Proteomics Analysis Reveals That Caspase-Like and Metacaspase-Like Activities Are Dispensable for Activation of Proteases Involved in Early Response to Biotic Stress in Triticum aestivum L.

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
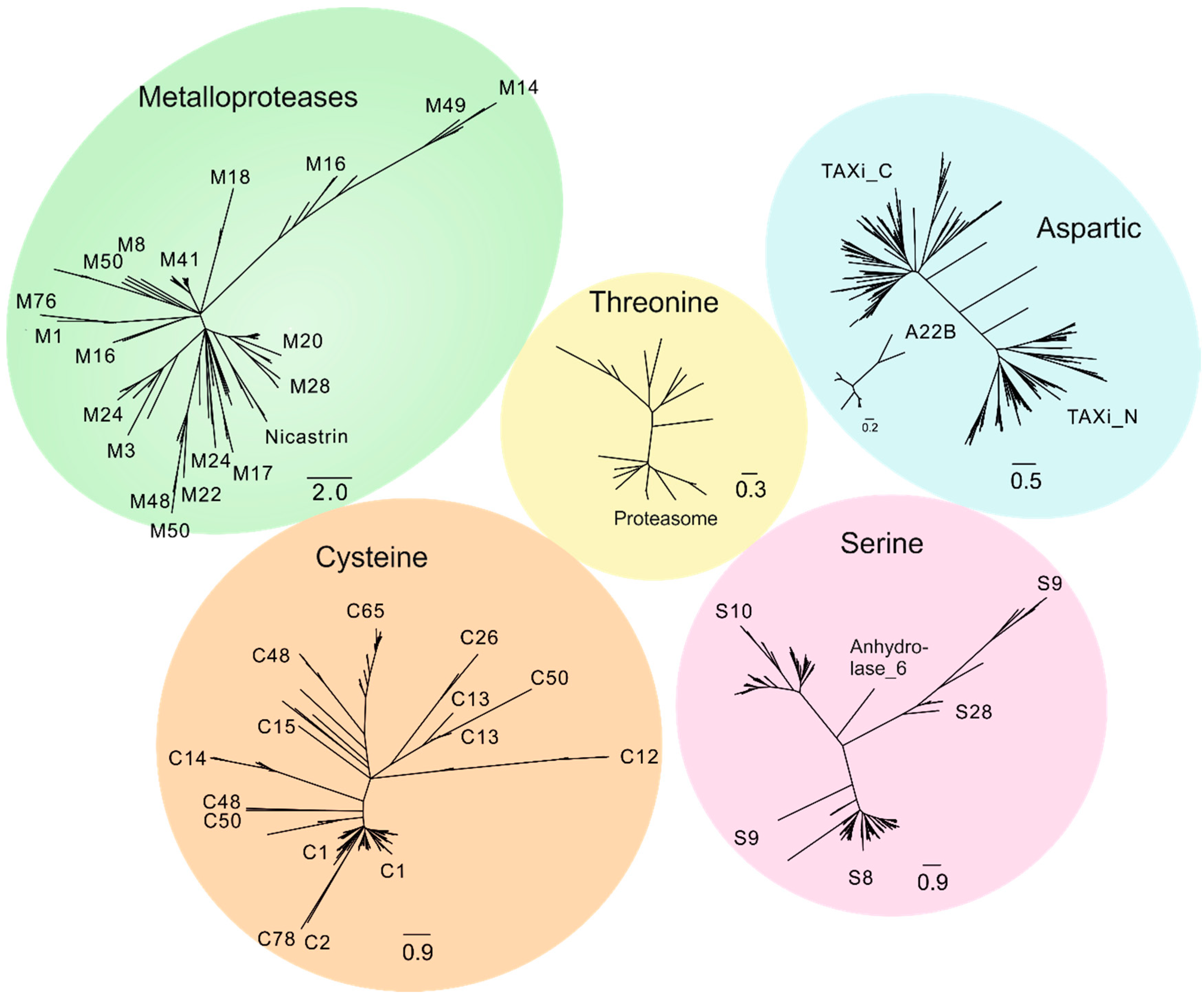
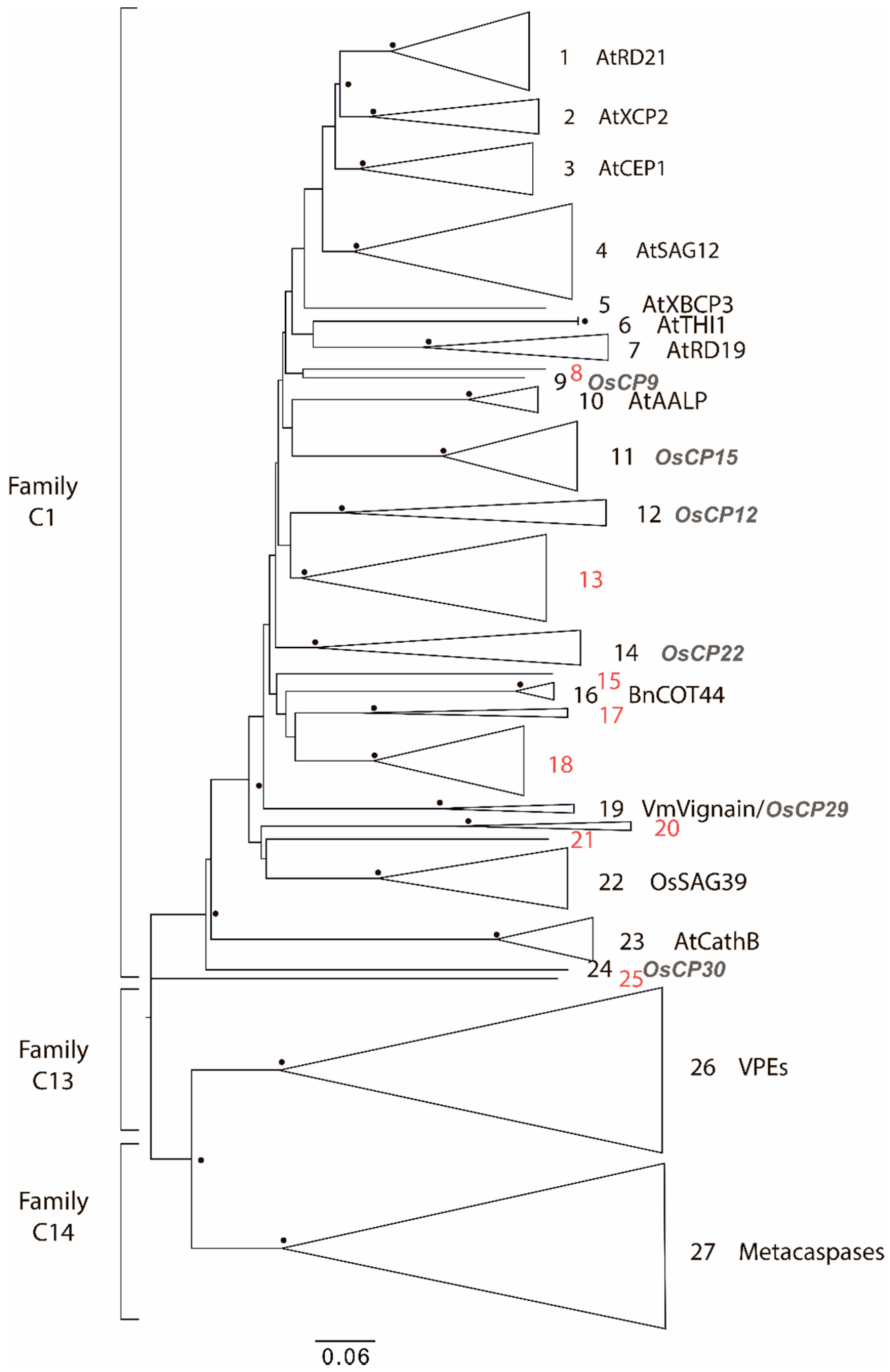
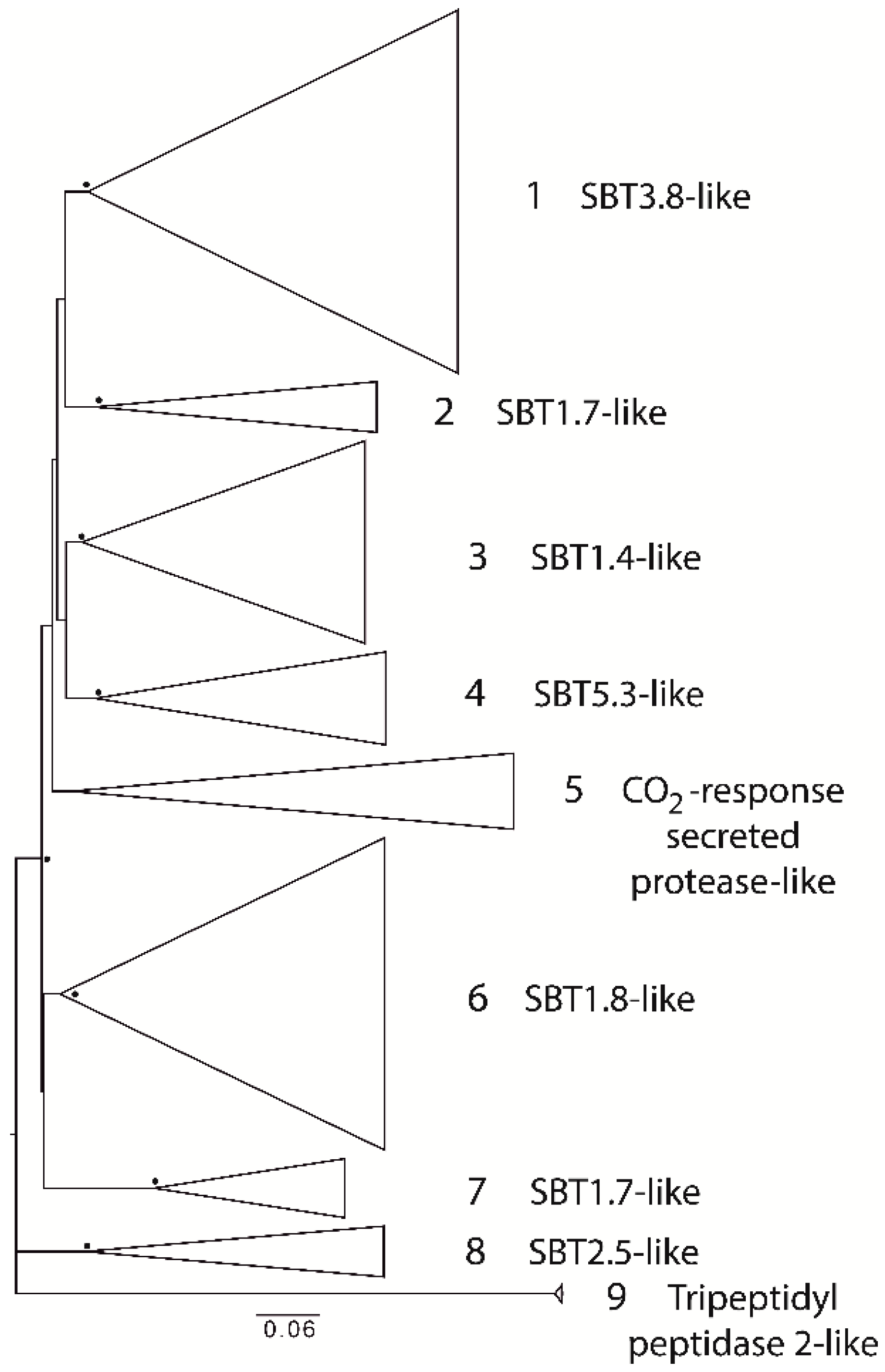
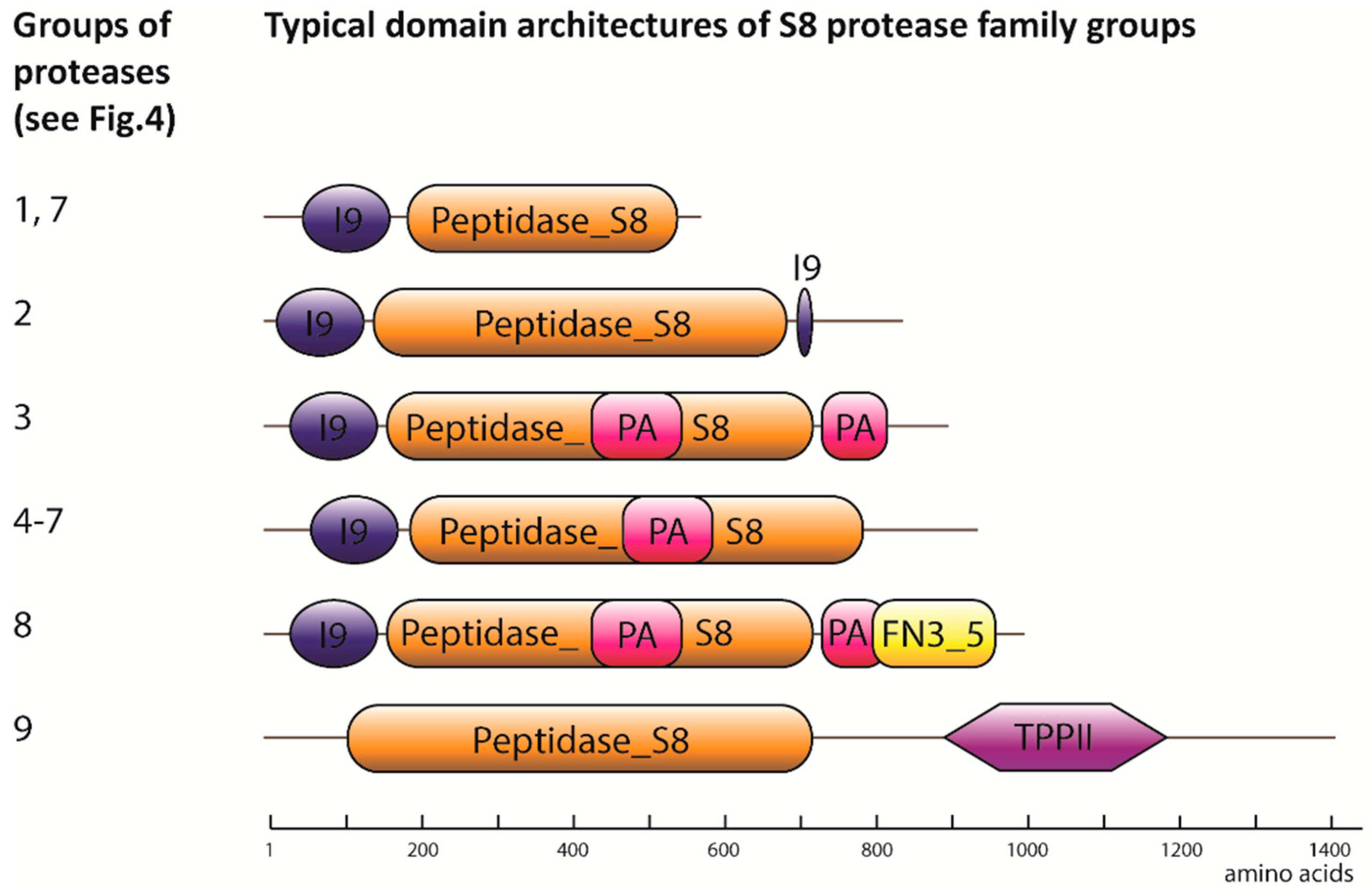
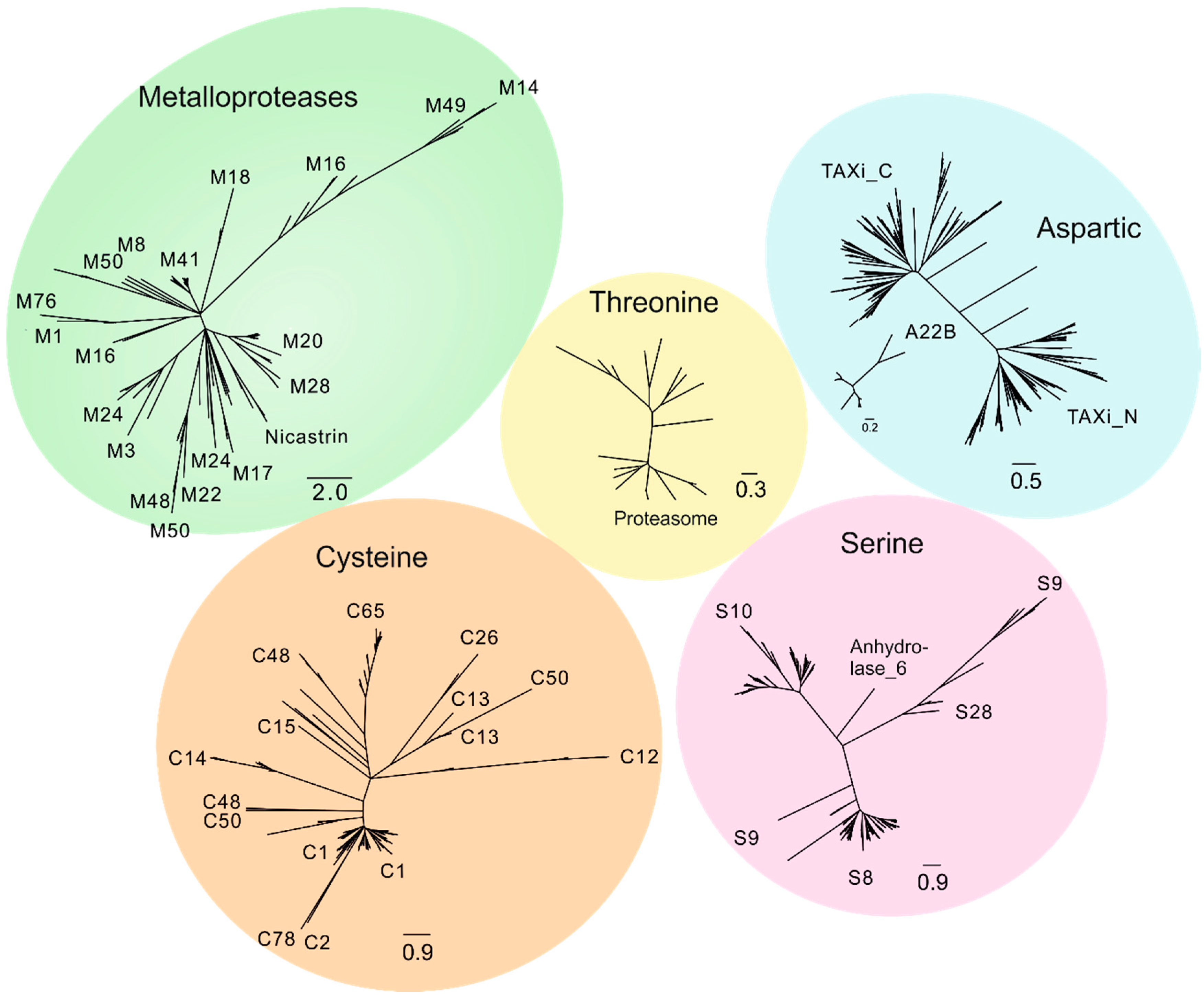
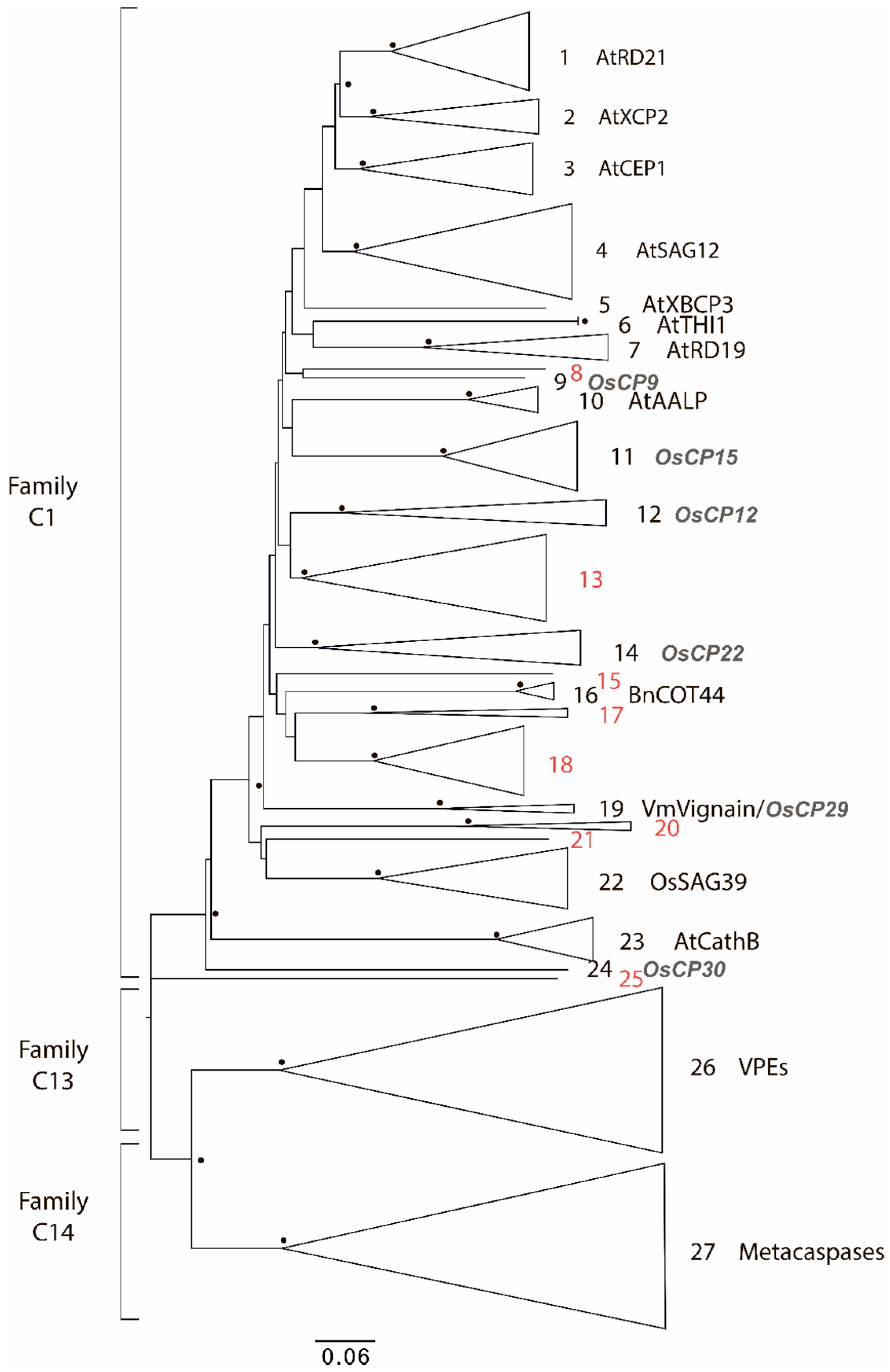
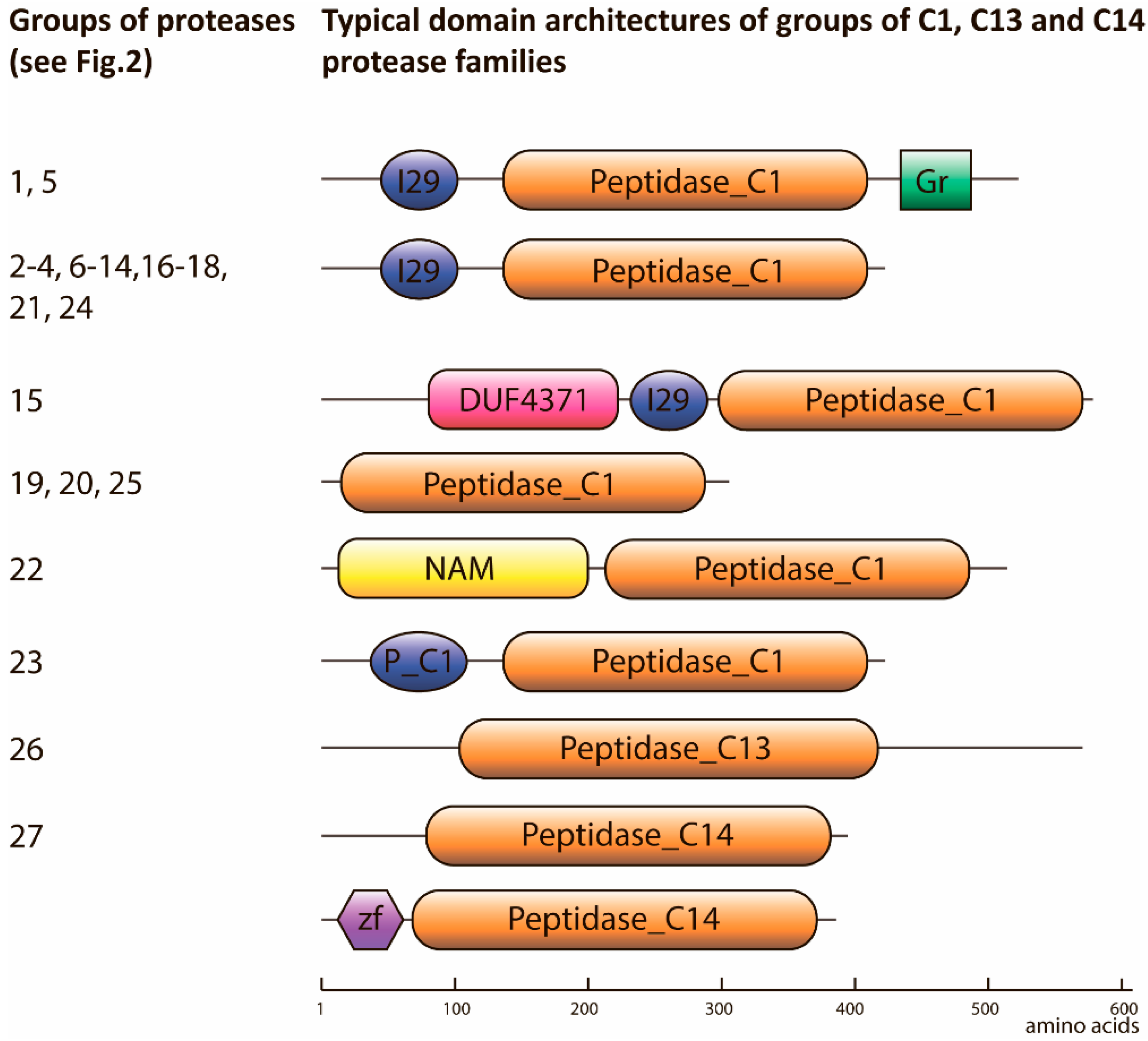
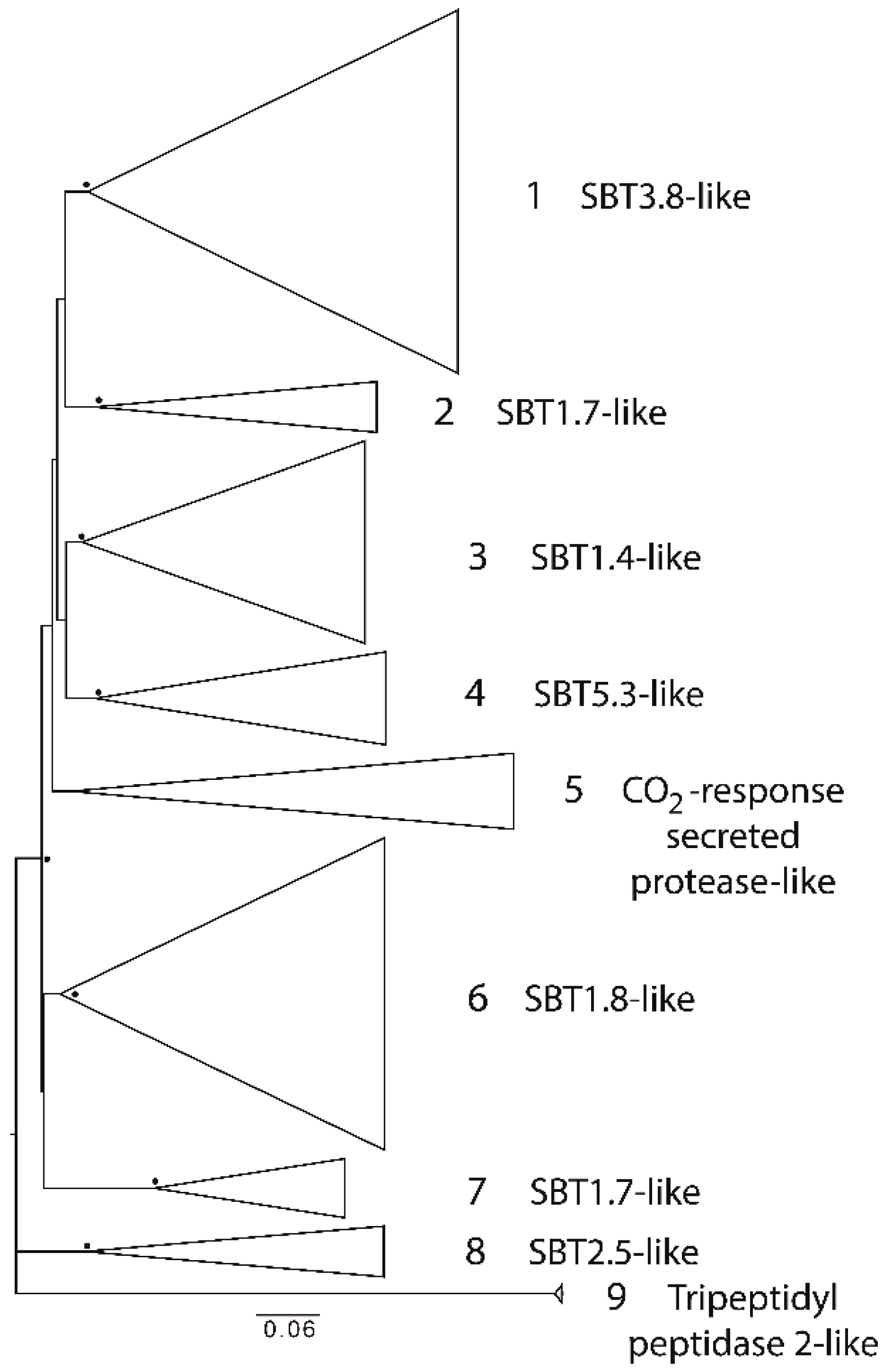
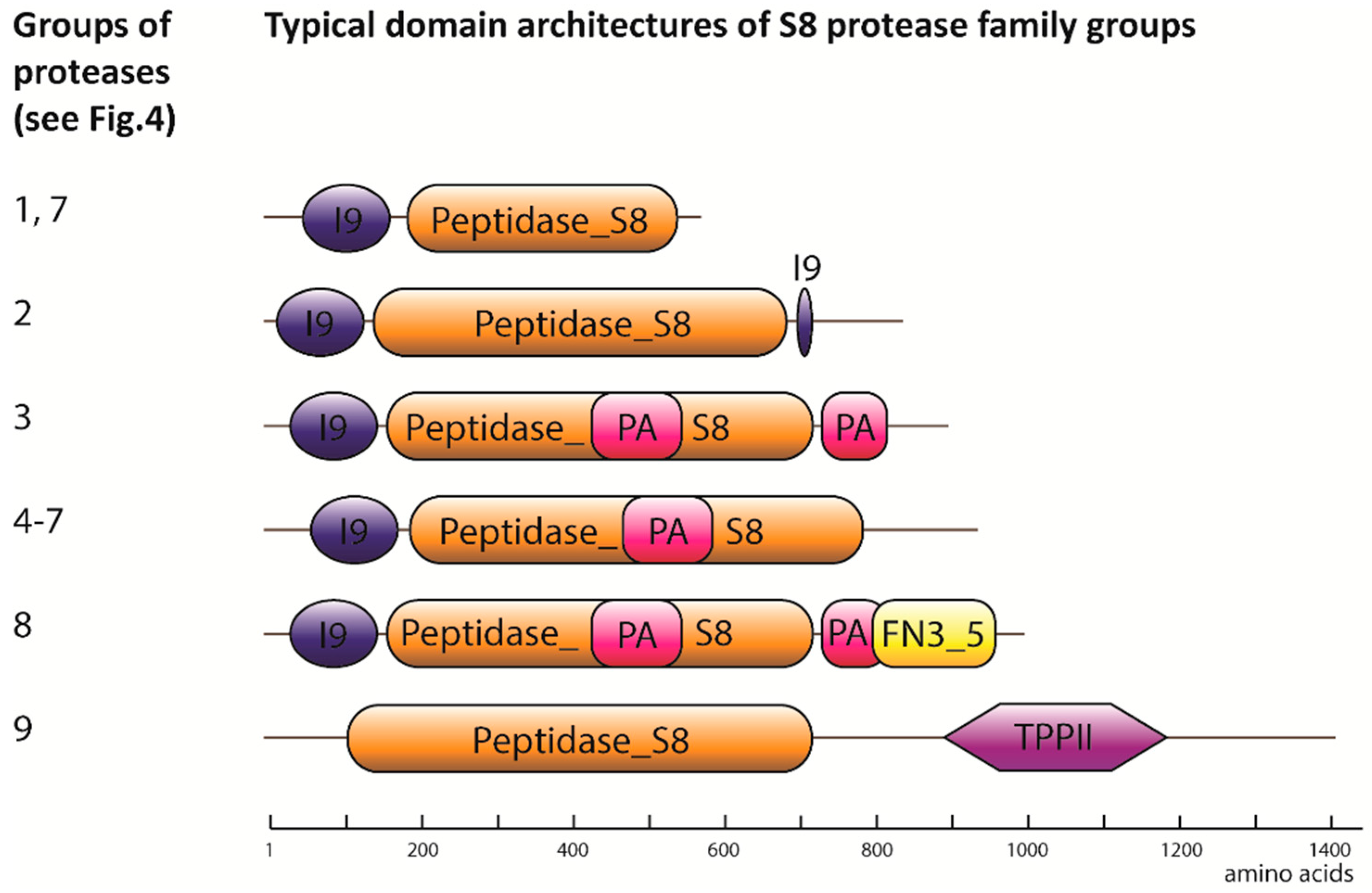
2.1. Degradome of T. aestivum Is Represented by Diverse Protease Families
- Check the common node for two leaves. If the bootstrap value for this node is more than 70, then merge these leaves into one subgroup. Otherwise, these leaves should be considered as distinct, independent groups. Repeat this step as many times as needed.
- Check the common node for the node with a low bootstrap value. If the bootstrap value for the upper node is more than 70, then subgroups should be considered as members of a larger group. However, the topology of tree nodes with a low bootstrap value cannot be resolved, although a highly credible, common node clearly indicates the shared origin of these subgroups.
- If uniquely specific features are shown as representative of some subgroups, they should be considered as independent.
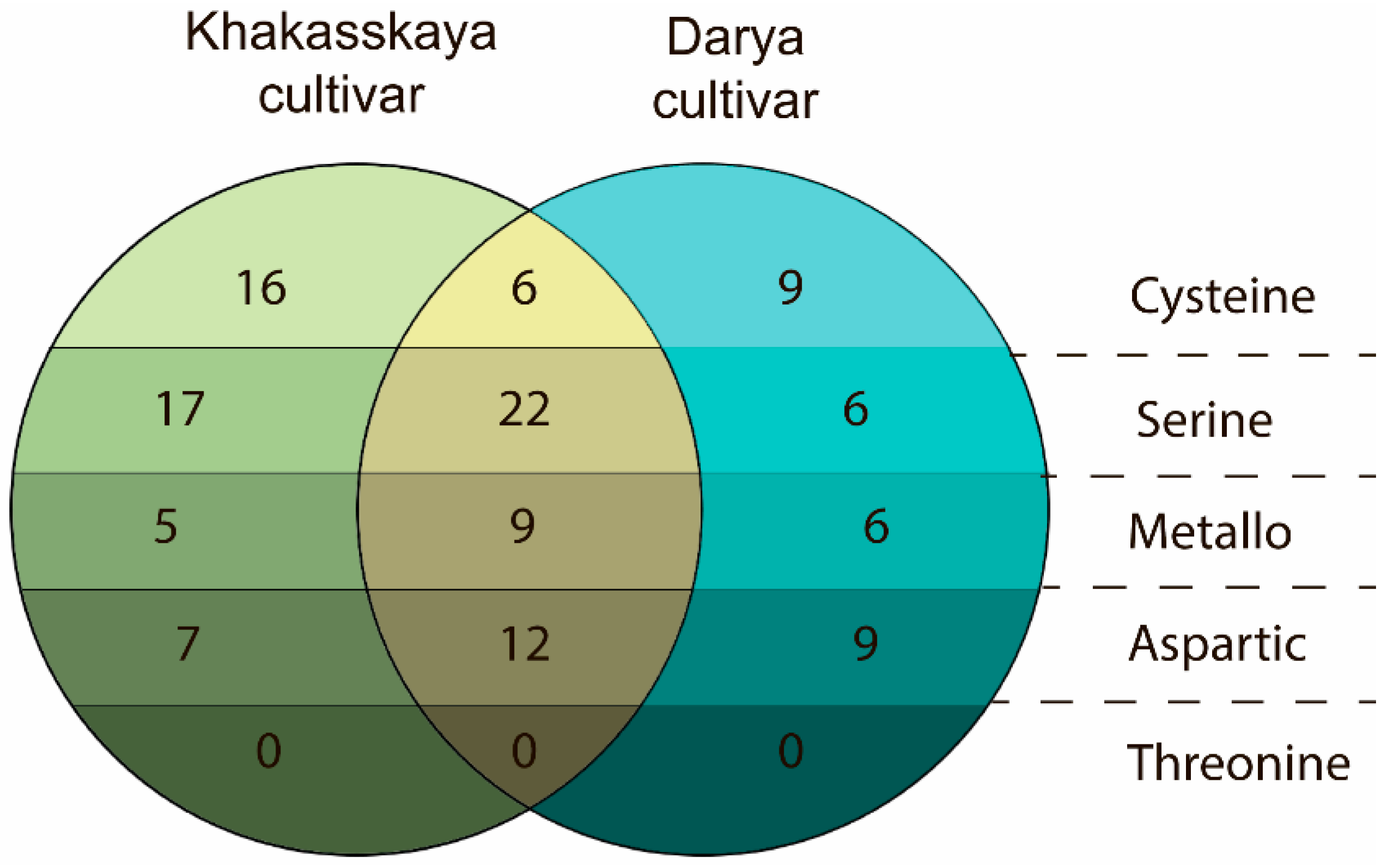
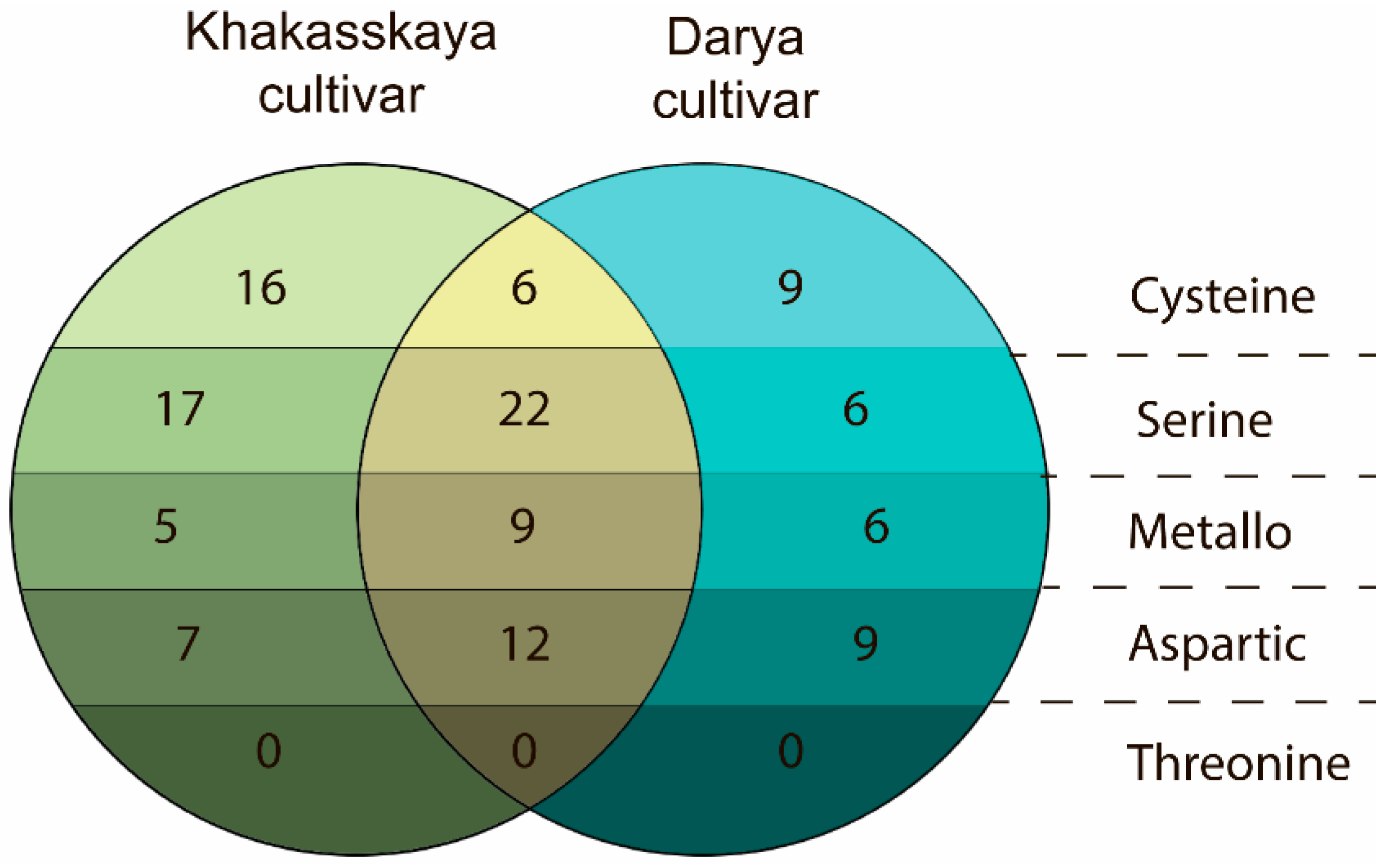
2.2. Differences in Degradomes of Two Wheat Cultivars Were Revealed
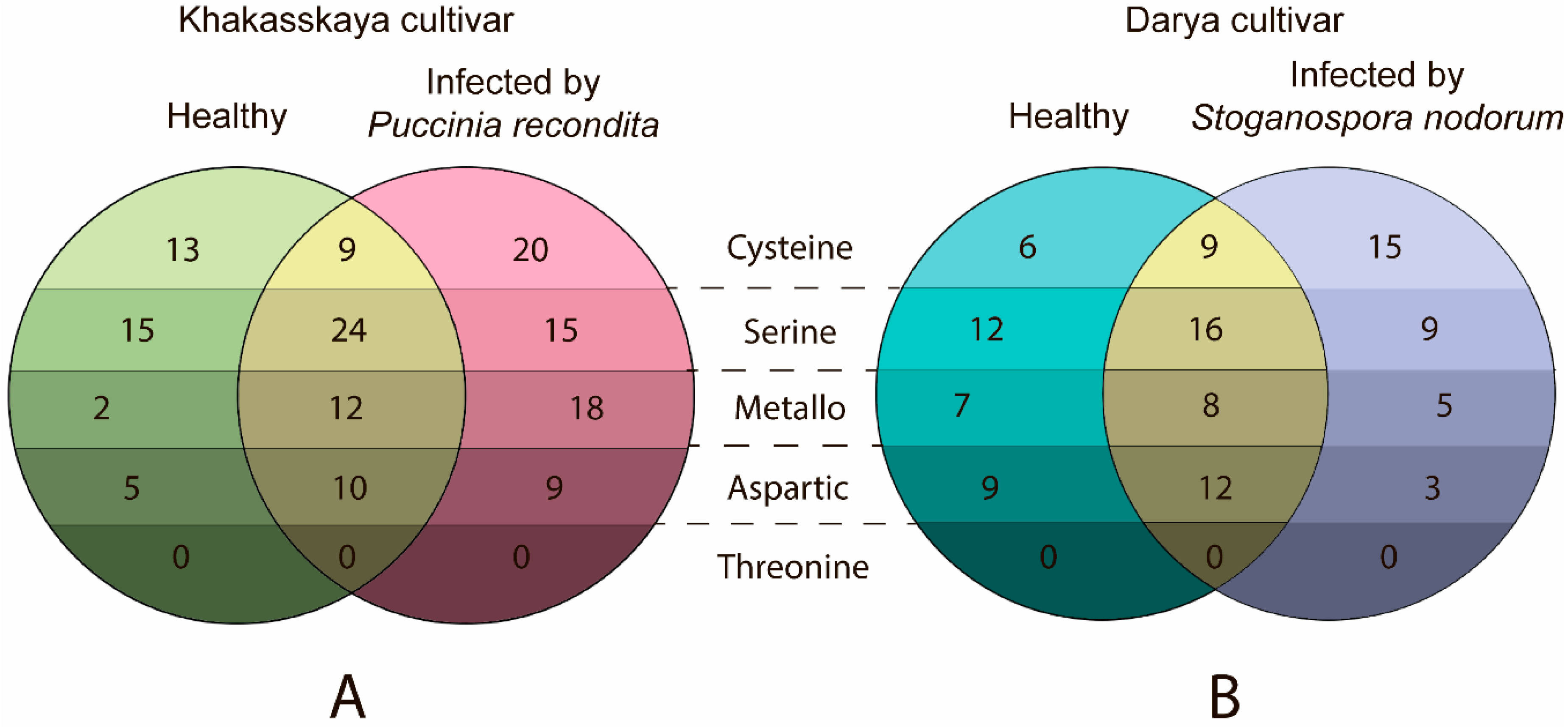
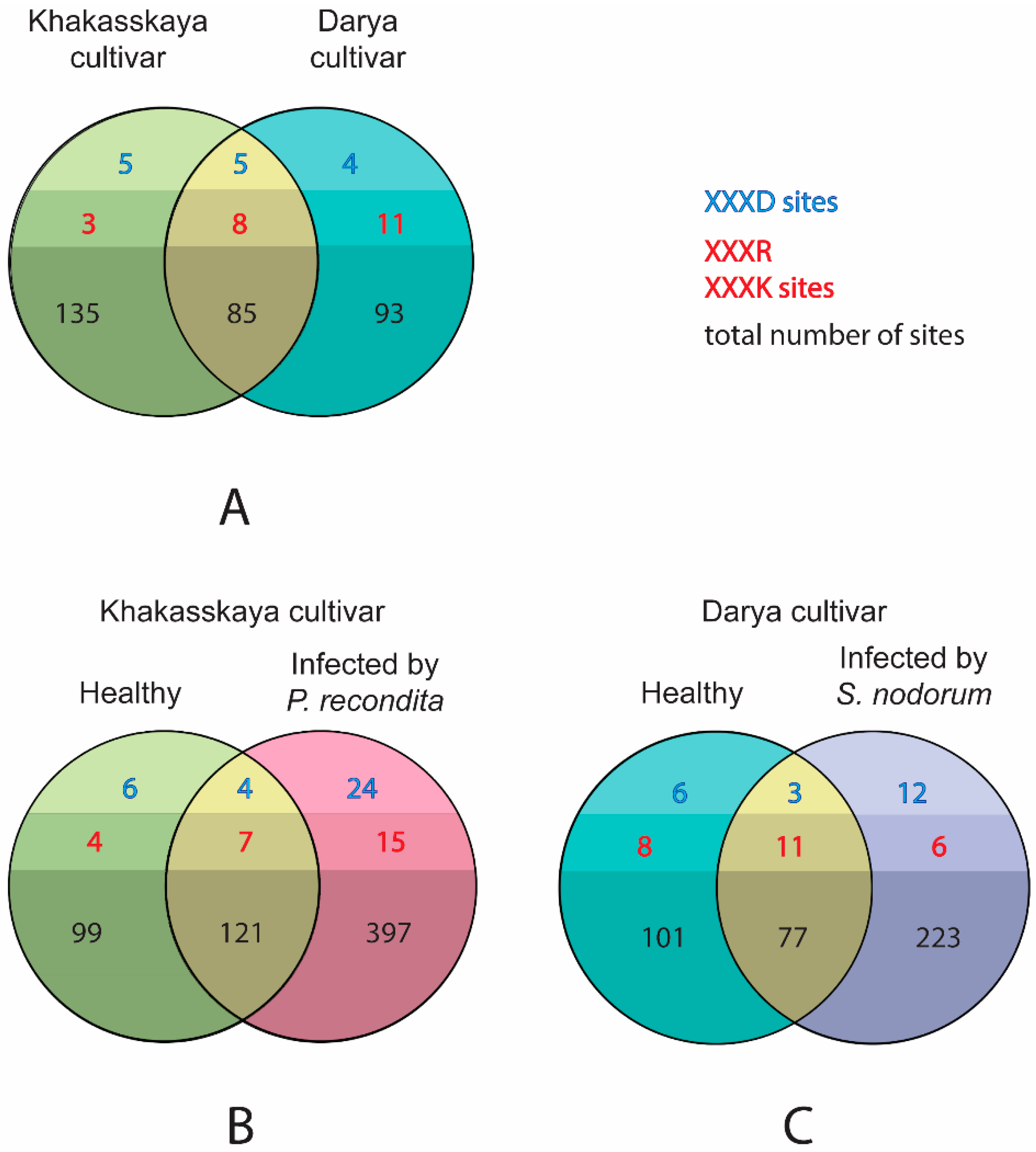
2.3. Infection by Pathogens Leads to an Increase in the Number of Expressed Proteases
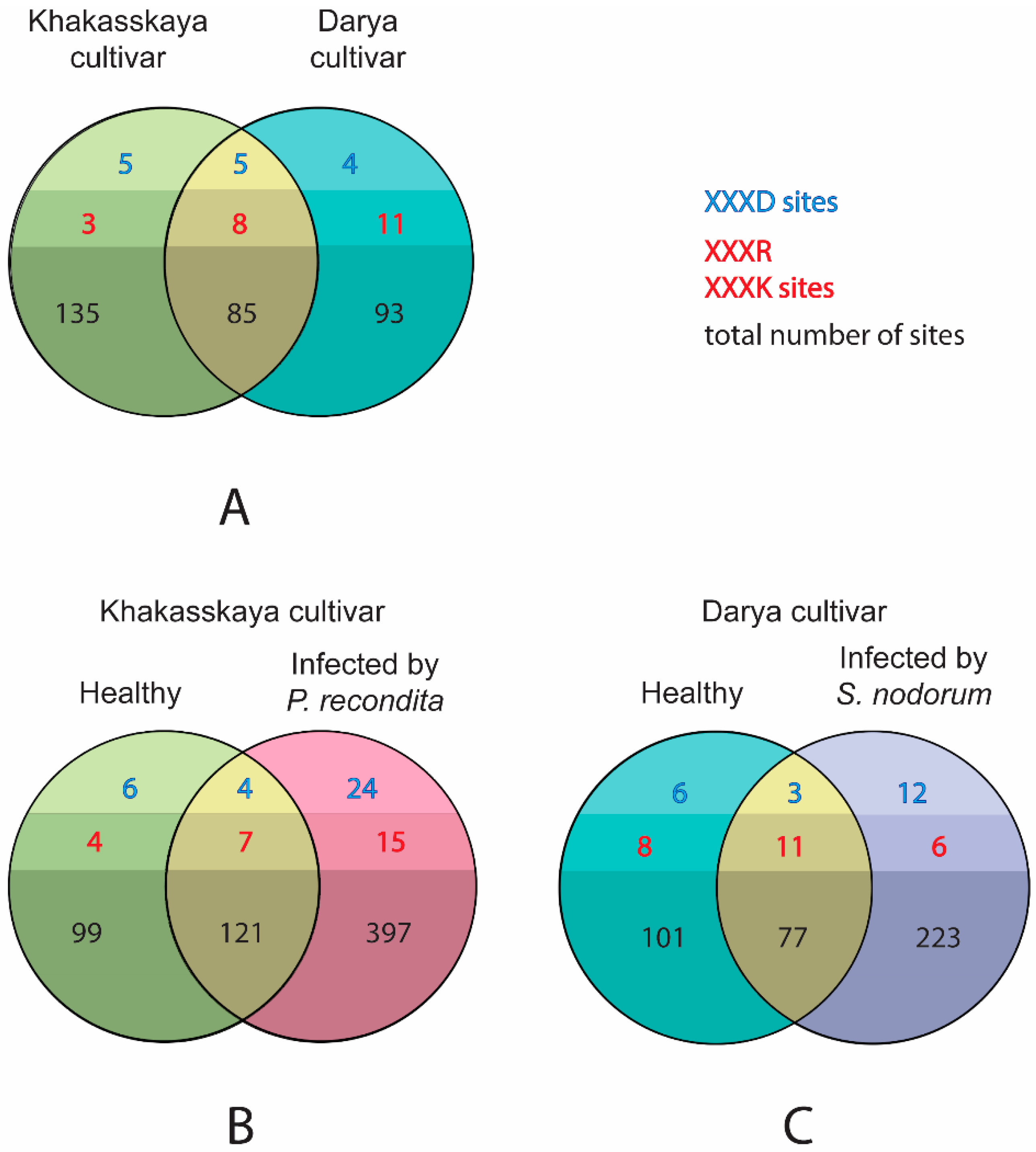
2.4. The Pool of Substrates Cleaved by Proteases In Vivo Expands upon Both Types of Infection
2.5. Infection Induces Synthesis and/or Proteolytic Activation of Proteases through Recruitment of Proteases Other Than Caspase-Like and Metacaspase-Like Proteases
3. Discussion
4. Materials and Methods
4.1. Phylogenetic Analysis of the Wheat Protease Families
4.2. Plant Growth and Infection by Different Pathogens
4.3. Plant Sample Preparation for Mass Spectrometry
4.4. LC-MS/MS Analysis
4.5. Protein Identification and Determination of Sites Hydrolyzed In Vivo by Endogenous Proteases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AALP | Arabidopsis Aleurain-Like Protease |
| AGC | Automatic Gain Control |
| CEP1 | Cysteine EndoPeptidase 1 |
| COT44 | COTyledon abundunt protease 44 |
| CYP1 | CYsteine Protease 1 |
| DUF | Domain of Unknown Function |
| EPF2 | Epidermal Patterning Factor 2 |
| ESI | Electrospray Ionization |
| ET | Ethylene |
| FDR | False Discovery Rate |
| HCD | High-energy Collisional Dissociation |
| HMMER | Hidden Markov Models |
| ISF | In-Source Fragmentation |
| JA | Jasmonic Acid |
| LC-MS | Liquid Chromatography-Mass Spectrometry |
| MCA1 | Metacaspase 1 |
| MMP | Matrix Metallo-protease |
| NAM | No Apical Meristem |
| NCE | Normalized Collision Energy |
| PA | Protease Associated |
| PBA1 | Proteasome Subunit Type |
| PCD | Programmed Cell Death |
| PLCP | Papain-Like Cysteine Protease |
| RD19 | Responsive To Dehydration 19 |
| RD21 | Responsive To Dehydration 21 |
| RISC | RNA-Induced Silencing Complex |
| SAG12 | Senescence-Associated Gene 12 |
| SAG39 | Senescence-Associated Gene 39 |
| SBT | Subtilase |
| THI1 | Thiol Protease 1 |
| TPP2 | Tripeptidyl Peptidase 2 |
| TSN | Tudor Staphylococcal Nuclease |
| VPE | Vacuolar Processing Enzyme |
| XBCP3 | Xylem Bark Cysteine Peptidase 3 |
| XCP2 | Xylem Cysteine Peptidase 2 |
References
- Zimin, A.V.; Puiu, D.; Hall, R.; Kingan, S.; Clavijo, B.J.; Salzberg, S.L. The first near-complete assembly of the hexaploid bread wheat genome, Triticum aestivum. Gigascience 2017, 6, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vu, L.D.; Verstraeten, I.; Stes, E.; Van Bel, M.; Coppens, F.; Gevaert, K.; De Smet, I. Proteome Profiling of Wheat Shoots from Different Cultivars. Front. Plant Sci. 2017, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Faghani, E.; Gharechahi, J.; Komatsu, S.; Mirzaei, M.; Khavarinejad, R.A.; Najafi, F.; Farsad, L.K.; Salekdeh, G.H. Comparative physiology and proteomic analysis of two wheat genotypes contrasting in drought tolerance. J. Proteom. 2015, 114, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Mauchline, T.H.; Chedom-Fotso, D.; Chandra, G.; Samuels, T.; Greenaway, N.; Backhaus, A.; McMillan, V.; Canning, G.; Powers, S.J.; Hammond-Kosack, K.E.; et al. An analysis of Pseudomonas genomic diversity in take-all infected wheat fields reveals the lasting impact of wheat cultivars on the soil microbiota. Environ. Microbiol. 2015, 17, 4764–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gardiner, D.M.; Upadhyaya, N.M.; Stiller, J.; Ellis, J.G.; Dodds, P.N.; Kazan, K.; Manners, J.M. Genomic analysis of Xanthomonas translucens pathogenic on wheat and barley reveals cross-kingdom gene transfer events and diverse protein delivery systems. PLoS ONE 2014, 9, e84995. [Google Scholar] [CrossRef]
- Duba, A.; Goriewa-Duba, K.; Wachowska, U. A Review of the Interactions between Wheat and Wheat Pathogens: Zymoseptoria tritici, Fusarium spp. and Parastagonospora nodorum. Int. J. Mol. Sci. 2018, 19, 1138. [Google Scholar] [CrossRef]
- Zhang, K.; Zhang, Y.; Yang, M.; Liu, S.; Li, Z.; Wang, X.; Han, C.; Yu, J.; Li, D. The Barley stripe mosaic virus gammab protein promotes chloroplast-targeted replication by enhancing unwinding of RNA duplexes. PLoS Pathog. 2017, 13, e1006319. [Google Scholar] [CrossRef]
- Lee, S.; Kim, J.H.; Choi, J.Y.; Jang, W.C. Loop-mediated Isothermal Amplification Assay to Rapidly Detect Wheat Streak Mosaic Virus in Quarantined Plants. Plant Pathol. J. 2015, 31, 438–440. [Google Scholar] [CrossRef] [Green Version]
- Ogasawara, Y.; Kaya, H.; Hiraoka, G.; Yumoto, F.; Kimura, S.; Kadota, Y.; Hishinuma, H.; Senzaki, E.; Yamagoe, S.; Nagata, K.; et al. Synergistic activation of the Arabidopsis NADPH oxidase AtrbohD by Ca2+ and phosphorylation. J. Biol. Chem. 2008, 283, 8885–8892. [Google Scholar] [CrossRef]
- Li, F.; Kong, L.; Liu, Y.; Wang, H.; Chen, L.; Peng, J. Response of wheat germplasm to infestation of English grain aphid (Hemiptera: Aphididae). J. Econ. Entomol. 2013, 106, 1473–1478. [Google Scholar] [CrossRef]
- Kong, L.A.; Wu, D.Q.; Huang, W.K.; Peng, H.; Wang, G.F.; Cui, J.K.; Liu, S.M.; Li, Z.G.; Yang, J.; Peng, D.L. Large-scale identification of wheat genes resistant to cereal cyst nematode Heterodera avenae using comparative transcriptomic analysis. BMC Genom. 2015, 16, 801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jia, H.; Zhang, C.; Pervaiz, T.; Zhao, P.; Liu, Z.; Wang, B.; Wang, C.; Zhang, L.; Fang, J.; Qian, J. Jasmonic acid involves in grape fruit ripening and resistant against Botrytis cinerea. Funct. Integr. Genom. 2016, 16, 79–94. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Wright, L.P.; Gershenzon, J.; Wasternack, C.; Hause, B.; Schaller, A.; Stintzi, A. Jasmonic acid and its precursor 12-oxophytodienoic acid control different aspects of constitutive and induced herbivore defenses in tomato. Plant Physiol. 2014, 166, 396–410. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, D.K.; Prakash, A.; Johri, B.N. Induced systemic resistance (ISR) in plants: Mechanism of action. Indian J. Microbiol. 2007, 47, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Kappelhoff, R.; Puente, X.S.; Wilson, C.H.; Seth, A.; López-Otín, C.; Overall, C.M. Overview of Transcriptomic Analysis of all Human Proteases, Non-Proteolytic Homologs and Inhibitors: Organ, Tissue and Ovarian Cancer Cell Line Expression Profiling of the Human Protease Degradome by the CLIP-CHIPTM DNA Microarray; Elsevier, B.V.: Amsterdam, The Netherlands, 2017; Volume 1864, ISBN 1604822295. [Google Scholar]
- García-Lorenzo, M.; Sjödin, A.; Jansson, S.; Funk, C. Protease gene families in Populus and Arabidopsis. BMC Plant Biol. 2006, 6, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Zhou, X.; Xiong, H.; Mao, W.; Zhao, P.; Sun, M. Papain-like and legumain-like proteases in rice: Genome-wide identification, comprehensive gene feature characterization and expression analysis. BMC Plant Biol. 2018, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, L.P.; Sowdhamini, R. Cross genome comparisons of serine proteases in Arabidopsis and rice. BMC Genom. 2006, 7, 1–31. [Google Scholar] [CrossRef]
- Chen, J.; Ouyang, Y.; Wang, L.; Xie, W.; Zhang, Q. Aspartic proteases gene family in rice: Gene structure and expression, predicted protein features and phylogenetic relation. Gene 2009, 442, 108–118. [Google Scholar] [CrossRef]
- Grosse-Holz, F.; Kelly, S.; Blaskowski, S.; Kaschani, F.; Kaiser, M.; van der Hoorn, R.A.L. The transcriptome, extracellular proteome and active secretome of agroinfiltrated N. benthamiana uncover a large, diverse protease repertoire. Plant Biotechnol. J. 2018, 16, 1068–1084. [Google Scholar] [CrossRef]
- Zamyatnin, A.A., Jr. Plant Proteases Involved in Regulated Cell Death. Biochemtry 2015, 80, 1701–1715. [Google Scholar] [CrossRef]
- Meyer, M.; Huttenlocher, F.; Cedzich, A.; Procopio, S.; Stroeder, J.; Pau-Roblot, C.; Lequart-Pillon, M.; Pelloux, J.; Stintzi, A.; Schaller, A. The subtilisin-like protease SBT3 contributes to insect resistance in tomato. J. Exp. Bot. 2016, 67, 4325–4338. [Google Scholar] [CrossRef] [PubMed]
- Hatsugai, N.; Iwasaki, S.; Tamura, K.; Kondo, M.; Fuji, K.; Ogasawara, K.; Nishimura, M.; Hara-Nishimura, I. A novel membrane fusion-mediated plant immunity against bacterial pathogens. Genes Dev. 2009, 23, 2496–2506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chichkova, N.V.; Shaw, J.; Galiullina, R.A.; Drury, G.E.; Tuzhikov, A.I.; Kim, S.H.; Kalkum, M.; Hong, T.B.; Gorshkova, E.N.; Torrance, L.; et al. Phytaspase, a relocalisable cell death promoting plant protease with caspase specificity. EMBO J. 2010, 29, 1149–1161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zheng, X.; Zhang, Z. The role of vacuolar processing enzymes in plant immunity. Plant Signal. Behav. 2010, 5, 1565–1567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, R.; Schaller, A.; Ottmann, C. Structural features of plant subtilases. Plant Signal. Behav. 2010, 5, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hatsugai, N.; Kuroyanagi, M.; Yamada, K.; Meshi, T.; Tsuda, S.; Kondo, M.; Nishimura, M.; Hara-Nishimura, I. A plant vacuolar protease, VPE, mediates virus-induced hypersensitive cell death. Science 2004, 305, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, J.F.; Vaculova, A.; Smertenko, A.P.; Savenkov, E.I.; Golovko, A.; Minina, E.; Tiwari, B.S.; Rodriguez-Nieto, S.; Zamyatnin, A.A., Jr.; Valineva, T.; et al. Tudor staphylococcal nuclease is an evolutionarily conserved component of the programmed cell death degradome. Nat. Cell Biol. 2009, 11, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Bozhkov, P.V.; Suarez, M.F.; Filonova, L.H.; Daniel, G.; Zamyatnin, A.A., Jr.; Rodriguez-Nieto, S.; Zhivotovsky, B.; Smertenko, A. Cysteine protease mcII-Pa executes programmed cell death during plant embryogenesis. Proc. Natl. Acad. Sci. USA 2005, 102, 14463–14468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coll, N.S.; Vercammen, D.; Smidler, A.; Clover, C.; Van Breusegem, F.; Dangl, J.L.; Epple, P. Arabidopsis type I metacaspases control cell death. Science 2010, 330, 1393–1397. [Google Scholar] [CrossRef] [PubMed]
- Bollhoner, B.; Zhang, B.; Stael, S.; Denance, N.; Overmyer, K.; Goffner, D.; Van Breusegem, F.; Tuominen, H. Post mortem function of AtMC9 in xylem vessel elements. New Phytol. 2013, 200, 498–510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollhoner, B.; Jokipii-Lukkari, S.; Bygdell, J.; Stael, S.; Adriasola, M.; Muniz, L.; Van Breusegem, F.; Ezcurra, I.; Wingsle, G.; Tuominen, H. The function of two type II metacaspases in woody tissues of Populus trees. New Phytol. 2018, 217, 1551–1565. [Google Scholar] [CrossRef] [PubMed]
- Fagundes, D.; Bohn, B.; Cabreira, C.; Leipelt, F.; Dias, N.; Bodanese-Zanettini, M.H.; Cagliari, A. Caspases in plants: Metacaspase gene family in plant stress responses. Funct. Integr. Genom. 2015, 15, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Balakireva, A.V.; Zamyatnin, A.A. Indispensable role of proteases in plant innate immunity. Int. J. Mol. Sci. 2018, 19, 629. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Wang, X.; Peng, H.; Yao, Y.; Xie, C.; Han, Y.; Ni, Z.; Sun, Q. Transcriptome Comparison of Susceptible and Resistant Wheat in Response to Powdery Mildew Infection. Genom. Proteom. Bioinf. 2012, 10, 94–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erayman, M.; Turktas, M.; Akdogan, G.; Gurkok, T.; Inal, B.; Ishakoglu, E.; Ilhan, E.; Unver, T. Transcriptome analysis of wheat inoculated with Fusarium graminearum. Front. Plant Sci. 2015, 6, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, S.; Kamal, A.H.M.; Hossain, Z. Wheat proteomics: Proteome modulation and abiotic stress acclimation. Front. Plant Sci. 2014, 5, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Wang, X.; Wang, K.; Li, H.; Duan, X.; Tang, C.; Kang, Z. TaMCA1, a regulator of cell death, is important for the interaction between wheat and Puccinia striiformis. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Liu, Z.; Xu, H.; Zhang, X.; Zhao, A.; Liang, F.; Xin, M.; Peng, H.; Yao, Y.; Sun, Q.; et al. Transcriptome analysis reveals potential mechanisms for different grain size between natural and resynthesized allohexaploid wheats with near-identical AABB genomes. BMC Plant Biol. 2018, 18, 1–15. [Google Scholar] [CrossRef]
- Yu, Y.; Zhu, D.; Ma, C.; Cao, H.; Wang, Y.; Xu, Y.; Zhang, W.; Yan, Y. Transcriptome analysis reveals key differentially expressed genes involved in wheat grain development. Crop J. 2016, 4, 92–106. [Google Scholar] [CrossRef]
- Wan, Y.; Poole, R.L.; Huttly, A.K.; Toscano-Underwood, C.; Feeney, K.; Welham, S.; Gooding, M.J.; Mills, C.; Edwards, K.J.; Shewry, P.R.; et al. Transcriptome analysis of grain development in hexaploid wheat. BMC Genom. 2008, 9, 1–16. [Google Scholar] [CrossRef]
- Wheeler, T.J.; Eddy, S.R. Nhmmer: DNA homology search with profile HMMs. Bioinformatics 2013, 29, 2487–2489. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Sharma, A.; Niewiara, M.J.; Singh, R.; Ming, R.; Yu, Q. Papain-like cysteine proteases in Carica papaya: Lineage-specific gene duplication and expansion. BMC Genom. 2018, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Mistry, J.; Mitchell, A.L.; Potter, S.C.; Punta, M.; Qureshi, M.; Sangrador-Vegas, A.; et al. The Pfam protein families database: Towards a more sustainable future. Nucleic Acids Res. 2016, 44, D279–D285. [Google Scholar] [CrossRef] [PubMed]
- Rawlings, N.D.; Waller, M.; Barrett, A.J.; Bateman, A. MEROPS: The database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 2014, 42, D503–D509. [Google Scholar] [CrossRef] [PubMed]
- Tsiatsiani, L.; Van Breusegem, F.; Gallois, P.; Zavialov, A.; Lam, E.; Bozhkov, P.V. Metacaspases. Cell. Death Differ. 2011, 18, 1279–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitsky, L.I.; Ivanov, M.V.; Lobas, A.A.; Bubis, J.A.; Tarasova, I.A.; Solovyeva, E.M.; Pridatchenko, M.L.; Gorshkov, M.V. IdentiPy: An Extensible Search Engine for Protein Identification in Shotgun Proteomics. J. Proteome Res. 2018, 17, 2249–2255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Monroe, M.E.; Camp, D.G.; Smith, R.D.; Qian, W.J. In-source fragmentation and the sources of partially tryptic peptides in shotgun proteomics. J. Proteome Res. 2013, 12, 910–916. [Google Scholar] [CrossRef]
- Alves, P.; Arnold, R.J.; Clemmer, D.E.; Li, Y.; Reilly, J.P.; Sheng, Q.; Tang, H.; Xun, Z.; Zeng, R.; Radivojac, P. Fast and accurate identification of semi-tryptic peptides in shotgun proteomics. Bioinformatics 2008, 24, 102–109. [Google Scholar] [CrossRef]
- Carapito, C.; Kuhn, L.; Karim, L.; Rompais, M.; Rabilloud, T.; Schwenzer, H.; Sissler, M. Two proteomic methodologies for defining N-termini of mature human mitochondrial aminoacyl-tRNA synthetases. Methods 2017, 113, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Coll, N.S.; Epple, P.; Dangl, J.L. Programmed cell death in the plant immune system. Cell Death Differ. 2011, 18, 1247–1256. [Google Scholar] [CrossRef] [Green Version]
- Richau, K.H.; Kaschani, F.; Verdoes, M.; Pansuriya, T.C.; Niessen, S.; Stuber, K.; Colby, T.; Overkleeft, H.S.; Bogyo, M.; Van der Hoorn, R.A. Subclassification and biochemical analysis of plant papain-like cysteine proteases displays subfamily-specific characteristics. Plant Physiol. 2012, 158, 1583–1599. [Google Scholar] [CrossRef]
- Zou, Z.; Xie, G.; Yang, L. Papain-like cysteine protease encoding genes in rubber (Hevea brasiliensis): Comparative genomics, phylogenetic, and transcriptional profiling analysis. Planta 2017, 246, 999–1018. [Google Scholar] [CrossRef] [PubMed]
- Golldack, D.; Vera, P.; Dietz, K.J. Expression of subtilisin-like serine proteases in Arabidopsis thaliana is cell-specific and responds to jasmonic acid and heavy metals with developmental differences. Physiol. Plant 2003, 118, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Martinez, D.E.; Borniego, M.L.; Battchikova, N.; Aro, E.M.; Tyystjärvi, E.; Guiamét, J.J. SASP, a Senescence-Associated Subtilisin Protease, is involved in reproductive development and determination of silique number in Arabidopsis. J. Exp. Bot. 2015, 66, 161–174. [Google Scholar] [CrossRef] [PubMed]
- Neuteboom, L.W.; Veth-Tello, L.M.; Clijdesdale, O.R.; Hooykaas, P.J.J.; Van Der Zaal, B.J. A novel subtilisin-like protease gene from Arabidopsis thaliana is expressed at sites of lateral root emergence. DNA Res. 1999, 6, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Engineer, C.B.; Ghassemian, M.; Anderson, J.C.; Peck, S.C.; Hu, H.; Schroeder, J.I. Carbonic anhydrases, EPF2 and a novel protease mediate CO2control of stomatal development. Nature 2014, 513, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Book, A.J.; Yang, P.; Scalf, M.; Smith, L.M.; Vierstra, R.D. Tripeptidyl peptidase II. An oligomeric protease complex from Arabidopsis. Plant Physiol. 2005, 138, 1046–1057. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, N.; Lam, E. Arabidopsis metacaspase 2d is a positive mediator of cell death induced during biotic and abiotic stresses. Plant J. 2011, 66, 969–982. [Google Scholar] [CrossRef] [Green Version]
- Petutschnig, E.K.; Stolze, M.; Lipka, U.; Kopischke, M.; Horlacher, J.; Valerius, O.; Rozhon, W.; Gust, A.A.; Kemmerling, B.; Poppenberger, B.; et al. A novel Arabidopsis CHITIN ELICITOR RECEPTOR KINASE 1 (CERK1) mutant with enhanced pathogen-induced cell death and altered receptor processing. New Phytol. 2014, 204, 955–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, R. Arabidopsis FHY3/FAR1 Gene Family and Distinct Roles of Its Members in Light Control of Arabidopsis Development. Plant Physiol. 2004, 136, 4010–4022. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsiatsiani, L.; Timmerman, E.; De Bock, P.J.; Vercammen, D.; Stael, S.; van de Cotte, B.; Staes, A.; Goethals, M.; Beunens, T.; Van Damme, P.; et al. The Arabidopsis metacaspase9 degradome. Plant Cell 2013, 25, 2831–2847. [Google Scholar] [CrossRef] [PubMed]
- Kersey, P.J.; Allen, J.E.; Armean, I.; Boddu, S.; Bolt, B.J.; Carvalho-Silva, D.; Christensen, M.; Davis, P.; Falin, L.J.; Grabmueller, C.; et al. Ensembl Genomes 2016: More genomes, more complexity. Nucleic Acids Res. 2016, 44, D574–D580. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuraku, S.; Zmasek, C.M.; Nishimura, O.; Katoh, K. aLeaves facilitates on-demand exploration of metazoan gene family trees on MAFFT sequence alignment server with enhanced interactivity. Nucleic Acids Res. 2013, 41, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2017, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Jenkyn, J.F.; King, J.E. Observations on the Origins of Septoria nodorum Infection of Winter Wheat. Plant Pathol. 1977, 26, 153–160. [Google Scholar] [CrossRef]
- Browder, L.E. Pathogenic Specialization in Cereal Rust Fungi, Especially Puccinia recondita f.sp. Tritici: Concepts, Methods of Study and Application; United States Department of Agriculture (USDA): Washington, DC, USA, 1971; pp. 37–51.
- Wiśniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359. [Google Scholar] [CrossRef] [PubMed]
- Kiran, K.; Rawal, H.C.; Dubey, H.; Jaswal, R.; Devanna, B.N.; Gupta, D.K.; Bhardwaj, S.C.; Prasad, P.; Pal, D.; Chhuneja, P.; et al. Draft genome of the wheat rust pathogen (Puccinia triticina) unravels genome-wide structural variations during evolution. Genome Biol. Evol. 2016, 8, 2702–2721. [Google Scholar] [CrossRef] [PubMed]
- Syme, R.A.; Hane, J.K.; Friesen, T.L.; Oliver, R.P. Resequencing and Comparative Genomics of Stagonospora nodorum: Sectional Gene Absence and Effector Discovery. Genes Genomes Genet. 2013, 3, 959–969. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. TANDEM: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef]
- Ivanov, M.V.; Levitsky, L.I.; Lobas, A.A.; Panic, T.; Laskay, Ü.A.; Mitulovic, G.; Schmid, R.; Pridatchenko, M.L.; Tsybin, Y.O.; Gorshkov, M.V. Empirical multidimensional space for scoring peptide spectrum matches in shotgun proteomics. J. Proteome Res. 2014, 13, 1911–1920. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinf. 2009, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protease Family | Protease Families and Subfamilies (According to Pfam Nomenclature) |
|---|---|
| Cysteine | C1, C2, C12, C13, C14, C15, C26, C48, C50, C54, C65, C78 |
| Serine | S8, S9, S10, S15, S28 |
| Aspartic | A1, A22B |
| Metallo-proteases | M1, M3, M8, M10, M14, M16_M, M16_C, M17, M18, M20, M22, M24, M28, M41, M48, M49, M50B, M50 |
| Threonine | T1 |
| Protein | Site of Hydrolysis |
|---|---|
| Potential substrates of caspase-like proteases | |
| Uncharacterized protein | VPTD↓AQLE |
| Protein Translocon at the inner envelope membrane of chloroplasts 22 (TIC 22), chloroplastic-like | ITLD↓QVYM |
| Disease resistance protein Rho-type GTPase-activating protein 2-like (RGA2) | VSAD↓GVTR |
| 5′-3′ exoribonuclease 2-like | ILRD↓MVPL |
| Homeobox-DDT (DNA-binding homeobox-containing proteins and the different transcription and chromatin remodeling factors) domain protein Ringlet 3-like (RLT3) | KPED↓LTEY |
| Protein Fatty Acyl-CoA Reductase 1 (FAR1)-Related Sequence 5-like | LAAD↓HPRR |
| Constitutive Photomorphogenic1 (COP1)-interacting protein 7 | IDID↓AELG |
| Potential substrates of metacaspase-like proteases | |
| Adenine/guanine permease Azaguanine Resistant 2 (AZG2) | CLAR↓TKSD |
| Wall-associated receptor kinase 5-like | LSTR↓NELI |
| Protein FAR1-Related Sequence 5-like | LFKK↓GVGA |
| Catalytic Type of proteases | Family | Number of Detected by LC-MS Sites of Cleavage * (Number of XXXD Sites, If Any), [Number of XXXR or XXXK Sites, If Any] | |||
|---|---|---|---|---|---|
| Healthy Khakasskaya | Khakasskaya infected with P. recondita | Healthy Darya | Darya infected with S. nodorum | ||
| Cysteine | Peptidase_C1 | 2 | 1 | 1 | 2 |
| Peptidase_C13 | 1 (1) | 1 (1) | |||
| Serine | Peptidase_S8 | 2 | 1 | 1 | |
| Peptidase_S10 | 1 | ||||
| Peptidase_S49 | 1 | ||||
| Metallo- | Peptidase_M17 | 1 | |||
| Peptidase_M20 | 1 | 1 | |||
| Peptidase_M24 | 1 | 1 | |||
| Peptidase_M41 | 1 | 2 | 2 | 2 | |
| Aspartic | A1 (TAXi_C) | 4 | 6 (2) | 2 (1) | 1 |
| Total | 7 | 16 (3) | 7 (1) | 9 (1) | |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Balakireva, A.V.; Deviatkin, A.A.; Zgoda, V.G.; Kartashov, M.I.; Zhemchuzhina, N.S.; Dzhavakhiya, V.G.; Golovin, A.V.; Zamyatnin, A.A., Jr. Proteomics Analysis Reveals That Caspase-Like and Metacaspase-Like Activities Are Dispensable for Activation of Proteases Involved in Early Response to Biotic Stress in Triticum aestivum L. Int. J. Mol. Sci. 2018, 19, 3991. https://doi.org/10.3390/ijms19123991
Balakireva AV, Deviatkin AA, Zgoda VG, Kartashov MI, Zhemchuzhina NS, Dzhavakhiya VG, Golovin AV, Zamyatnin AA Jr. Proteomics Analysis Reveals That Caspase-Like and Metacaspase-Like Activities Are Dispensable for Activation of Proteases Involved in Early Response to Biotic Stress in Triticum aestivum L. International Journal of Molecular Sciences. 2018; 19(12):3991. https://doi.org/10.3390/ijms19123991
Chicago/Turabian StyleBalakireva, Anastasia V., Andrei A. Deviatkin, Victor G. Zgoda, Maxim I. Kartashov, Natalia S. Zhemchuzhina, Vitaly G. Dzhavakhiya, Andrey V. Golovin, and Andrey A. Zamyatnin, Jr. 2018. "Proteomics Analysis Reveals That Caspase-Like and Metacaspase-Like Activities Are Dispensable for Activation of Proteases Involved in Early Response to Biotic Stress in Triticum aestivum L." International Journal of Molecular Sciences 19, no. 12: 3991. https://doi.org/10.3390/ijms19123991
APA StyleBalakireva, A. V., Deviatkin, A. A., Zgoda, V. G., Kartashov, M. I., Zhemchuzhina, N. S., Dzhavakhiya, V. G., Golovin, A. V., & Zamyatnin, A. A., Jr. (2018). Proteomics Analysis Reveals That Caspase-Like and Metacaspase-Like Activities Are Dispensable for Activation of Proteases Involved in Early Response to Biotic Stress in Triticum aestivum L. International Journal of Molecular Sciences, 19(12), 3991. https://doi.org/10.3390/ijms19123991