosimo, M.G.; Raina, G.B.; Pecci, C.; Pellene, A.; Calandra, C.R.; Gutiérrez, C.; Micheli, F.E.; Benarroch, E.E. Gastrointestinal manifestations in Parkinson’s disease: Prevalence and occurrence before motor symptoms. J. Neurol. 2013, 260, 1332–1338. [Google Scholar] [CrossRef]
- Edwards, L.L.; Quigley, E.M.; Pfeifer, R.F. Gastrointestinal dysfunction in Parkinson’s disease: Frequency and pathophysiology. Neurology 1992, 42, 726–732. [Google Scholar] [CrossRef]
- Reichmann, H.; Schneider, C.; Löhle, M. Non-motor features of Parkinson’s disease: Depression and dementia. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S87–S92. [Google Scholar] [CrossRef]
- Braak, H.; de Vos, R.A.I.; Bohl, J.; Del Tredici, K. Gastric alpha-synuclein immunoreactive inclusions in Meissner’s and Auerbach‘s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci. Lett. 2006, 396, 67–72. [Google Scholar] [CrossRef] [PubMed]
- Reichmann, H. View point: Etiology in Parkinson’s disease. Dual hit or spreading intoxication. J. Neurol. Sci. 2011, 310, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Rüb, U.; Gai, W.P.; Del Tredici, K. Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J. Neural Transm. (Vienna) 2003, 110, 517–536. [Google Scholar] [CrossRef] [PubMed]
- Hawkes, C.H.; Del Tredici, K.; Braak, H. Parkinson’s disease: A dual-hit hypothesis. Neuropathol. Appl. Neurobiol. 2007, 33, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Svensson, E.; Horváth-Puhó, E.; Thomsen, R.W.; Djurhuus, J.C.; Pedersen, L.; Borghammer, P.; Sørensen, H.T. Vagotomy and subsequent risk of Parkinson’s disease. Ann. Neurol. 2015, 78, 522–529. [Google Scholar] [CrossRef]
- Liu, B.; Fang, F.; Pedersen, N.L.; Tillander, A.; Ludvigsson, J.F.; Ekbom, A.; Svenningsson, P.; Chen, H.; Wirdefeldt, K. Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 2017, 88, 1996–2002. [Google Scholar] [CrossRef]
- Tysnes, O.-B.; Kenborg, L.; Herlofson, K.; Steding-Jessen, M.; Horn, A.; Olsen, J.H.; Reichmann, H. Does vagotomy reduce the risk of Parkinson’s disease? Ann. Neurol. 2015, 78, 1011–1012. [Google Scholar] [CrossRef]
- Adler, C.H.; Beach, T.G. Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord. 2016, 31, 1114–1119. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K.A. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol. Appl Neurobiol. 2008, 34, 466–467. [Google Scholar]
- Beach, T.G.; Adler, C.H.; Sue, L.I.; Vedders, L.; Lue, L.; White Iii, C.L.; Akiyama, H.; Caviness, J.N.; Shill, H.A.; Sabbagh, M.N.; Walker, D.G. Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol. 2010, 119, 689–702. [Google Scholar] [CrossRef]
- Kalaitzakis, M.E.; Graeber, M.B.; Gentleman, S.M.; Pearce, R.K.B. The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: A critical analysis of α-synuclein staging. Neuropathol. Appl. Neurobiol. 2008, 34, 284–295. [Google Scholar] [CrossRef] [PubMed]
- Holmqvist, S.; Chutna, O.; Bousset, L.; Aldrin-Kirk, P.; Li, W.; Björklund, T.; Wang, Z.Y.; Roybon, L.; Melki, R.; Li, J.Y. Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol. 2014, 128, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Ulusoy, A.; Phillips, R.J.; Helwig, M.; Klinkenberg, M.; Powley, T.L.; Di Monte, D.A. Brain-to-stomach transfer of α-synuclein via vagal preganglionic projections. Acta Neuropathol. 2017, 133, 381–393. [Google Scholar] [CrossRef] [PubMed]
- Sampson, T.R.; Debelius, J.W.; Thron, T.; Janssen, S.; Shastri, G.G.; Ilhan, Z.E.; Challis, C.; Schretter, C.E.; Rocha, S.; Gradinaru, V.; Chesselet, M.F.; Keshavarzian, A.; Shannon, K.M.; Krajmalnik-Brown, R.; Wittung-Stafshede, P.; Knight, R.; Mazmanian, S.K. Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 2016, 167, 1469–1480. [Google Scholar] [CrossRef] [PubMed]
- Pan-Montojo, F.; Anichtchik, O.; Dening, Y.; Knels, L.; Pursche, S.; Jung, R.; Jackson, S.; Gille, G.; Spillantini, M.G.; Reichmann, H.; Funk, R.H. Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS ONE 2010, 5, e8762. [Google Scholar] [CrossRef] [PubMed]
- Tasselli, M.; Chaumette, T.; Paillusson, S.; Monnet, Y.; Lafoux, A.; Huchet-Cadiou, C.; Aubert, P.; Hunot, S.; Derkinderen, P.; Neunlist, M. Effects of oral administration of rotenone on gastrointestinal functions in mice. Neurogastroenterol. Motil. 2013, 25, e183–e193. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Oertel, W.H.; Hirsch, E.C. The rotenone model of parkinsonism–the five years inspection. J. Neural Transm. Suppl. 2006, 70, 269–272. [Google Scholar]
- Killinger, B.A.; Madaj, Z.; Sikora, J.W.; Rey, N.; Haas, A.J.; Vepa, Lindqvist, Y.D.; Chen, H.; Thomas, P.M.; Brundin, P.; Brundin, L.; et al. The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci. Transl. Med. 2018, 10, eaar5280. [Google Scholar] [CrossRef]
- Pouclet, H.; Lebouvier, T.; Coron, E.; Rouaud, T.; Flamant, M.; Toulgoat, F.; Roy, M.; Vavasseur, F.; Bruley des Varannes, S.; Neunlist, M.; Derkinderen, P. Analysis of colonic alpha-synuclein pathology in multiple system atrophy. Park. Relat. Disord. 2012, 18, 893–895. [Google Scholar] [CrossRef]
- Wyatt, A.R.; Yerbury, J.J.; Ecroyd, H.; Wilson, M.R. Extracellular Chaperones and Proteostasis. Annu. Rev. Biochem. 2013, 82, 295–322. [Google Scholar] [CrossRef]
- Tatebe, H.; Watanabe, Y.; Kasai, T.; Mizuno, T.; Nakagawa, M.; Tanaka, M.; Tokuda, T. Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci. Res. 2010, 67, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Iwata, A.; Maruyama, M.; Akagi, T.; Hashikawa, T.; Kanazawa, I.; Tsuji, S.; Nukina, N. Alpha-synuclein degradation by serine protease neurosin: Implication for pathogenesis of synucleinopathies. Hum. Mol. Genet. 2003, 12, 2625–2635. [Google Scholar] [CrossRef] [PubMed]
- Kasai, T.; Tokuda, T.; Yamaguchi, N.; Watanabe, Y.; Kametani, F.; Nakagawa, M.; Mizuno, T. Cleavage of normal and pathological forms of α-synuclein by neurosin in vitro. Neurosci. Lett. 2008, 436, 52–56. [Google Scholar] [CrossRef]
- Kim, K.S.; Choi, Y.R.; Park, J.Y.; Lee, J.H.; Kim, D.K.; Lee, S.J.; Paik, S.R.; Jou, I.; Park, S.M. Proteolytic Cleavage of Extracellular α-Synuclein by Plasmin. J. Biol. Chem. 2012, 287, 24862–24872. [Google Scholar] [CrossRef] [PubMed]
- Gottschall, P.E.; Deb, S. Regulation of Matrix Metalloproteinase Expression in Astrocytes, Microglia and Neurons. Neuroimmunomodulation 1996, 3, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Park, S.M.; Lee, C.H.; Um, J.W.; Lee, H.J.; Kim, J.; Oh, Y.J.; Lee, S.T.; Paik, S.R.; Chung, K.C. Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J. Biol. Chem. 2005, 280, 25216–25224. [Google Scholar] [CrossRef]
- Vega, V.L.; Rodríguez-Silva, M.; Frey, T.; Gehrmann, M.; Diaz, J.C.; Steinem, C.; Multhoff, G.; Arispe, N.; De Maio, A. Hsp70 translocates into the plasma membrane after stress and is released into the extracellular environment in a membrane-associated form that activates macrophages. J. Immunol. 2008, 180, 4299–4307. [Google Scholar] [CrossRef]
- Klucken, J.; Shin, Y.; Masliah, E.; Hyman, B.T.; McLean, P.J. Hsp70 Reduces α-Synuclein Aggregation and Toxicity. J. Biol. Chem. 2004, 279, 25497–25502. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Källstig, E.; Limorenko, G.; Vendruscolo, M.; Ron, D.; Hansen, C. The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci. Rep. 2017, 7, 9039. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, J.P.; Shi, M.; Quinn, T.; Bradner, J.; Beyer, R.; Chen, S.; Zhang, J. Rab11a and HSP90 regulate recycling of extracellular alpha-synuclein. J. Neurosci. 2009, 29, 1480–1485. [Google Scholar] [CrossRef]
- Poon, S.; Treweek, T.M.; Wilson, M.R.; Easterbrook-Smith, S.B.; Carver, J.A. Clusterin is an extracellular chaperone that specifically interacts with slowly aggregating proteins on their off-folding pathway. FEBS Lett. 2002, 513, 259–266. [Google Scholar] [CrossRef]
- Yerbury, J.J.; Poon, S.; Meehan, S.; Thompson, B.; Kumita, J.R.; Dobson, C.M.; Wilson, M.R. The extracellular chaperone clusterin influences amyloid formation and toxicity by interacting with prefibrillar structures. FASEB J. 2007, 21, 2312–2322. [Google Scholar] [CrossRef]
- Sasaki, K.; Doh-ura, K.; Wakisaka, Y.; Iwaki, T. Clusterin/apolipoprotein J is associated with cortical Lewy bodies: Immunohistochemical study in cases with alpha-synucleinopathies. Acta Neuropathol. 2002, 104, 225–230. [Google Scholar]
- Yun, S.P.; Kam, T.I.; Panicker, N.; Kim, S.; Oh, Y.; Park, J.S.; Kwon, S.H.; Park, Y.J.; Karuppagounder, S.S.; Park, H.; et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat. Med. 2018, 24, 931–938. [Google Scholar] [CrossRef]
- Tong, J.; Wong, H.; Guttman, M.; Ang, L.C.; Forno, L.S.; Shimadzu, M.; Rajput, A.H.; Muenter, M.D.; Kish, S.J.; Hornykiewicz, O.; et al. Brain alpha-synuclein accumulation in multiple system atrophy, Parkinson’s disease and progressive supranuclear palsy: A comparative investigation. Brain 2010, 133, 172–188. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| MSA | PD | DLB | |
|---|---|---|---|
| Biochemistry | |||
| SDS soluble |  | variable | n.d. |
| CSFα-syn | |||
| Total |  | | |
| Oligomeric | | | |
| Neurons | |||
| Neuronal Loss | + | + | + |
| α-syn pathology | ± | + | + |
| Astrocytes | |||
| Gliosis | + | +/− activation | Some activation |
| α-syn pathology | ± | + | ± |
| Astrocyte subtype | Subpial, ventricular and perivascular | Protoplasmic | Subpial, ventricular and perivascular |
| Oligodendrocytes | +/− | ||
| α-syn pathology | + | +/− | |
| Oligodendrocyte subtype | Myelinating | Non-myelinating | |
| Pathological staging | Primary event | Late Stage | |
| Microglia | Activation | Activation | Activation |
| Prion Transmission | |||
| Fetal grafts | ? | + | ? |
| Rodent models | + | ± | ? |
| Primate models | ? | ± | ? |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valdinocci, D.; Radford, R.A.W.; Goulding, M.; Hayashi, J.; Chung, R.S.; Pountney, D.L. Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy. Int. J. Mol. Sci. 2018, 19, 4129. https://doi.org/10.3390/ijms19124129
Valdinocci D, Radford RAW, Goulding M, Hayashi J, Chung RS, Pountney DL. Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy. International Journal of Molecular Sciences. 2018; 19(12):4129. https://doi.org/10.3390/ijms19124129
Chicago/Turabian StyleValdinocci, Dario, Rowan A. W. Radford, Michael Goulding, Junna Hayashi, Roger S. Chung, and Dean L. Pountney. 2018. "Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy" International Journal of Molecular Sciences 19, no. 12: 4129. https://doi.org/10.3390/ijms19124129
APA StyleValdinocci, D., Radford, R. A. W., Goulding, M., Hayashi, J., Chung, R. S., & Pountney, D. L. (2018). Extracellular Interactions of Alpha-Synuclein in Multiple System Atrophy. International Journal of Molecular Sciences, 19(12), 4129. https://doi.org/10.3390/ijms19124129