Genetic and Epigenetic Modifiers of Alcoholic Liver Disease



Abstract
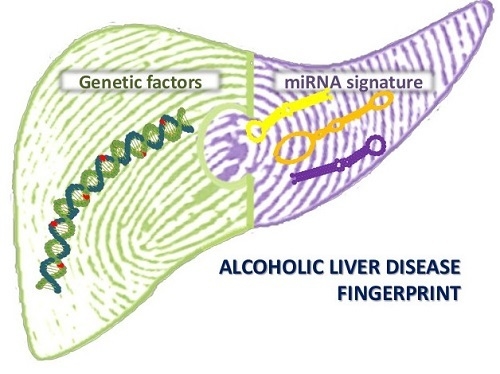
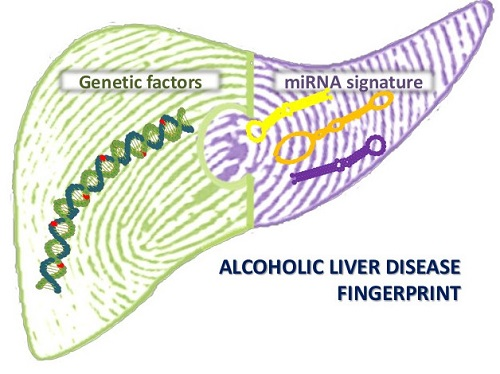
1. Introduction
2. Pathogenesis, Natural History, and Clinical Aspects of Alcoholic Liver Disease (ALD)
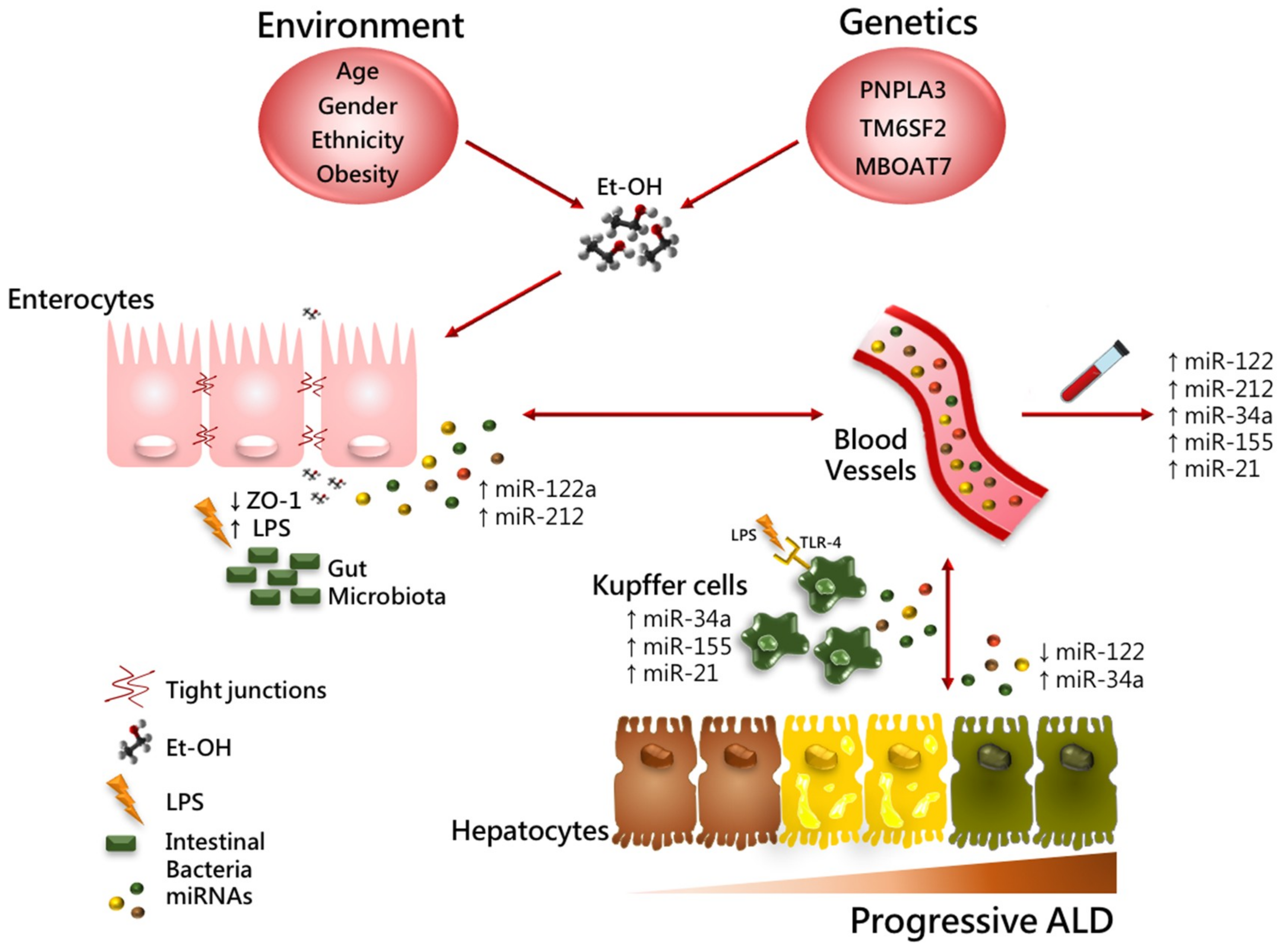
3. Environmental Risk Factors Involved in ALD Pathogenesis
3.1. Alcohol Consumption
3.2. Age
3.3. Gender
3.4. Ethnicity
3.5. Obesity
4. Genetic Factors Involved in ALD Pathogenesis
4.1. PNPLA3
4.2. TM6SF2
4.3. MBOAT7
4.4. Other Genetic Factors Involved in Alcohol-Related Liver Injury
5. Epigenetic Factors Involved in ALD Pathogenesis
5.1. miR-155
5.2. miR-34a
5.3. miR-122-miR-212
5.4. miR-21
5.5. Other miRNAs Implicated in Alcohol-Induced Liver Injury
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ACC1 | Acetyl-CoA Carboxylase Alpha |
| ADH | Alcohol Dehydrogenase |
| ALD | Alcoholic Liver Disease |
| ALDH | Aldehyde Dehydrogenase |
| ASH | Alcoholic Steatohepatitis |
| AUD | Alcohol Use Disorder |
| BMI | Body Mass Index |
| CYP2E1 | Cytochrome P450 2E1 |
| DR5 | Death receptor 5 |
| ER | Endoplasmic reticulum |
| Et-OH | Ethanol |
| FABP4 | Fatty Acid Binding Protein 4 |
| FASLG | Fas Cell Surface Death Receptor Ligand G |
| FFAs | FFAs Free Fatty Acids |
| GST | Glutathione- S-Transferases |
| GWAS | Genome-Wide Association Studies |
| HCC | Hepatocellular Carcinoma |
| HIF-1α | Hypoxia Inducible Factor 1 Subunit Alpha |
| HNF4α | Hepatocyte Nuclear Factor 4 Alpha |
| HSCs | Hepatic Stellate Cells |
| IL1β | Interleukin-1 Beta |
| IL6/STAT3IL10 | Interleukin-6/ Signal Transducer and Activator Of Transcription 3Interleukin-10 |
| LDLR | Low Density Lipoprotein Receptor |
| LPIAT1 | Lysophosphatidylinositol acyl- transferase 1 |
| LPS | Lipopolysaccharides |
| MBOAT7/TMC4 | Membrane bound O-acyltransferase domain containing 7-Transmembrane channel-like 4 |
| MCP1 | Monocyte chemoattractant protein 1 |
| MetS | Metabolic Syndrome |
| miRNAs | microRNAs |
| MMP-3 | Matrix Metalloproteinase-3 |
| MnSOD | Manganese-Dependent Superoxide Dismutase |
| NAFLD | Nonalcoholic Fatty Liver Disease |
| NASH | Nonalcoholic Steatohepatitis |
| NCAN | Neurocan |
| NF-κB | Nuclear Factor Kappa-Light-Chain-Enhancer Of Activated B Cells |
| NOD | Nucleotide-binding Oligomerization Domain-like |
| OR [CI 95%] | Odds Ratio [Confidence Interval 95%] |
| p47phox | Phagocytic oxidase p47 |
| PNPLA3 | Patatin-like phospholipase domain containing-3 |
| PPARα | Peroxisome Proliferator Activated Receptor-alpha |
| PPARγ | Peroxisome Proliferator Activated Receptor-gamma |
| ROS | Reactive Oxygen Species |
| SIRT1 | Sirtuin 1 |
| SNP | Single Nucleotide Polymorphism |
| SOD2 | Superoxide Dismutase 2 |
| SREBP-1c | Sterol regulatory element-binding protein |
| TG | Triglycerides |
| TGFβ | Transforming Growth Factor-Beta |
| TLR4 | Toll-like receptor4 |
| TM6SF2 | Transmembrane 6 superfamily member 2 |
| TNFα | Tumor Necrosis Factor alpha |
| UTR | Untranslated region |
| VLDL | Very Low-Density Lipoprotein |
| WHO | World Health Organization |
| ZO-1 | Zonula Occludens-1 |
References
- Rehm, J.; Samokhvalov, A.; Shield, K. Global burden of alcoholic liver diseases. J. Hepatol. 2013, 59, 160–168. [Google Scholar] [CrossRef] [PubMed]
- World Health Organisation Global Status Report on Alcohol and Health 2018; Global Information System on Alcohol and Health: Geneva, Switzerland, 2018; ISBN 978-92-4-156563-9.
- Asrani, S.K.; Saracino, G.; O’Leary, J.G.; Gonzales, S.; Kim, P.T.; McKenna, G.J.; Klintmalm, G.; Trotter, J. Recipient characteristics and morbidity and mortality after liver transplantation. J. Hepatol. 2018, 69, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Mathurin, P.; Bataller, R. Trends in the management and burden of alcoholic liver disease. J. Hepatol. 2015, 62, S38–S46. [Google Scholar] [CrossRef] [PubMed]
- Diehl, A.M.; Goodman, Z.; Ishak, K.G. Alcohollike liver disease in nonalcoholics. A clinical and histologic comparison with alcohol-induced liver injury. Gastroenterology 1988, 95, 1056–1062. [Google Scholar] [CrossRef]
- Völzke, H. Multicausality in fatty liver disease: Is there a rationale to distinguish between alcoholic and non-alcoholic origin? World J. Gastroenterol. 2012, 18, 3492–3501. [Google Scholar] [CrossRef]
- Toshikuni, N.; Tsutsumi, M.; Arisawa, T. Clinical differences between alcoholic liver disease and nonalcoholic fatty liver disease. World J. Gastroenterol. 2014, 20, 8393–8406. [Google Scholar] [CrossRef] [PubMed]
- Celli, R.; Zhang, X. Pathology of Alcoholic Liver Disease. J. Clin. Transl. Hepatol. 2014, 2, 103–109. [Google Scholar] [CrossRef] [PubMed]
- Singal, A.K.; Bataller, R.; Ahn, J.; Kamath, P.S.; Shah, V.H. ACG clinical guideline: Alcoholic liver disease. Am. J. Gastroenterol. 2018, 113, 175–194. [Google Scholar] [CrossRef]
- Teli, M.; James, O.; Burt, A.; Bennett, M.; Day, C. The natural history of nonalcoholic fatty liver: A follow-up study. Hepatology 1995, 22, 1714–1719. [Google Scholar] [CrossRef]
- Stickel, F. Alcoholic cirrhosis and hepatocellular carcinoma. Adv. Exp. Med. Biol. 2015, 815, 113–130. [Google Scholar] [CrossRef]
- Fattovich, G.; Stroffolini, T.; Zagni, I.; Donato, F. Hepatocellular carcinoma in cirrhosis: Incidence and risk factors. Gastroenterology 2004, 127, S35–S50. [Google Scholar] [CrossRef] [PubMed]
- Ganne-Carrie, N.; Chaffaut, C.; Bourcier, V.; Archambeaud, I.; Perarnau, J.-M.; Oberti, F.; Roulot, D.; Moreno, C.; Louvet, A.; Dao, T.; et al. Estimate of hepatocellular carcinoma incidence in patients with alcoholic cirrhosis. J. Hepatol. 2018, 69, 1274–1283. [Google Scholar] [CrossRef] [PubMed]
- Donohue, T.M. Alcohol-induced steatosis in liver cells. World J. Gastroenterol. 2007, 13, 4974–4978. [Google Scholar]
- Donohue, T.M.; Curry-McCoy, T.V.; Todero, S.L.; White, R.L.; Kharbanda, K.K.; Nanji, A.A.; Osna, N.A. L-Buthionine (S,R) sulfoximine depletes hepatic glutathione but protects against ethanol-induced liver injury. Alcohol. Clin. Exp. Res. 2007, 31, 1053–1060. [Google Scholar] [CrossRef] [PubMed]
- Crabb, D.W.; Galli, A.; Fischer, M.; You, M. Molecular mechanisms of alcoholic fatty liver: Role of peroxisome proliferator-activated receptor alpha. Alcohol 2004, 34, 35–38. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-G.; Dou, X.-B.; Zhou, Z.-X.; Song, Z.-Y. Adipose tissue-liver axis in alcoholic liver disease. World J. Gastrointest. Pathophysiol. 2016, 7, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Addolorato, G.; Capristo, E.; Greco, A.V.; Stefanini, G.F.; Gasbarrini, G. Energy expenditure, substrate oxidation, and body composition in subjects with chronic alcoholism: New findings from metabolic assessment. Alcohol. Clin. Exp. Res. 1997, 21, 962–967. [Google Scholar] [CrossRef]
- Loria, P.; Marchesini, G.; Nascimbeni, F.; Ballestri, S.; Maurantonio, M.; Carubbi, F.; Ratziu, V.; Lonardo, A. Cardiovascular risk, lipidemic phenotype and steatosis. A comparative analysis of cirrhotic and non-cirrhotic liver disease due to varying etiology. Atherosclerosis 2014, 232, 99–109. [Google Scholar] [CrossRef]
- Casini, A.; Ceni, E.; Salzano, R.; Schuppan, D.; Milani, S.; Pellegrini, G.; Surrenti, C. Regulation of undulin synthesis and gene expression in human fat-storing cells by acetaldehyde and transforming growth factor-beta 1: Comparison with fibronectin. Biochem. Biophys. Res. Commun. 1994, 199, 1019–1026. [Google Scholar] [CrossRef]
- Svegliati-Baroni, G.; Inagaki, Y.; Rincon-Sanchez, A.R.; Else, C.; Saccomanno, S.; Benedetti, A.; Ramirez, F.; Rojkind, M. Early response of α2(I) collagen to acetaldehyde in human hepatic stellate cells is TGF-β independent. Hepatology 2005, 42, 343–352. [Google Scholar] [CrossRef]
- Schwartz, J.M.; Reinus, J.F. Prevalence and Natural History of Alcoholic Liver Disease. Clin. Liver Dis. 2012, 16, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Deleuran, T.; Grønbæk, H.; Vilstrup, H.; Jepsen, P. Cirrhosis and mortality risks of biopsy-verified alcoholic pure steatosis and steatohepatitis: A nationwide registry-based study. Aliment. Pharmacol. Ther. 2012, 35, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef] [PubMed]
- Lelbach, W.K. Cirrhosis in the alcoholic and its relations to the volume of alcohol abuse. Ann. N. Y. Acad. Sci. 2015, 252, 85–105. [Google Scholar] [CrossRef]
- Lelbach, W.K. Cirrhosis in the alcoholic and its relations to the volume of alcohol abuse. Ann. N. Y. Acad. Sci. 1975, 252, 85–105. [Google Scholar] [CrossRef] [PubMed]
- Kamper-Jørgensen, M.; Grønbæk, M.; Tolstrup, J.; Becker, U. Alcohol and cirrhosis: Dose-response or threshold effect? J. Hepatol. 2004, 41, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Rehm, J.; Taylor, B.; Mohapatra, S.; Irving, H.; Baliunas, D.; Patra, J.; Roerecke, M. Alcohol as a risk factor for liver cirrhosis: A systematic review and meta-analysis. Drug Alcohol Rev. 2010, 29, 437–445. [Google Scholar] [CrossRef]
- Bellentani, S.; Saccoccio, G.; Costa, G.; Tiribelli, C.; Manenti, F.; Sodde, M.; Croce’, L.S.; Sasso, F.; Pozzato, G.; Cristianini, G.; Brandi and the Dionysos Study Group, G. Drinking habits as cofactors of risk for alcohol induced liver damage. Gut 1997, 41, 845–850. [Google Scholar] [CrossRef]
- Becker, U.; Gronbak, M.; Johansen, D.; Sorensen, T.I.A. Lower risk for alcohol-induced cirrhosis in wine drinkers. Hepatology 2002, 35, 868–875. [Google Scholar] [CrossRef]
- Johansen, D.; Friis, K.; Skovenborg, E.; Grønbæk, M. Food buying habits of people who buy wine or beer: Cross sectional study. Br. Med. J. 2006, 332, 519–522. [Google Scholar] [CrossRef]
- Mäkelä, K.; Mustonen, H. Relationships of drinking behaviour, gender and age with reported negative and positive experiences related to drinking. Addiction 2000, 95, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Meier, P.; Seitz, H.K. Age, alcohol metabolism and liver disease. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Poikolainen, K. Risk factors for alcohol dependence: A case-control study. Alcohol Alcohol. 2000, 35, 190–196. [Google Scholar] [CrossRef] [PubMed]
- Mumenthaler, M.S.; Taylor, J.L.; O’Hara, R.; Yesavage, J.A. Gender differences in moderate drinking effects. Alcohol Res. Health 1999, 23, 55–64. [Google Scholar] [PubMed]
- Ikejima, K.; Enomoto, N.; Iimuro, Y.; Ikejima, A.; Fang, D.; Xu, J.; Forman, D.T.; Brenner, D.A.; Thurman, R.G. Estrogen increases sensitivity of hepatic Kupffer cells to endotoxin. Am. J. Physiol. Gastrointest. Liver Physiol. 1998, 22, 768–769. [Google Scholar] [CrossRef]
- Fulham, M.A.; Mandrekar, P. Sexual dimorphism in alcohol induced adipose inflammation relates to liver injury. PLoS ONE 2016, 11, e0164225. [Google Scholar] [CrossRef] [PubMed]
- Ballestri, S.; Nascimbeni, F.; Baldelli, E.; Marrazzo, A.; Romagnoli, D.; Lonardo, A. NAFLD as a Sexual Dimorphic Disease: Role of Gender and Reproductive Status in the Development and Progression of Nonalcoholic Fatty Liver Disease and Inherent Cardiovascular Risk. Adv. Ther. 2017, 34, 1291–1326. [Google Scholar] [CrossRef]
- Levy, R.E.; Catana, A.M.; Durbin-Johnson, B.; Halsted, C.H.; Medici, V. Ethnic differences in presentation and severity of alcoholic liver disease. Alcohol. Clin. Exp. Res. 2015, 39, 566–574. [Google Scholar] [CrossRef]
- Stinson, F.S.; Grant, B.F.; Dufour, M.C. The critical dimension of ethnicity in liver cirrhosis mortality statistics. Alcohol. Clin. Exp. Res. 2001. [Google Scholar] [CrossRef]
- Lazo, M.; Hernaez, R.; Eberhardt, M.S.; Bonekamp, S.; Kamel, I.; Guallar, E.; Koteish, A.; Brancati, F.L.; Clark, J.M. Prevalence of nonalcoholic fatty liver disease in the United States: The third national health and nutrition examination survey, 1988–1994. Am. J. Epidemiol. 2013, 178, 38–45. [Google Scholar] [CrossRef]
- Landen, M.; Roeber, J.; Naimi, T.; Nielsen, L.; Sewell, M. Alcohol-attributable mortality among American Indians and Alaska Natives in the United States, 1999–2009. Am. J. Public Health 2014, 104, S343–S349. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Balkwill, A.; Reeves, G.; Beral, V. Body mass index and risk of liver cirrhosis in middle aged UK women: Prospective study. BMJ 2010, 340, c912. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Thaury, J.; Barri-Ova, N.; Balian, A.; Dauvois, B.; Njiké-Nakseu, M.; Prévot, S.; Agostini, H.; Perlemuter, G. Predictive factors for pure steatosis in alcoholic patients. Alcohol. Clin. Exp. Res. 2009, 33, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Chaput, J.C.; Fallik, D.; Balian, A.; Raynard, B.; Capron, F.; Bedossa, P. Risk factors of fibrosis in alcohol-induced liver disease. Hepatology 2002, 35, 635–638. [Google Scholar] [CrossRef]
- Diehl, A.M. Obesity and alcoholic liver disease. Alcohol 2004, 34, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Hart, C.L.; Morrison, D.S.; Batty, G.D.; Mitchell, R.J.; Davey Smith, G. Effect of body mass index and alcohol consumption on liver disease: Analysis of data from two prospective cohort studies. BMJ 2010, 340, c1240. [Google Scholar] [CrossRef] [PubMed]
- Naveau, S.; Giraud, V.; Borotto, E.; Aubert, A.; Capron, F.; Chaput, J. Excess weight risk factor for alcoholic liver disease. Hepatology 1997, 25, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Bellentani, S.; Saccoccio, G.; Masutti, F.; Crocè, L.S.; Brandi, G.; Sasso, F.; Cristanini, G.; Tiribelli, C. Prevalence of and risk factors for hepatic steatosis in Northern Italy. Ann. Intern. Med. 2000, 132, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Bierut, L.J.; Dinwiddie, S.H.; Begleiter, H.; Crowe, R.R.; Hesselbrock, V.; Nurnberger, J.I.; Porjesz, B.; Schuckit, M.A.; Reich, T. Familial transmission of substance dependence: Alcohol, marijuana, cocaine, and habitual smoking: A report from the collaborative study on the genetics of alcoholism. Arch. Gen. Psychiatry 1998, 55, 982–988. [Google Scholar] [CrossRef]
- Walters, G.D. The heritability of alcohol abuse and dependence: A meta-analysis of behavior genetic research. Am. J. Drug Alcohol Abuse 2002, 28, 557–584. [Google Scholar] [CrossRef]
- Verhulst, B.; Neale, M.C.; Kendler, K.S. The heritability of alcohol use disorders: A meta-analysis of twin and adoption studies. Psychol. Med. 2015, 45, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Hrubec, Z.; Omenn, G.S. Evidence of genetic predisposition to alcholic cirrhosis and psychosis: Twin concordances for alcholism and its biological end points by zygosity among male veterans. Alcohol. Clin. Exp. Res. 1981, 5, 207–215. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Kozlitina, J.; Smagris, E.; Stender, S.; Nordestgaard, B.G.; Zhou, H.H.; Tybjærg-Hansen, A.; Vogt, T.F.; Hobbs, H.H.; Cohen, J.C. Exome-wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat. Genet. 2014, 46, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Yerges-Armstrong, L.M.; Wu, J.; Hernaez, R.; Kim, L.J.; Palmer, C.D.; Gudnason, V.; Eiriksdottir, G.; Garcia, M.E.; Launer, L.J.; et al. Genome-wide association analysis identifies variants associated with nonalcoholic fatty liver disease that have distinct effects on metabolic traits. PLoS Genet. 2011, 7, e1001324. [Google Scholar] [CrossRef] [PubMed]
- Mancina, R.M.; Dongiovanni, P.; Petta, S.; Pingitore, P.; Meroni, M.; Rametta, R.; Borén, J.; Montalcini, T.; Pujia, A.; Wiklund, O.; et al. The MBOAT7-TMC4 Variant rs641738 Increases Risk of Nonalcoholic Fatty Liver Disease in Individuals of European Descent. Gastroenterology 2016, 150, 1219–1230.e6. [Google Scholar] [CrossRef] [PubMed]
- Buch, S.; Stickel, F.; Trépo, E.; Way, M.; Herrmann, A.; Nischalke, H.D.; Brosch, M.; Rosendahl, J.; Berg, T.; Ridinger, M.; et al. A genome-wide association study confirms PNPLA3 and identifies TM6SF2 and MBOAT7 as risk loci for alcohol-related cirrhosis. Nat. Genet. 2015, 47, 1443–1448. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Burgueño, A.L.; Gianotti, T.F.; Rosselli, M.S.; Pirola, C.J. A nonsynonymous gene variant in the adiponutrin gene is associated with nonalcoholic fatty liver disease severity. J. Lipid Res. 2009, 50, 2111–2116. [Google Scholar] [CrossRef] [PubMed]
- Valenti, L.; Al-Serri, A.; Daly, A.K.; Galmozzi, E.; Rametta, R.; Dongiovanni, P.; Nobili, V.; Mozzi, E.; Roviaro, G.; Vanni, E.; et al. Homozygosity for the patatin-like phospholipase-3/adiponutrin I148M polymorphism influences liver fibrosis in patients with nonalcoholic fatty liver disease. Hepatology 2010, 51, 1209–1217. [Google Scholar] [CrossRef]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C >g polymorphism confers an increased risk of non-alcoholic fatty liver disease associated hepatocellular carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef]
- Di Costanzo, A.; D’Erasmo, L.; Polimeni, L.; Baratta, F.; Coletta, P.; Di Martino, M.; Loffredo, L.; Perri, L.; Ceci, F.; Montali, A.; et al. Non-alcoholic fatty liver disease and subclinical atherosclerosis: A comparison of metabolically- versus genetically-driven excess fat hepatic storage. Atherosclerosis 2017, 257, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Lonardo, A.; Ballestri, S.; Targher, G. “Not all forms of NAFLD were created equal”. Do metabolic syndrome-related NAFLD and PNPLA3-related NAFLD exert a variable impact on the risk of early carotid atherosclerosis? Atherosclerosis 2017, 257, 253–255. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Stokowski, R.P.; Kershenobich, D.; Ballinger, D.G.; Hinds, D.A. Variant in PNPLA3 is associated with alcoholic liver disease. Nat. Genet. 2010, 42, 21–23. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Buch, S.; Lau, K.; Zu Schwabedissen, H.M.; Berg, T.; Ridinger, M.; Rietschel, M.; Schafmayer, C.; Braun, F.; Hinrichsen, H.; et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology 2011, 58, 86–95. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Buch, S.; Lau, K.; Zu Schwabedissen, H.M.; Berg, T.; Ridinger, M.; Rietschel, M.; Schafmayer, C.; Braun, F.; Hinrichsen, H.; et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology 2011, 53, 86–95. [Google Scholar] [CrossRef]
- Falleti, E.; Fabris, C.; Cmet, S.; Cussigh, A.; Bitetto, D.; Fontanini, E.; Fornasiere, E.; Bignulin, S.; Fumolo, E.; Bignulin, E.; et al. PNPLA3 rs738409 C/G polymorphism in cirrhosis: Relationship with the aetiology of liver disease and hepatocellular carcinoma occurrence. Liver Int. 2011, 31, 1137–1143. [Google Scholar] [CrossRef]
- Friedrich, K.; Wannhoff, A.; Kattner, S.; Brune, M.; Hov, J.R.; Weiss, K.H.; Antoni, C.; Dollinger, M.; Neumann-Haefelin, C.; Seufferlein, T.; et al. PNPLA3 in end-stage liver disease: Alcohol consumption, hepatocellular carcinoma development, and transplantation-free survival. J. Gastroenterol. Hepatol. 2014, 29, 1477–1484. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Trepo, E.; Nahon, P.; Cao, Q.; Moreno, C.; Letouze, E.; Imbeaud, S.; Gustot, T.; Deviere, J.; Debette, S.; et al. PNPLA3 and TM6SF2 variants as risk factors of hepatocellular carcinoma across various etiologies and severity of underlying liver diseases. Int. J. Cancer 2018. [Google Scholar] [CrossRef] [PubMed]
- Guyot, E.; Sutton, A.; Rufat, P.; Laguillier, C.; Mansouri, A.; Moreau, R.; Ganne-Carrié, N.; Beaugrand, M.; Charnaux, N.; Trinchet, J.C.; Nahon, P. PNPLA3 rs738409, hepatocellular carcinoma occurrence and risk model prediction in patients with cirrhosis. J. Hepatol. 2013, 58, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Trépo, E.; Nahon, P.; Bontempi, G.; Valenti, L.; Falleti, E.; Nischalke, H.-D.; Hamza, S.; Corradini, S.G.; Burza, M.A.; Guyot, E.; et al. Association between the PNPLA3 (rs738409 C>G) variant and hepatocellular carcinoma: Evidence from a meta-analysis of individual participant data. Hepatology 2014, 59, 2170–2177. [Google Scholar] [CrossRef]
- He, S.; McPhaul, C.; Li, J.Z.; Garuti, R.; Kinch, L.; Grishin, N.V.; Cohen, J.C.; Hobbs, H.H. A sequence variation (I148M) in PNPLA3 associated with nonalcoholic fatty liver disease disrupts triglyceride hydrolysis. J. Biol. Chem. 2010, 285, 6706–6715. [Google Scholar] [CrossRef] [PubMed]
- BasuRay, S.; Smagris, E.; Cohen, J.C.; Hobbs, H.H. The PNPLA3 variant associated with fatty liver disease (I148M) accumulates on lipid droplets by evading ubiquitylation. Hepatology 2017, 66, 1111–1124. [Google Scholar] [CrossRef] [PubMed]
- Pirazzi, C.; Adiels, M.; Burza, M.A.; Mancina, R.M.; Levin, M.; Ståhlman, M.; Taskinen, M.R.; Orho-Melander, M.; Perman, J.; Pujia, A.; et al. Patatin-like phospholipase domain-containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J. Hepatol. 2012, 57, 127–1282. [Google Scholar] [CrossRef] [PubMed]
- Pingitore, P.; Dongiovanni, P.; Motta, B.M.; Meroni, M.; Lepore, S.M.; Mancina, R.M.; Pelusi, S.; Russo, C.; Caddeo, A.; Rossi, G.; et al. PNPLA3 overexpression results in reduction of proteins predisposing to fibrosis. Hum. Mol. Genet. 2016, 25, 5212–5222. [Google Scholar] [CrossRef]
- Dongiovanni, P.; Petta, S.; Maglio, C.; Fracanzani, A.L.; Pipitone, R.; Mozzi, E.; Motta, B.M.; Kaminska, D.; Rametta, R.; Grimaudo, S.; et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015, 61, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Lu, H.; Guo, Y.; Zhu, T.; Garcia-Barrio, M.T.; Jiang, Z.; Willer, C.J.; Zhang, J.; Eugene Chen, Y. Hepatic Transmembrane 6 Superfamily Member 2 Regulates Cholesterol Metabolism in Mice. Gastroenterology 2016, 150, 1208–1218. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Wong, G.L.-H.; Tse, C.-H.; Chan, H.L.-Y. Prevalence of the TM6SF2 variant and non-alcoholic fatty liver disease in Chinese. J. Hepatol. 2014, 61, 708–709. [Google Scholar] [CrossRef]
- Sookoian, S.; Castaño, G.O.; Scian, R.; Mallardi, P.; Fernández Gianotti, T.; Burgueño, A.L.; San Martino, J.; Pirola, C.J. Genetic variation in transmembrane 6 superfamily member 2 and the risk of nonalcoholic fatty liver disease and histological disease severity. Hepatology 2015, 61, 515–525. [Google Scholar] [CrossRef]
- Goffredo, M.; Caprio, S.; Feldstein, A.E.; D’Adamo, E.; Shaw, M.M.; Pierpont, B.; Savoye, M.; Zhao, H.; Bale, A.E.; Santoro, N. Role of TM6SF2 rs58542926 in the pathogenesis of nonalcoholic pediatric fatty liver disease: A multiethnic study. Hepatology 2016, 63, 117–125. [Google Scholar] [CrossRef]
- Liu, Y.-L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.D.; Alexander, G.J.; Piguet, A.-C.; Anty, R.; et al. TM6SF2 rs58542926 influences hepatic fibrosis progression in patients with non-alcoholic fatty liver disease. Nat. Commun. 2014, 5, 4305. [Google Scholar] [CrossRef]
- Milano, M.; Aghemo, A.; Mancina, R.M.; Fischer, J.; Dongiovanni, P.; De Nicola, S.; Fracanzani, A.L.; D’Ambrosio, R.; Maggioni, M.; De Francesco, R.; et al. Transmembrane 6 Superfamily Member 2 Gene E167K Variant Impacts on Steatosis and Liver Damage in Chronic Hepatitis C Patients. Hepatology 2015, 62, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Falleti, E.; Cussigh, A.; Cmet, S.; Fabris, C.; Toniutto, P. PNPLA3 rs738409 and TM6SF2 rs58542926 variants increase the risk of hepatocellular carcinoma in alcoholic cirrhosis. Dig. Liver Dis. 2016, 48, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Simons, N.; Isaacs, A.; Koek, G.H.; Kuč, S.; Schaper, N.C.; Brouwers, M.C. PNPLA3, TM6SF2, and MBOAT7 Genotypes and Coronary Artery Disease. Gastroenterology 2017, 152, 912–913. [Google Scholar] [CrossRef] [PubMed]
- D’Souza, K.; Nzirorera, C.; Kienesberger, P.C. Lipid metabolism and signaling in cardiac lipotoxicity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 1513–1524. [Google Scholar] [CrossRef] [PubMed]
- Gijón, M.A.; Riekhof, W.R.; Zarini, S.; Murphy, R.C.; Voelker, D.R. Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils. J. Biol. Chem. 2008, 283, 30235–30245. [Google Scholar] [CrossRef] [PubMed]
- Donati, B.; Dongiovanni, P.; Romeo, S.; Meroni, M.; McCain, M.; Miele, L.; Petta, S.; Maier, S.; Rosso, C.; De Luca, L.; et al. MBOAT7 rs641738 variant and hepatocellular carcinoma in non-cirrhotic individuals. Sci. Rep. 2017, 7, 4492. [Google Scholar] [CrossRef]
- Stickel, F.; Hampe, J. Genetic determinants of alcoholic liver disease. Gut 2012, 61, 150–159. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Daly, A.K.; Day, C.P. Genetics of Alcoholic Liver Disease. Semin. Liver Dis. 2015, 35, 361–374. [Google Scholar] [CrossRef]
- Williams, J.T.; Begleiter, H.; Porjesz, B.; Edenberg, H.J.; Foroud, T.; Reich, T.; Goate, A.; Van Eerdewegh, P.; Almasy, L.; Blangero, J. Joint Multipoint Linkage Analysis of Multivariate Qualitative and Quantitative Traits. II. Alcoholism and Event-Related Potentials. Am. J. Hum. Genet. 1999, 65, 1148–1160. [Google Scholar] [CrossRef]
- Homann, N.; Stickel, F.; König, I.R.; Jacobs, A.; Junghanns, K.; Benesova, M.; Schuppan, D.; Himsel, S.; Zuber-Jerger, I.; Hellerbrand, C.; et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int. J. Cancer 2006, 118, 1998–2002. [Google Scholar] [CrossRef]
- Zintzaras, E.; Stefanidis, I.; Santos, M.; Vidal, F. Do alcohol-metabolizing enzyme gene polymorphisms increase the risk of alcoholism and alcoholic liver disease? Hepatology 2006, 43, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Zeng, T.; Guo, F.F.; Zhang, C.L.; Song, F.Y.; Zhao, X.L.; Xie, K.Q. Roles of Cytochrome P4502E1 Gene Polymorphisms and the Risks of Alcoholic Liver Disease: A Meta-Analysis. PLoS ONE 2013, 8, e54188. [Google Scholar] [CrossRef] [PubMed]
- Degoul, F.; Sutton, A.; Mansouri, A.; Cepanec, C.; Degott, C.; Fromenty, B.; Beaugrand, M.; Valla, D.; Pessayre, D. Homozygosity for alanine in the mitochondrial targeting sequence of superoxide dismutase and risk for severe alcoholic liver disease. Gastroenterology 2001, 120, 1468–1474. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.F.; Leathart, J.B.; Chen, Y.; Daly, A.K.; Rolla, R.; Vay, D.; Mottaran, E.; Vidali, M.; Albano, E.; Day, C.P. Valine-alanine manganese superoxide dismutase polymorphism is not associated with alcohol-induced oxidative stress or liver fibrosis. Hepatology 2002, 36, 1355–1360. [Google Scholar] [CrossRef]
- Lucena, M.I.; Andrade, R.J.; Martínez, C.; Ulzurrun, E.; García-Martín, E.; Borraz, Y.; Fernández, M.C.; Romero-Gomez, M.; Castiella, A.; Planas, R.; et al. Glutathione S-transferase m1 and t1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology 2008, 48, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Brind, A.M.; Hurlstone, A.; Edrisinghe, D.; Gilmore, I.; Fisher, N.; Pirmohamed, M.; Fryer, A.A. The role of polymorphisms of glutathione S-transferases GSTM1, M3, P1, T1 and A1 in susceptibility to alcoholic liver disease. Alcohol Alcohol. 2004, 39, 478–483. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Daly, A.K.; Bassendine, M.F.; Gilvarry, E.; Day, C.P. Interleukin 10 promoter region polymorphisms and susceptibility to advanced alcoholic liver disease. Gut 2000, 46, 540–545. [Google Scholar] [CrossRef]
- Lazarus, R.; Vercelli, D.; Palmer, L.J.; Klimecki, W.J.; Silverman, E.K.; Richter, B.; Riva, A.; Ramoni, M.; Martinez, F.D.; Weiss, S.T.; et al. Single nucleotide polymorphisms in innate immunity genes: Abundant variation and potential role in complex human disease. Immunol. Rev. 2002, 190, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.; Mukhopadhyay, I.; Das, K.; Pandit, P.; Majumder, P.P.; Santra, A.; Datta, S.; Banerjee, S.; Chowdhury, A. Genetic variants of TNFα, IL10, IL1β, CTLA4 and TGFβ1 modulate the indices of alcohol-induced liver injury in East Indian population. Gene 2012, 509, 178–188. [Google Scholar] [CrossRef]
- Grove, J.; Daly, A.K.; Bassendine, M.F.; Day, C.P. Association of a tumor necrosis factor promoter polymorphism with susceptibility to alcoholic steatohepatitis. Hepatology 1997, 26, 143–146. [Google Scholar] [CrossRef]
- Pastor, I.J.; Laso, F.J.; Romero, A.; Gonzalez-Sarmiento, R. −238 G>A polymorphism of tumor necrosis factor alpha gene (TNFA) is associated with alcoholic liver cirrhosis in alcoholic Spanish men. Alcohol. Clin. Exp. Res. 2005, 29, 1928–1931. [Google Scholar] [CrossRef] [PubMed]
- Österreicher, C.H.; Halangk, J.; Berg, T.; Patsenker, E.; Homann, N.; Hellerbrand, C.; Seitz, H.K.; Eurich, D.; Stickel, F. Evaluation of the transforming growth factor β1 codon 25 (Arg → Pro) polymorphism in alcoholic liver disease. Cytokine 2008, 42, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Stickel, F.; Österreicher, C.H.; Halangk, J.; Berg, T.; Homann, N.; Hellerbrand, C.; Patsenker, E.; Schneider, V.; Kolb, A.; Friess, H.; et al. No role of matrixmetalloproteinase-3 genetic promoter polymorphism 1171 as a risk factor for cirrhosis in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2008, 32, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Nischalke, H.D.; Lutz, P.; Krämer, B.; Söhne, J.; Müller, T.; Rosendahl, J.; Fischer, J.; Berg, T.; Hittatiya, K.; Fischer, H.P.; et al. A common polymorphism in the NCAN gene is associated with hepatocellular carcinoma in alcoholic liver disease. J. Hepatol. 2014, 61, 1073–1079. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Friso, S.; Choi, S.-W. Epigenetics in non-alcoholic fatty liver disease. Mol. Asp. Med. 2016, 54, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, C.; Sharma, A.R.; Sharma, G.; Doss, C.G.P.; Lee, S.S. Therapeutic miRNA and siRNA: Moving from Bench to Clinic as Next Generation Medicine. Mol. Ther.-Nucleic Acids 2017, 8, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Bala, S. Emerging role of microRNAs in liver diseases. World J. Gastroenterol. 2009, 15, 5633–5640. [Google Scholar] [CrossRef] [PubMed]
- Starkey Lewis, P.J.; Dear, J.; Platt, V.; Simpson, K.J.; Craig, D.G.N.; Antoine, D.J.; French, N.S.; Dhaun, N.; Webb, D.J.; Costello, E.M.; et al. Circulating microRNAs as potential markers of human drug-induced liver injury. Hepatology 2011, 54, 1767–1776. [Google Scholar] [CrossRef]
- Xu, T.; Li, L.; Hu, H.Q.; Meng, X.M.; Huang, C.; Zhang, L.; Qin, J.; Li, J. MicroRNAs in alcoholic liver disease: Recent advances and future applications. J. Cell. Physiol. 2018, 234, 382–394. [Google Scholar] [CrossRef]
- Bala, S.; Szabo, G. MicroRNA Signature in Alcoholic Liver Disease. Int. J. Hepatol. 2012, 2012, 498232. [Google Scholar] [CrossRef] [PubMed]
- Dolganiuc, A.; Petrasek, J.; Kodys, K.; Catalano, D.; Mandrekar, P.; Velayudham, A.; Szabo, G. MicroRNA expression profile in lieber-decarli diet-induced alcoholic and methionine choline deficient diet-induced nonalcoholic steatohepatitis models in mice. Alcohol. Clin. Exp. Res. 2009, 33, 1704–1710. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Satishchandran, A. MicroRNAs in alcoholic liver disease. Semin. Liver Dis. 2015, 35, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Marcos, M.; Kodys, K.; Csak, T.; Catalano, D.; Mandrekar, P.; Szabo, G. Up-regulation of microRNA-155 in macrophages contributes to increased Tumor Necrosis Factor α (TNFα) production via increased mRNA half-life in alcoholic liver disease. J. Biol. Chem. 2011, 286, 1436–1444. [Google Scholar] [CrossRef]
- Hritz, I.; Mandrekar, P.; Velayudham, A.; Catalano, D.; Dolganiuc, A.; Kodys, K.; Kurt-Jones, E.; Szabo, G. The critical role of toll-like receptor (TLR) 4 in alcoholic liver disease is independent of the common TLR adapter MyD88. Hepatology 2008, 48, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Bala, S.; Csak, T.; Saha, B.; Zatsiorsky, J.; Kodys, K.; Catalano, D.; Satishchandran, A.; Szabo, G. The pro-inflammatory effects of miR-155 promote liver fibrosis and alcohol-induced steatohepatitis. J. Hepatol. 2016, 64, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Glaser, S.S.; Francis, H.; Yang, F.; Han, Y.; Stokes, A.; Staloch, D.; McCarra, J.; Liu, J.; Venter, J.; et al. Epigenetic regulation of miR-34a expression in alcoholic liver injury. Am. J. Pathol. 2012, 181, 804–817. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Hu, M.; Liang, X.; Ajmo, J.M.; Li, X.; Bataller, R.; Odena, G.; Stevens, S.M.; You, M. Deletion of SIRT1 from hepatocytes in mice disrupts lipin-1 signaling and aggravates alcoholic fatty liver. Gastroenterology 2014, 146, 801–811. [Google Scholar] [CrossRef] [PubMed]
- Dippold, R.P.; Vadigepalli, R.; Gonye, G.E.; Patra, B.; Hoek, J.B. Chronic Ethanol Feeding Alters miRNA Expression Dynamics During Liver Regeneration. Alcohol. Clin. Exp. Res. 2013, 37, E59–E69. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Padhye, A.; Sharma, A.; Song, G.; Miao, J.; Mo, Y.Y.; Wang, L.; Kemper, J.K. A pathway involving farnesoid X receptor and small heterodimer partner positively regulates hepatic sirtuin 1 levels via MicroRNA-34a inhibition. J. Biol. Chem. 2010, 285, 12604–12611. [Google Scholar] [CrossRef] [PubMed]
- Satishchandran, A.; Ambade, A.; Rao, S.; Hsueh, Y.C.; Iracheta-Vellve, A.; Tornai, D.; Lowe, P.; Gyongyosi, B.; Li, J.; Catalano, D.; et al. MicroRNA 122, Regulated by GRLH2, Protects Livers of Mice and Patients From Ethanol-Induced Liver Disease. Gastroenterology 2018, 154, 238–252.e7. [Google Scholar] [CrossRef] [PubMed]
- Ambade, A.; Satishchandran, A.; Szabo, G. Alcoholic hepatitis accelerates early hepatobiliary cancer by increasing stemness and MIR-122-mediated HIF-1α activation. Sci. Rep. 2016, 6, 21340. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Duan, K.; Wang, C.; McClain, C.; Feng, W. Probiotics and alcoholic liver disease: Treatment and potential mechanisms. Gastroenterol. Res. Pract. 2016, 2016, 5491465. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Banan, A.; Forsyth, C.B.; Fields, J.Z.; Lau, C.K.; Zhang, L.J.; Keshavarzian, A. Effect of alcohol on miR-212 expression in intestinal epithelial cells and its potential role in alcoholic liver disease. Alcohol. Clin. Exp. Res. 2008, 32, 355–364. [Google Scholar] [CrossRef] [PubMed]
- Ng, R.; Song, G.; Roll, G.R.; Frandsen, N.M.; Willenbring, H. A microRNA-21 surge facilitates rapid cyclin D1 translation and cell cycle progression in mouse liver regeneration. J. Clin. Investig. 2012, 122, 1097–1108. [Google Scholar] [CrossRef] [PubMed]
- Dippold, R.P.; Vadigepalli, R.; Gonye, G.E.; Hoek, J.B. Chronic ethanol feeding enhances miR-21 induction during liver regeneration while inhibiting proliferation in rats. AJP Gastrointest. Liver Physiol. 2012, 303, G733–G743. [Google Scholar] [CrossRef] [PubMed]
- Francis, H.; McDaniel, K.; Han, Y.; Liu, X.; Kennedy, L.; Yang, F.; McCarra, J.; Zhou, T.; Glaser, S.; Venter, J.; et al. Regulation of the extrinsic apoptotic pathway by microRNA-21 in alcoholic liver injury. J. Biol. Chem. 2014, 289, 27526–27539. [Google Scholar] [CrossRef]
- Roderburg, C.; Urban, G.W.; Bettermann, K.; Vucur, M.; Zimmermann, H.; Schmidt, S.; Janssen, J.; Koppe, C.; Knolle, P.; Castoldi, M.; et al. Micro-RNA-profiling reveals a role for MIR-29 in human and murine liver fibrosis. Hepatology 2010, 53, 209–218. [Google Scholar] [CrossRef]
- Huang, G.H.; Shan, H.; Li, D.; Zhou, B.; Pang, P.F. MiR-199a-5p suppresses tumorigenesis by targeting clathrin heavy chain in hepatocellular carcinoma. Cell Biochem. Funct. 2017, 35, 98–104. [Google Scholar] [CrossRef]
- Tili, E.; Michaille, J.-J.; Cimino, A.; Costinean, S.; Dumitru, C.D.; Adair, B.; Fabbri, M.; Alder, H.; Liu, C.G.; Calin, G.A.; et al. Modulation of miR-155 and miR-125b Levels following Lipopolysaccharide/TNF-α Stimulation and Their Possible Roles in Regulating the Response to Endotoxin Shock. J. Immunol. 2007, 179, 5082–5089. [Google Scholar] [CrossRef]
- Alpini, G.; Glaser, S.S.; Zhang, J.P.; Francis, H.; Han, Y.; Gong, J.; Stokes, A.; Francis, T.; Hughart, N.; Hubble, L.; et al. Regulation of placenta growth factor by microRNA-125b in hepatocellular cancer. J. Hepatol. 2011, 55, 1339–1345. [Google Scholar] [CrossRef]
- Ladeiro, Y.; Couchy, G.; Balabaud, C.; Bioulac-Sage, P.; Pelletier, L.; Rebouissou, S.; Zucman-Rossi, J. MicroRNA profiling in hepatocellular tumors is associated with clinical features and oncogene/tumor suppressor gene mutations. Hepatology 2008, 47, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.J.; Pan, Q.; Xiong, H.; Qiao, Y.Q.; Bian, Z.L.; Zhong, W.; Sheng, L.; Li, H.; Shen, L.; Hua, J.; et al. Dynamic expression of miR-126*and its effects on proliferation and contraction of hepatic stellate cells. FEBS Lett. 2013, 587, 3792–3801. [Google Scholar] [CrossRef]
- Saikia, P.; Bellos, D.; McMullen, M.R.; Pollard, K.A.; de la Motte, C.; Nagy, L.E. MicroRNA 181b-3p and its target importin α5 regulate toll-like receptor 4 signaling in Kupffer cells and liver injury in mice in response to ethanol. Hepatology 2017, 66, 602–615. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Liang, X.; Jogasuria, A.; Davidson, N.O.; You, M. miR-217 regulates ethanol-induced hepatic inflammation by disrupting sirtuin 1–Lipin-1 signaling. Am. J. Pathol. 2015, 185, 1286–1296. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; He, Y.; Zhou, Z.; Ramirez, T.; Gao, Y.; Gao, Y.; Ross, R.A.; Cao, H.; Cai, Y.; Xu, M.; et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL6-p47phox-oxidative stress pathway in neutrophils. Gut 2017, 66, 705–715. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Zhang, J. Decreased expression of microRNA-223 promotes cell proliferation in hepatocellular carcinoma cells via the insulin-like growth factor-1 signaling pathway. Exp. Ther. Med. 2018, 15, 4325–4331. [Google Scholar] [CrossRef] [PubMed]
- Hartmann, P.; Tacke, F. Tiny RNA with great effects: MiR-155 in alcoholic liver disease. J. Hepatol. 2016, 64, 1214–1216. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Yang, Y.; Xu, C.; Peng, Z.; Zhang, M.; Lei, L.; Gao, W.; Dong, Y.; Shi, Z.; Sun, X.; et al. Upregulation of miR-181a impairs hepatic glucose and lipid homeostasis. Oncotarget 2017, 8, 91362–91378. [Google Scholar] [CrossRef] [PubMed]
- Raver-Shapira, N.; Marciano, E.; Meiri, E.; Spector, Y.; Rosenfeld, N.; Moskovits, N.; Bentwich, Z.; Oren, M. Transcriptional Activation of miR-34a Contributes to p53-Mediated Apoptosis. Mol. Cell 2007, 26, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zalzala, M.; Xu, J.; Li, Y.; Yin, L.; Zhang, Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015, 6, 7466. [Google Scholar] [CrossRef] [PubMed]
- Szabo, G.; Bala, S.; Petrasek, J.; Gattu, A. Gut-liver axis and sensing microbes. Dig. Dis. 2010, 28, 737–744. [Google Scholar] [CrossRef] [PubMed]
- Janssen, H.L.A.; Reesink, H.W.; Lawitz, E.J.; Zeuzem, S.; Rodriguez-Torres, M.; Patel, K.; van der Meer, A.J.; Patick, A.K.; Chen, A.; Zhou, Y.; et al. Treatment of HCV Infection by Targeting MicroRNA. N. Engl. J. Med. 2013, 368, 1685–1694. [Google Scholar] [CrossRef] [PubMed]
- Thursz, M.; Gual, A.; Lackner, C.; Mathurin, P.; Moreno, C.; Spahr, L.; Sterneck, M.; Cortez-Pinto, H. EASL Clinical Practice Guidelines: Management of alcohol-related liver disease. J. Hepatol. 2018, 69, 154–181. [Google Scholar] [CrossRef] [PubMed]
- Leggio, L.; Lee, M.R. Treatment of Alcohol Use Disorder in Patients with Alcoholic Liver Disease. Am. J. Med. 2017, 130, 124–134. [Google Scholar] [CrossRef] [PubMed]
- McPherson, S.; Lucey, M.R.; Moriarty, K.J. Decompensated alcohol related liver disease: Acute management. BMJ 2016, 352, i124. [Google Scholar] [CrossRef]
- Addolorato, G.; Bataller, R.; Burra, P.; DiMartini, A.; Graziadei, I.; Lucey, M.R.; Mathurin, P.; O’Grady, J.; Pageaux, G.; Berenguer, M. Liver transplantation for alcoholic liver disease. Transplantation 2016, 65, 618–630. [Google Scholar] [CrossRef]
- Addolorato, G.; Mirijello, A.; Barrio, P.; Gual, A. Treatment of alcohol use disorders in patients with alcoholic liver disease. J. Hepatol. 2016, 65, 618–630. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| miRNAs | Epigenetic Mechanism | Function | Experimental Model | ALD Severity | Ref. |
|---|---|---|---|---|---|
| miR-155 | Upregulated | Induced TNFα and PPARα and hampered PPARγ expression | Hepatocytes, macrophages, Et-OH fed mice | Steatosis, inflammation, oxidative stress, fibrosis, cirrhosis | [112,115,116,117] |
| miR-34a | Upregulated | Reduced SIRT1 gene | Et-OH fed mice and rats | Steatosis, NASH, Fibrosis | [118,119,120,121] |
| miR-122 | Downregulated | Induced genes involved in lipid metabolism, cholesterol homeostasis, cytokines and HIF1α. Reduced tight junction proteins | ALD patients, Et-OH fed mice | Steatosis, inflammation, altered gut permeability, fibrosis, cirrhosis | [112,114,122,123,124] |
| miR-212 | Upregulated | Dampened tight junction proteins | ALD patients | Altered gut permeability | [125] |
| miR-21 | Upregulated | Modulation of cell proliferation, apoptosis and survival | Hepatocytes and HSCs treated with alcohol Et-OH fed rats and mice, | Liver Injury and HCC | [126,127,128] |
| miR-29 | Downregulated | Modulation of hepatic inflammation and HSCs activation | ALD patients, CCl4-induced fibrosis in rodents | Liver Injury, altered gut permeability fibrosis | [129] |
| miR-199a | Downregulated | Regulation of cell cycle and apoptosis through HIF1α induction | ALD patients, HCC cell lines and AML-12, Et-OH fed mice | Inflammation, cirrhosis, HCC | [112,130] |
| miR-125b | Downregulated | Increased TNFα production | Et-OH fed mice, macrophages | Liver Injury, HCC | [131,132] |
| miR-126 | Downregulated | Activation of HSCs and induction of Et-OH related HCC | ALD patients, HSCs, CCl4-induced fibrosis in rodents | Fibrosis, HCC | [133,134] |
| miR-181a | Upregulated | Increased LPS sensitivity via TLR4/NF-kB | Et-OH fed mice | Steatosis, altered gut permeability, Inflammation | [135] |
| miR-200a | Downregulated | Induction of hepatocytic apoptosis | ALD patients, Et-OH fed mice | HCC | [113,133] |
| miR-217 | Upregulated | Inhibition of SIRT1 | Hepatocytes, macrophages, Et-OH fed mice | Steatosis, Inflammation | [136] |
| miR-375 | Downregulated | Regulation of HIF1α and association with beta-catenin genes mutations | ALD patients | Inflammation, HCC | [133] |
| miR-223 | Upregulated | Hepatocytic apoptosis, neutrophils hepatic infiltration | ALD patients, Et-OH fed mice, CCl4-induced acute liver injury | Liver injury, inflammation, HCC development | [137,138] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meroni, M.; Longo, M.; Rametta, R.; Dongiovanni, P. Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. Int. J. Mol. Sci. 2018, 19, 3857. https://doi.org/10.3390/ijms19123857
Meroni M, Longo M, Rametta R, Dongiovanni P. Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. International Journal of Molecular Sciences. 2018; 19(12):3857. https://doi.org/10.3390/ijms19123857
Chicago/Turabian StyleMeroni, Marica, Miriam Longo, Raffaela Rametta, and Paola Dongiovanni. 2018. "Genetic and Epigenetic Modifiers of Alcoholic Liver Disease" International Journal of Molecular Sciences 19, no. 12: 3857. https://doi.org/10.3390/ijms19123857
APA StyleMeroni, M., Longo, M., Rametta, R., & Dongiovanni, P. (2018). Genetic and Epigenetic Modifiers of Alcoholic Liver Disease. International Journal of Molecular Sciences, 19(12), 3857. https://doi.org/10.3390/ijms19123857