Sulfonamide Inhibition Studies of a New β-Carbonic Anhydrase from the Pathogenic Protozoan Entamoeba histolytica

 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
- (i)
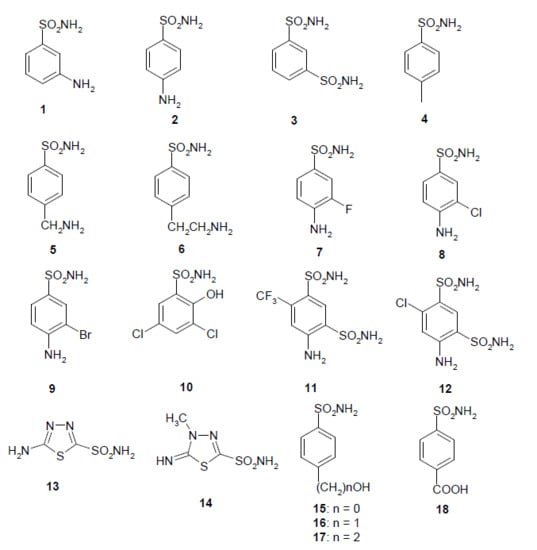
- The most effective EhiCA inhibitors were the two simple compounds 16 and 17, 4-hydroxymethyl/ethyl-benzenesulfonamides, which showed KIs ranging between 36 and 89 nM, with the longer linker derivative (17) being a more effective CAI compared to the hydroxymethyl one 16. It should also be noted that 17 is a weaker hCA II inhibitor (KI of 125 nM) and a quite ineffective hCA I inhibitor (KI of 21 µM), making it a slightly ameba-CA—selective compound.
- (ii)
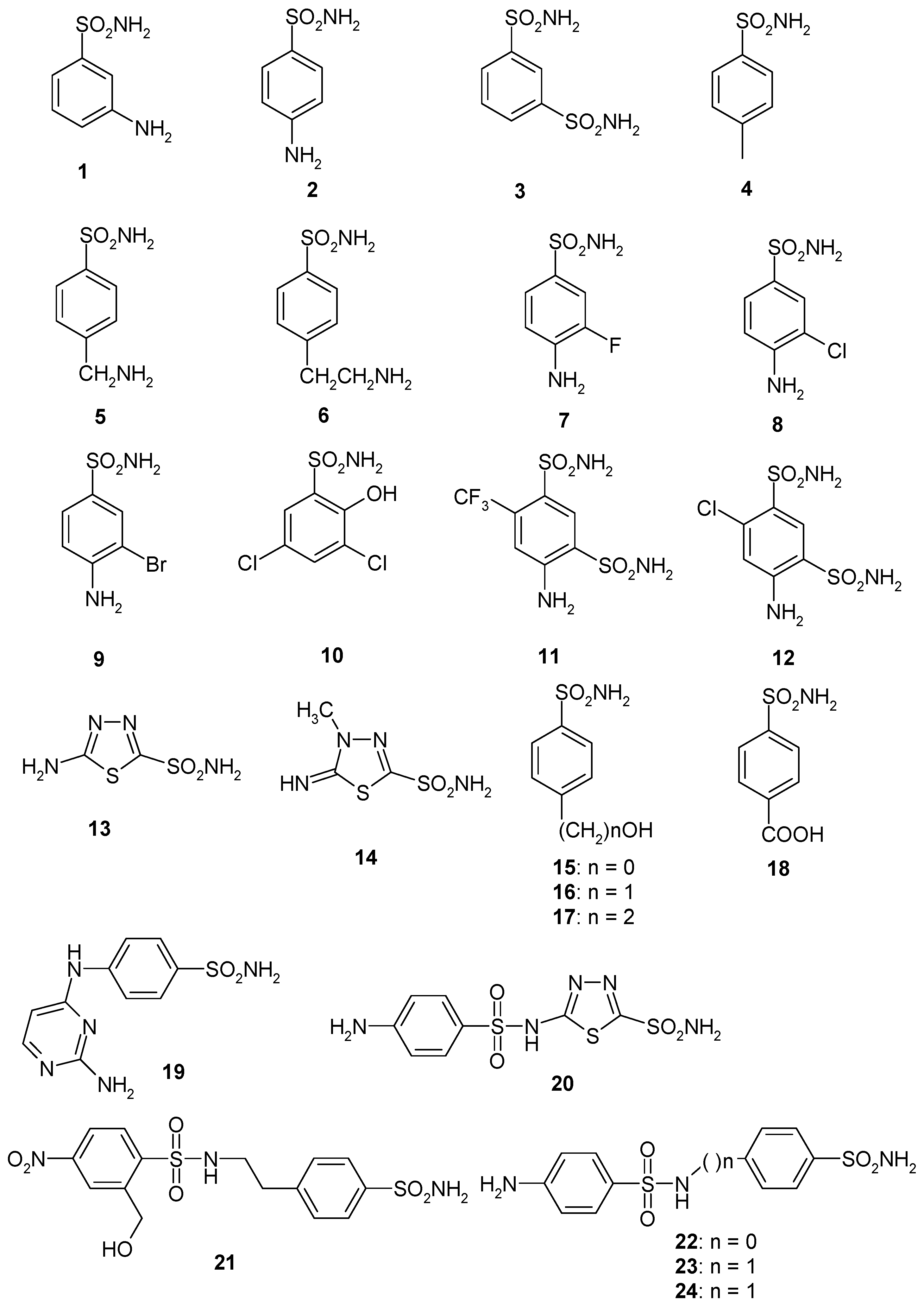
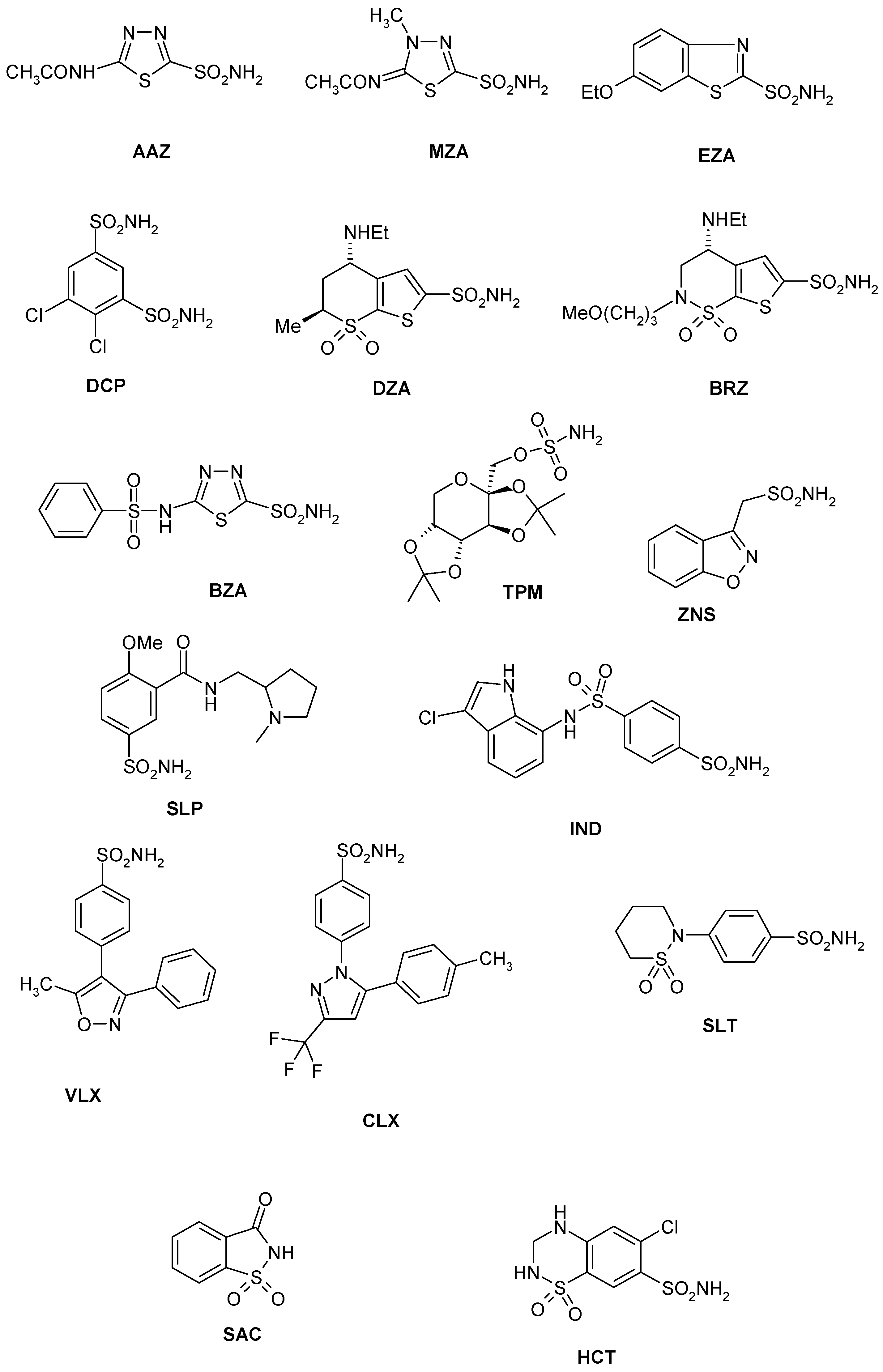
- Several sulfonamides were slightly less effective as EhiCA inhibitors, with KIs ranging between 285 and 521 nM. They include 18–24 and acetazolamide AAZ (Table 2). Apart from 18 (4-carboxy-benzenesulfonamide) and 19 (a pyrimidinylamino-benzenesulfonamide), the remaining derivatives 20–24 belong to the sulfanilyl-sulfonamide class of CAIs, which possess an elongated molecule, shown to interact favorably with many other CAs belonging to the β-class [15,20,21] and, thus, leading to effective inhibitors. For the homologous series of 22–24, the efficacy as EhiCA inhibitors increases with the increase of the linker between the two aromatic rings. AAZ and 20 contain the 1,3,4-thiadiazole-2-sulfonamide motif present in many potent CAIs. In this case, aminobenzolamide 20 is a more effective EhiCA inhibitor compared to AAZ. It is interesting to note that BZA, lacking the amino moiety present in 20, but with an identical scaffold, is a very weak CAI, with a KI of 2471 nM (whereas it is a very potent hCA I and II inhibitor). Thus, minor structural changes in the molecule of the inhibitor lead to drastic effects on their inhibitory profiles against various CAs, including the one form the parasitic protozoan investigated here.
- (iii)
- The following compounds showed modest EhiCA inhibitory properties: 3–6, 11, 13–15, MZA, EZA, DCP, and IND, with KIs ranging between 567 and 951 nM. They belong to heterogeneous classes of sulfonamides, most of them being benzenesulfonamides (apart 13 and 14 which are the deacetylated precursors of AAZ and MZA, thus, heterocyclic derivatives). A special mention regards 15, which is structurally related to the most effective EhiCA inhibitors detected here, compounds 16 and 17. Indeed, 15 is 9–20 times a weaker EhiCA inhibitor compared to 16 and 17, although they differ only by one or two CH2 functionalities. From these data, it is again obvious that SAR is very sensitive to small changes in the molecule of the inhibitor and that the 4-hydroxyalkyl-substituted-benzenesulfonamides may lead to highly effective and isoform-selective CAIs targeting the enzyme from this parasite.
- (iv)
- Weak, micromolar inhibition against EhiCA was observed with 1, 2, 10, 12, DZA, BRZ, BZA, TPM, ZNZ, SLT, and HCT (KIs ranging between 1.91–9.59 µM) as discussed earlier. In addition, these derivatives belong to heterogeneous classes of derivatives, but overall one may observe that they possess a bulkier scaffold and more substituents on the aromatic/heterocyclic ring compared to the effective EhiCA inhibitors described above.
- (v)
- The ineffective compounds as EhiCA inhibitors (KI > 10 µM) detected here were 7–9 (halogenated sulfanilamide derivatives), sulpiride SLP, the COX-2 inhibitors CLX and VLX (possessing a bulky, Y-shaped molecule), and saccharin SAC, the only acylated, secondary sulfonamide included in the study.
- (vi)
- The inhibition profile of EhiCA with sulfonamides/sulfamates is very different from those of the human isoforms hCA I and II, but only two compounds, 16 and 17 showed selectivity for the protozoan over the human isoforms (Table 2).
3. Experimental
3.1. Vector Construction
3.2. Production of the Protein
3.3. CA Activity and Inhibition Measurements
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sánchez, C.; López, M.C.; Galeano, L.A.; Qvarnstrom, Y.; Houghton, K.; Ramírez, J.D. Molecular detection and genotyping of pathogenic protozoan parasites in raw and treated water samples from southwest Colombia. Parasit. Vectors 2018, 11, 563. [Google Scholar] [CrossRef] [PubMed]
- Domazetovska, A.; Lee, R.; Adhikari, C.; Watts, M.; Gilroy, N.; Stark, D.; Sivagnanam, S. A 12-Year Retrospective Study of Invasive Amoebiasis in Western Sydney: Evidence of Local Acquisition. Trop. Med. Infect. Dis. 2018, 3, 73. [Google Scholar] [CrossRef]
- Costa, J.O.; Resende, J.A.; Gil, F.F.; Santos, J.F.G.; Gomes, M.A. Prevalence of Entamoeba histolytica and other enteral parasitic diseases in the metropolitan region of Belo Horizonte, Brazil. A cross-sectional study. Sao Paulo Med. J. 2018, 136, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Shirley, D.T.; Farr, L.; Watanabe, K.; Moonah, S. A Review of the Global Burden, New Diagnostics, and Current Therapeutics for Amebiasis. Open Forum Infect. Dis. 2018, 5, ofy161. [Google Scholar] [CrossRef] [PubMed]
- Hashmey, N.; Genta, N.; White, N., Jr. Parasites and Diarrhea. I: Protozoans and Diarrhea. J. Travel. Med. 1997, 4, 17–31. [Google Scholar] [CrossRef] [PubMed]
- Quach, J.; St-Pierre, J.; Chadee, K. The future for vaccine development against Entamoeba histolytica. Hum. Vaccin. Immunother. 2014, 10, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, E.V.; Bricker-Ford, R.; Rogers, M.J.; McKerrow, J.H.; Reed, S.L. Phase I Clinical Trial Results of Auranofin, a Novel Antiparasitic Agent. Antimicrob. Agents Chemother. 2016, 61, e01947-16. [Google Scholar] [CrossRef]
- Gonzales, M.L.; Dans, L.F.; Martinez, E.G. Antiamoebic drugs for treating amoebic colitis. Cochrane Database Syst. Rev. 2009, 2, CD006085. [Google Scholar] [CrossRef]
- Andrade, R.M.; Reed, S.L. New drug target in protozoan parasites: The role of thioredoxin reductase. Front. Microbiol. 2015, 6, 975. [Google Scholar] [CrossRef]
- Leitsch, D.; Williams, C.F.; Hrdý, I. Redox Pathways as Drug Targets in Microaerophilic Parasites. Trends Parasitol. 2018, 34, 576–589. [Google Scholar] [CrossRef]
- Supuran, C.T. Structure and function of carbonic anhydrases. Biochem. J. 2016, 473, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Neri, D.; Supuran, C.T. Interfering with pH regulation in tumours as a therapeutic strategy. Nat. Rev. Drug Discov. 2011, 10, 767–777. [Google Scholar] [CrossRef] [PubMed]
- Capasso, C.; Supuran, C.T. An overview of the alpha-, beta-and gamma-carbonic anhydrases from Bacteria: Can bacterial carbonic anhydrases shed new light on evolution of bacteria? J. Enzym. Inhib. Med. Chem. 2015, 30, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Vullo, D.; Del Prete, S.; Di Fonzo, P.; Carginale, V.; Donald, W.A.; Supuran, C.T.; Capasso, C. Comparison of the Sulfonamide Inhibition Profiles of the β- and γ-Carbonic Anhydrases from the Pathogenic Bacterium Burkholderia Pseudomallei. Molecules 2017, 22, 421. [Google Scholar] [CrossRef] [PubMed]
- Berrino, E.; Bua, S.; Mori, M.; Botta, M.; Murthy, V.S.; Vijayakumar, V.; Tamboli, Y.; Bartolucci, G.; Mugelli, A.; Cerbai, E.; et al. Novel Sulfamide-Containing. Molecules 2017, 22, 1049. [Google Scholar] [CrossRef] [PubMed]
- Cau, Y.; Mori, M.; Supuran, C.T.; Botta, M. Mycobacterial carbonic anhydrase inhibition with phenolic acids and esters: Kinetic and computational investigations. Org. Biomol. Chem. 2016, 14, 8322–8330. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Advances in structure-based drug discovery of carbonic anhydrase inhibitors. Expert. Opin. Drug Discov. 2017, 12, 61–88. [Google Scholar] [CrossRef]
- Nishimori, I.; Onishi, S.; Takeuchi, H.; Supuran, C.T. The α and β Classes Carbonic Anhydrases from Helicobacter pylori as Novel Drug Targets. Curr. Pharm. Des. 2008, 14, 622–630. [Google Scholar]
- Supuran, C.T.; Capasso, C. An Overview of the Bacterial Carbonic Anhydrases. Metab. 2017, 7, 56. [Google Scholar] [CrossRef]
- Rowlett, R.S. Structure and catalytic mechanism of the β-carbonic anhydrases. Biochim. Biophys. Acta—Prot. Proteom. 2010, 1804, 362–373. [Google Scholar] [CrossRef]
- Zolfaghari Emameh, R.; Barker, H.; Hytönen, V.P.; Tolvanen, M.E.E.; Parkkila, S. Beta carbonic anhydrases: Novel targets for pesticides and anti-parasitic agents in agriculture and livestock husbandry. Parasites Vect. 2014, 7, 403. [Google Scholar] [CrossRef] [PubMed]
- Syrjänen, L.; Parkkila, S.; Scozzafava, A.; Supuran, C.T. Sulfonamide inhibition studies of the β carbonic anhydrase from Drosophila melanogaster. Bioorg. Med. Chem. Lett. 2014, 24, 2797–2801. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T.; Capasso, C. New light on bacterial carbonic anhydrases phylogeny based on the analysis of signal peptide sequences. J. Enzym. Inhib. Med. Chem. 2016, 2016. 31, 1254–1260. [Google Scholar] [CrossRef]
- Supuran, C.T.; Capasso, C. Biomedical applications of prokaryotic carbonic anhydrases. Expert Opin. Ther. Pat. 2018, 28, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, S.; De Luca, V.; Capasso, C.; Supuran, C.T.; Carginale, V. Recombinant thermoactive phosphoenolpyruvate carboxylase (PEPC) from Thermosynechococcus elongatus and its coupling with mesophilic/thermophilic bacterial carbonic anhydrases (CAs) for the conversion of CO2 to oxaloacetate. Bioorg. Med. Chem. 2016, 24, 220–225. [Google Scholar] [CrossRef] [PubMed]
- Bootorabi, F.; Jänis, J.; Smith, E.; Waheed, A.; Kukkurainen, S.; Hytönen, V.; Valjakka, J.; Supuran, C.T.; Vullo, D.; Sly, W.S.; Parkkila, S. Analysis of a shortened form of human carbonic anhydrase VII expressed in vitro compared to the full-length enzyme. Biochimie 2010, 92, 1072–1080. [Google Scholar] [CrossRef]
- Vermelho, A.B.; Capaci, G.R.; Rodrigues, I.A.; Cardoso, V.S.; Mazotto, A.M.; Supuran, C.T. Carbonic anhydrases from Trypanosoma and Leishmania as anti-protozoan drug targets. Bioorg. Med. Chem. 2017, 25, 1543–1555. [Google Scholar] [CrossRef] [PubMed]
- da Silva Cardoso, V.; Vermelho, A.B.; Ricci Junior, E.; Almeida Rodrigues, I.; Mazotto, A.M.; Supuran, C.T. Antileishmanial activity of sulphonamide nanoemulsions targeting the β-carbonic anhydrase from Leishmania species. J Enzym. Inhib Med. Chem. 2018, 33, 850–857. [Google Scholar] [CrossRef] [PubMed]
- Loftus, B.; Anderson, I.; Davies, R.; Alsmark, U.C.; Samuelson, J.; Amedeo, P.; Roncaglia, P.; Berriman, M.; Hirt, R.P.; Mann, B.J.; et al. The genome of the protist parasite Entamoeba Histolytica. Nature 2005, 433, 865–868. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, A.S.; Bergfors, T.; Jones, T.A.; Högbom, M. Structural mechanics of the pH-dependent activity of beta-carbonic anhydrase from Mycobacterium tuberculosis. J. Biol. Chem. 2006, 281, 4993–4999. [Google Scholar] [CrossRef] [PubMed]
- Nishimori, I.; Minakuchi, T.; Vullo, D.; Scozzafava, A.; Innocenti, A.; Supuran, C.T. Carbonic anhydrase inhibitors. Cloning, characterization, and inhibition studies of a new beta-carbonic anhydrase from Mycobacterium tuberculosis. J. Med. Chem. 2009, 52, 3116–3120. [Google Scholar] [CrossRef] [PubMed]
- Ferraroni, M.; Del Prete, S.; Vullo, D.; Capasso, C.; Supuran, C.T. Crystal structure and kinetic studies of a tetrameric type II β-carbonic anhydrase from the pathogenic bacterium Vibrio cholerae. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 2449–2456. [Google Scholar] [CrossRef] [PubMed]
- McGurn, L.D.; Moazami-Goudarzi, M.; White, S.A.; Suwal, T.; Brar, B.; Tang, J.Q.; Espie, G.S.; Kimber, M.S. The structure, kinetics and interactions of the β-carboxysomal β-carbonic anhydrase, CcaA. Biochem. J. 2016, 473, 4559–4572. [Google Scholar] [CrossRef] [PubMed]
- Khalifah, R.G. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J. Biol. Chem. 1971, 246, 2561–2573. [Google Scholar] [PubMed]
- Nishimori, I.; Minakuchi, T.; Vullo, D.; Scozzafava, A.; Supuran, C.T. Inhibition studies of the β-carbonic anhydrases from the bacterial pathogen Salmonella enterica serovar Typhimurium with sulfonamides and sulfamates. Bioorg. Med. Chem. 2011, 19, 5023–5030. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Carbonic anhydrase inhibitors and their potential in a range of therapeutic areas. Expert Opin. Ther. Pat. 2018, 28, 709–712. [Google Scholar] [CrossRef] [PubMed]
- Supuran, C.T. Applications of carbonic anhydrases inhibitors in renal and central nervous system diseases. Expert Opin Ther Pat. 2018, 28, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Nocentini, A.; Supuran, C.T. Carbonic anhydrase inhibitors as antitumor/antimetastatic agents: A patent review (2008–2018). Expert Opin Ther Pat. 2018, 28, 729–740. [Google Scholar] [CrossRef]
- Muñoz, W.; Lamm, A.; Poppers, D.; Lamm, S. Acetazolamide promotes decreased consumption of carbonated drinks and weight loss. Oxf. Med. Case Rep. 2018, 2018, Omy081. [Google Scholar] [CrossRef]
- Abbate, F.; Supuran, C.T.; Scozzafava, A.; Orioli, P.; Stubbs, M.T.; Klebe, G. Nonaromatic sulfonamide group as an ideal anchor for potent human carbonic anhydrase inhibitors: Role of hydrogen-bonding networks in ligand binding and drug design. J. Med. Chem. 2004, 47, 550–557. [Google Scholar] [CrossRef]
- Borras, J.; Scozzafava, A.; Menabuoni, L.; Mincione, F.; Briganti, F.; Mincione, G.; Supuran, C.T. Carbonic anhydrase inhibitors: Synthesis of water-soluble, topically effective intraocular pressure lowering aromatic/heterocyclic sulfonamides containing 8-quinoline-sulfonyl moieties: Is the tail more important than the ring? Bioorg. Med. Chem. 1999, 7, 2397–2406. [Google Scholar] [CrossRef]
- Supuran, C.T.; Clare, B.W. Carbonic anhydrase inhibitors–Part 57: Quantum chemical QSAR of a group of 1, 3, 4-thiadiazole-and 1,3,4-thiadiazoline disulfonamides with carbonic anhydrase inhibitory properties. Eur. J. Med. Chem. 1999, 34, 41–50. [Google Scholar] [CrossRef]
- Laitinen, O.H.; Airenne, K.J.; Hytönen, V.P.; Peltomaa, E.; Mähönen, A.J.; Wirth, T.; Lind, M.M.; Mäkelä, K.A.; Toivanen, P.I.; Schenkwein, D.; et al. A multipurpose vector system for the screening of libraries in bacteria, insect and mammalian cells and expression in vivo. Nucleic Acids Res. 2005, 33, e42. [Google Scholar] [CrossRef] [PubMed]
- Määttä, J.A.E.; Eisenberg-Domovich, Y.; Nordlund, H.R.; Hayouka, R.; Kulomaa, M.S.; Livnah, O. Chimeric avidin shows stability against harsh chemical conditions—Biochemical analysis and 3D structure. Biotechnol. Bioengin. 2011, 108, 481–490. [Google Scholar] [CrossRef]
- De Simone, G.; Supuran, C.T. (In)organic anions as carbonic anhydrase inhibitors. J. Inorg. Biochem. 2012, 111, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Murray, A.B.; Aggarwal, M.; Pinard, M.; Vullo, D.; Patrauchan, M.; Supuran, C.T.; McKenna, R. Structural Mapping of Anion Inhibitors to β-Carbonic Anhydrase psCA3 from Pseudomonas aeruginosa. ChemMedChem. 2018, 13, 2024–2029. [Google Scholar] [CrossRef] [PubMed]
- Zimmerman, S.A.; Ferry, J.G.; Supuran, C.T. Inhibition of the archaeal beta-class (Cab) and gamma-class (Cam) carbonic anhydrases. Curr. Top. Med. Chem. 2007, 7, 901–908. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Activity Level | Class | kcat | kcat/Km | KI (Acetazolamide) | Ref |
|---|---|---|---|---|---|---|
| (s−1) | (M−1 s−1) | (nM) | ||||
| hCA I | moderate | α | 2.0 × 105 | 5.0 × 107 | 250 | [12] |
| hCA II | very high | α | 1.4 × 106 | 1.5 × 108 | 12 | [12] |
| mtCA 1 | moderate | β | 3.9 × 105 | 3.7 × 107 | 480 | [32] |
| mtCA 2 | high | β | 9.6 × 105 | 9.3 × 107 | 9.8 | [32] |
| EhiCA | high | β | 6.7 × 105 | 8.9 × 107 | 509 | this work |
| Inhibitor/Enzyme Class | KI * (nM) | ||
|---|---|---|---|
| hCA I a | hCA II a | EhiCA | |
| α | α | β | |
| 1 | 28,000 | 300 | 2363 |
| 2 | 25,000 | 240 | 6011 |
| 3 | 79 | 8 | 951 |
| 4 | 78,500 | 320 | 833 |
| 5 | 25,000 | 170 | 567 |
| 6 | 21,000 | 160 | 798 |
| 7 | 8300 | 60 | >10,000 |
| 8 | 9800 | 110 | >10,000 |
| 9 | 6500 | 40 | >10,000 |
| 10 | 7300 | 54 | 4656 |
| 11 | 5800 | 63 | 742 |
| 12 | 8400 | 75 | 1911 |
| 13 | 8600 | 60 | 821 |
| 14 | 9300 | 19 | 579 |
| 15 | 5500 | 80 | 772 |
| 16 | 9500 | 94 | 89 |
| 17 | 21,000 | 125 | 36 |
| 18 | 164 | 46 | 383 |
| 19 | 109 | 33 | 521 |
| 20 | 6 | 2 | 385 |
| 21 | 69 | 11 | 368 |
| 22 | 164 | 46 | 331 |
| 23 | 109 | 33 | 290 |
| 24 | 95 | 30 | 285 |
| AAZ | 250 | 12 | 509 |
| MZA | 50 | 14 | 845 |
| EZA | 25 | 8 | 746 |
| DCP | 1200 | 38 | 790 |
| DZA | 50,000 | 9 | 6444 |
| BRZ | 45,000 | 3 | 3051 |
| BZA | 15 | 9 | 2471 |
| TPM | 250 | 10 | 3100 |
| ZNS | 56 | 35 | 9595 |
| SLP | 1200 | 40 | >10,000 |
| IND | 31 | 15 | 822 |
| VLX | 54,000 | 43 | >10,000 |
| CLX | 50,000 | 21 | >10,000 |
| SLT | 374 | 9 | 6727 |
| SAC | 18,540 | 5959 | >10,000 |
| HCT | 328 | 290 | 3402 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bua, S.; Haapanen, S.; Kuuslahti, M.; Parkkila, S.; Supuran, C.T. Sulfonamide Inhibition Studies of a New β-Carbonic Anhydrase from the Pathogenic Protozoan Entamoeba histolytica. Int. J. Mol. Sci. 2018, 19, 3946. https://doi.org/10.3390/ijms19123946
Bua S, Haapanen S, Kuuslahti M, Parkkila S, Supuran CT. Sulfonamide Inhibition Studies of a New β-Carbonic Anhydrase from the Pathogenic Protozoan Entamoeba histolytica. International Journal of Molecular Sciences. 2018; 19(12):3946. https://doi.org/10.3390/ijms19123946
Chicago/Turabian StyleBua, Silvia, Susanna Haapanen, Marianne Kuuslahti, Seppo Parkkila, and Claudiu T. Supuran. 2018. "Sulfonamide Inhibition Studies of a New β-Carbonic Anhydrase from the Pathogenic Protozoan Entamoeba histolytica" International Journal of Molecular Sciences 19, no. 12: 3946. https://doi.org/10.3390/ijms19123946
APA StyleBua, S., Haapanen, S., Kuuslahti, M., Parkkila, S., & Supuran, C. T. (2018). Sulfonamide Inhibition Studies of a New β-Carbonic Anhydrase from the Pathogenic Protozoan Entamoeba histolytica. International Journal of Molecular Sciences, 19(12), 3946. https://doi.org/10.3390/ijms19123946