Translocation-Related Sarcomas


Abstract

1. Introduction
1.1. What Is a Chromosomal Translocation?
1.2. Chromosomal Translocations in Soft Tissue Sarcoma
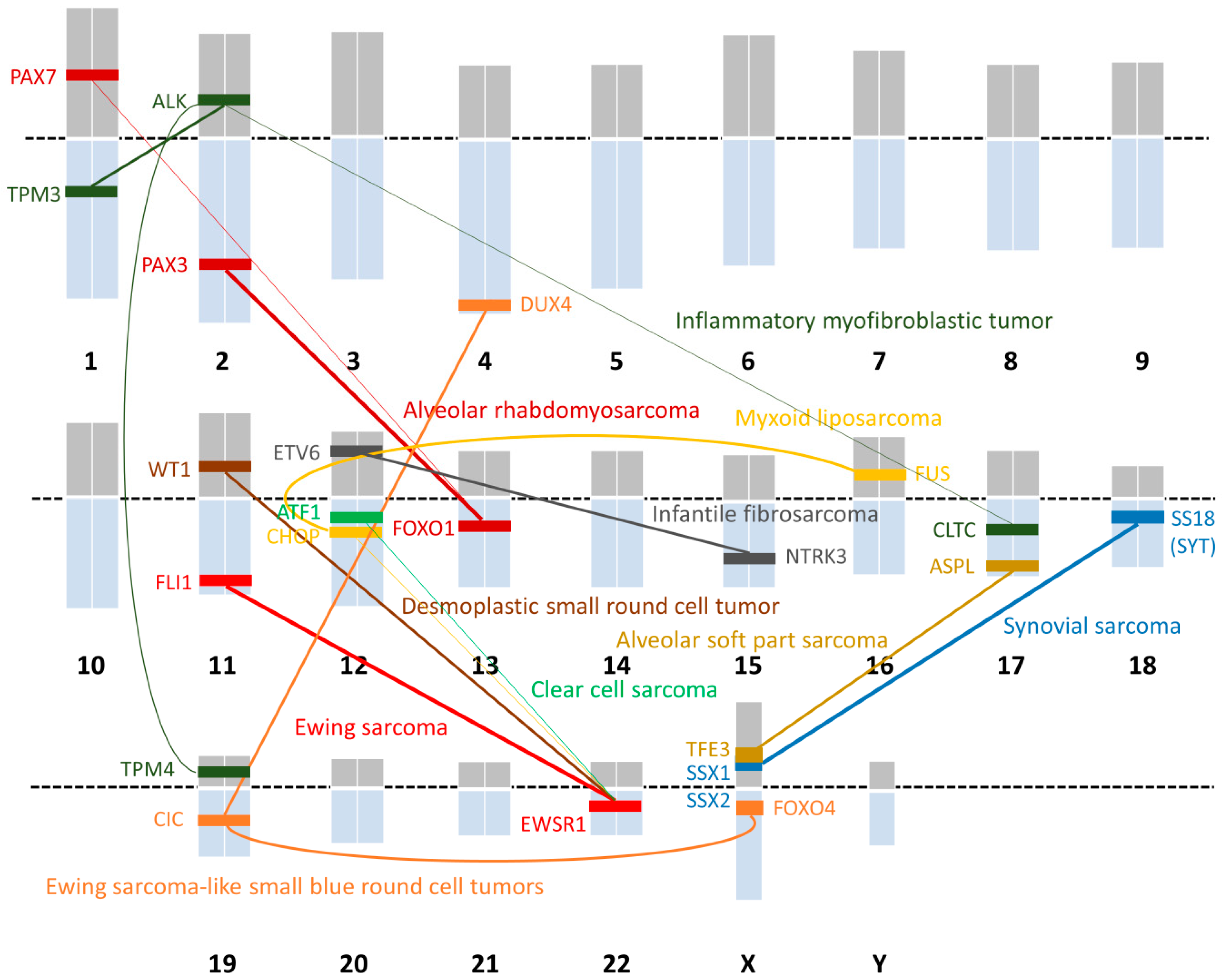
2. Subtypes of Translocation-Related Sarcomas: Clinical Significance
2.1. The Ewing Sarcoma Family of Tumors
2.1.1. Ewing Sarcoma
2.1.2. Ewing Sarcoma-like Small Blue Round Cell Tumors
2.1.3. Desmoplastic Small Round Cell Tumors (DSRCTs)
2.2. Alveolar Rhabdomyosarcomas
2.3. Alveolar Soft Part Sarcoma (ASPS)
2.4. Synovial Sarcoma
2.5. Myxoid Liposarcoma
2.6. Clear Cell Sarcoma (CCS)
2.7. Inflammatory Myofibroblastic Tumor (IMT)
2.8. Infantile Fibrosarcoma
2.9. Other TRS
3. Translocation Analysis Technology: The Detection and Induction of Fusion Genes
Author Contributions
Conflicts of Interest
Abbreviations
| ALK | anaplastic lymphoma kinase |
| ASPS | alveolar soft part sarcoma |
| BRCA | breast cancer susceptibility gene |
| BRD4 | bromodomain-containing protein 4 |
| CCS | clear cell sarcoma |
| CML | chronic myeloid leukemia |
| CTLA-4 | cytotoxic T-lymphocyte-associated protein 4 |
| FISH | fluorescence in situ hybridization |
| IGF1R | insulin-like growth factor-1 receptor |
| IMT | inflammatory myofibroblastic tumor |
| NGS | Next-generation sequencing |
| NSAID | non-steroidal anti-inflammatory drug |
| PARP | poly-ADP-ribose-polymerase |
| PCR | polymerase chain reaction |
| PD-1 | programmed death-1 |
| PD-L1 | programmed death-ligand 1 |
| PI3K | phosphatidylinositol 3-kinase |
| STS | soft tissue sarcoma |
| TFE3 | Transcription factor E3 |
| TRK | Tropomyosin receptor kinase |
| TRS | translocation-related sarcoma |
References
- Rowley, J.D.; Le Beau, M.M.; Rabbitts, T.H. Chromosomal Translocations and Genome Rearrangements in Cancer; Springer: Cham, Switzerland, 2015; pp. 3–14. ISBN 978-3-319-19982-5. [Google Scholar]
- Schiffer, C.A. BCR-ABL tyrosine kinase inhibitors for chronic myelogenous leukemia. N. Engl. J. Med. 2007, 357, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Solomon, B.J.; Mok, T.; Kim, D.W.; Wu, Y.L.; Nakagawa, K.; Mekhail, T.; Felip, E.; Cappuzzo, F.; Paolini, J.; Usari, T.; et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N. Engl. J. Med. 2014, 371, 2167–2177. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, T.M.; Crinò, L.; Gridelli, C.; Kiura, K.; Liu, G.; Novello, S.; Bearz, A.; Gautschi, O.; Mok, T.; et al. Ceritinib versus chemotherapy in patients with ALK-rearranged non-small-cell lung cancer previously given chemotherapy and crizotinib (ASCEND-5): A randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2017, 18, 874–886. [Google Scholar] [CrossRef]
- Peters, S.; Camidge, D.R.; Shaw, A.T.; Gadgeel, S.; Ahn, J.S.; Kim, D.W.; Ou, S.I.; Pérol, M.; Dziadziuszko, R.; Rosell, R.; et al. Alectinib versus crizotinib in untreated ALK-positive non-small-cell lung cancer. N. Engl. J. Med. 2017, 377, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, C.D.M.; Bridge, J.A.; Hogendoorn, P.C.W.; Mertens, F. WHO Classification of Tumours of Soft Tissue and Bone, 4th ed.; International Agency Research on Cancer: Lyon, France, 2013; ISBN 978-92-832-2434-1. [Google Scholar]
- Le Cesne, A.; Cresta, S.; Maki, R.G.; Blay, J.Y.; Verweij, J.; Poveda, A.; Casali, P.G.; Balaña, C.; Schöffski, P.; Grosso, F.; et al. A retrospective analysis of antitumour activity with trabectedin in translocation-related sarcomas. Eur. J. Cancer 2012, 48, 3036–3044. [Google Scholar] [CrossRef] [PubMed]
- Blay, J.Y.; Leahy, M.G.; Nguyen, B.B.; Patel, S.R.; Hohenberger, P.; Santoro, A.; Staddon, A.P.; Penel, N.; Piperno-Neumann, S.; Hendifar, A.; et al. Randomised phase III trial of trabectedin versus doxorubicin-based chemotherapy as first-line therapy in translocation-related sarcomas. Eur. J. Cancer 2014, 50, 1137–1147. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Araki, N.; Sugiura, H.; Ueda, T.; Yonemoto, T.; Takahashi, M.; Morioka, H.; Hiraga, H.; Hiruma, T.; Kunisada, T.; et al. Trabectedin monotherapy after standard chemotherapy versus best supportive care in patients with advanced, translocation-related sarcoma: A randomised, open-label, phase 2 study. Lancet Oncol. 2015, 16, 406–416. [Google Scholar] [CrossRef]
- Takebayashi, Y.; Pourquier, P.; Zimonjic, D.B.; Nakayama, K.; Emmert, S.; Ueda, T.; Urasaki, Y.; Kanzaki, A.; Akiyama, S.I.; Popescu, N.; et al. Antiproliferative activity of ecteinascidin 743 is dependent upon transcription-coupled nucleotide-excision repair. Nat. Med. 2001, 7, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Taron, M.; Jimeno, J.; Grosso, F.; Sanfilipio, R.; Casali, P.G.; Le Cesne, A.; Jones, R.L.; Blay, J.Y.; Poveda, A.; et al. Predictive impact of DNA repair functionality on clinical outcome of advanced sarcoma patients treated with trabectedin: A retrospective multicentric study. Eur. J. Cancer 2011, 47, 1006–1012. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Laurand, A.; Laroche, A.; Casali, P.; Sanfilippo, R.; Le Cesne, A.; Judson, I.; Blay, J.Y.; Ray-Coquard, I.; Bui, B.; et al. ERCC5/XPG, ERCC1, and BRCA1 gene status and clinical benefit of trabectedin in patients with soft tissue sarcoma. Cancer 2011, 117, 3445–3456. [Google Scholar] [CrossRef] [PubMed]
- Laroche-Clary, A.; Chaire, V.; Le Morvan, V.; Neuville, A.; Bertucci, F.; Salas, S.; Sanfilippo, R.; Pourquier, P.; Italiano, A. BRCA1 haplotype and clinical benefit of trabectedin in soft-tissue sarcoma patients. Br. J. Cancer 2015, 112, 688–692. [Google Scholar] [CrossRef] [PubMed]
- Choy, E.; Butrynski, J.E.; Harmon, D.C.; Morgan, J.A.; George, S.; Wagner, A.J.; D’Adamo, D.; Cote, G.M.; Flamand, Y.; Benes, C.H.; et al. Phase II study of olaparib in patients with refractory Ewing sarcoma following failure of standard chemotherapy. BMC Cancer 2014, 14, 813. [Google Scholar] [CrossRef] [PubMed]
- Grignani, G.; D’Ambrosio, L.; Pignochino, Y.; Palmerini, E.; Zucchetti, M.; Boccone, P.; Aliberti, S.; Stacchiotti, S.; Bertulli, R.; Piana, R.; Miano, S.; et al. Trabectedin and olaparib in patients with advanced and non-resectable bone and soft-tissue sarcomas (TOMAS): An open-label, phase 1b study from the Italian Sarcoma Group. Lancet Oncol. 2018, 19, 1360–1371. [Google Scholar] [CrossRef]
- D’Incalci, M.; Badri, N.; Galmarini, C.M.; Allavena, P. Trabectedin, a drug acting on both cancer cells and the tumour microenvironment. Br. J. Cancer 2014, 111, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current management and future approaches through collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [PubMed]
- Zoubek, A.; Dockhorn-Dworniczak, B.; Delattre, O.; Christiansen, H.; Niggli, F.; Gatterer-Menz, I.; Smith, T.L.; Jürgens, H.; Gadner, H.; Kovar, H. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing tumor patients? J. Clin. Oncol. 1996, 14, 1245–1251. [Google Scholar] [CrossRef] [PubMed]
- De Alava, E.; Kawai, A.; Healey, J.H.; Fligman, I.; Meyers, P.A.; Huvos, A.G.; Gerald, W.L.; Jhanwar, S.C.; Argani, P.; Antonescu, C.R.; et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. J. Clin. Oncol. 1998, 16, 1248–1255. [Google Scholar] [CrossRef] [PubMed]
- Le Deley, M.C.; Delattre, O.; Schaefer, K.L.; Burchill, S.A.; Koehler, G.; Hogendoorn, P.C.; Lion, T.; Poremba, C.; Marandet, J.; Ballet, S.; et al. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: Prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J. Clin. Oncol. 2010, 28, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Van Maldegem, A.M.; Bovée, J.V.; Peterse, E.F.; Hogendoorn, P.C.; Gelderblom, H. Ewing sarcoma: The clinical relevance of the insulin-like growth factor 1 and the poly-ADP-ribose-polymerase pathway. Eur. J. Cancer 2016, 53, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Kinsey, M.; Smith, R.; Iyer, A.K.; McCabe, E.R.; Lessnick, S.L. EWS/FLI and its downstream target NR0B1 interact directly to modulate transcription and oncogenesis in Ewing’s sarcoma. Cancer Res. 2009, 69, 9047–9055. [Google Scholar] [CrossRef] [PubMed]
- Grier, H.E.; Krailo, M.D.; Tarbell, N.J.; Link, M.P.; Fryer, C.J.; Pritchard, D.J.; Gebhardt, M.C.; Dickman, P.S.; Perlman, E.J.; Meyers, P.A.; et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N. Engl. J. Med. 2003, 348, 694–701. [Google Scholar] [CrossRef] [PubMed]
- Granowetter, L.; Womer, R.; Devidas, M.; Krailo, M.; Wang, C.; Bernstein, M.; Marina, N.; Leavey, P.; Gebhardt, M.; Healey, J.; et al. Dose-intensified compared with standard chemotherapy for nonmetastatic Ewing sarcoma family of tumors: A Children’s Oncology Group Study. J. Clin. Oncol. 2009, 27, 2536–2541. [Google Scholar] [CrossRef] [PubMed]
- Womer, R.B.; West, D.C.; Krailo, M.D.; Dickman, P.S.; Pawel, B.R.; Grier, H.E.; Marcus, K.; Sailer, S.; Healey, J.H.; Dormans, J.P.; et al. Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2012, 30, 4148–4154. [Google Scholar] [CrossRef] [PubMed]
- Whelan, J.; Le Deley, M.C.; Dirksen, U.; Le Teuff, G.; Brennan, B.; Gaspar, N.; Hawkins, D.S.; Amler, S.; Bauer, S.; Bielack, S.; et al. High-dose chemotherapy and blood autologous stem-cell rescue compared with standard chemotherapy in localized high-risk Ewing sarcoma: Results of Euro-E.W.I.N.G.99 and Ewing-2008. J. Clin. Oncol. 2018, 36, 3110. [Google Scholar] [CrossRef] [PubMed]
- Grohar, P.J.; Griffin, L.B.; Yeung, C.; Chen, Q.R.; Pommier, Y.; Khanna, C.; Khan, J.; Helman, L.J. Ecteinascidin 743 interferes with the activity of EWS-FLI1 in Ewing sarcoma cells. Neoplasia 2011, 13, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Ueda, T.; Kakunaga, S.; Ando, M.; Yonemori, K.; Sugiura, H.; Yamada, K.; Kawai, A. Phase I and pharmacokinetic study of trabectedin, a DNA minor groove binder, administered as a 24-h continuous infusion in Japanese patients with soft tissue sarcoma. Investig. New Drugs 2014, 32, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Olmos, D.; Postel-Vinay, S.; Molife, L.R.; Okuno, S.H.; Schuetze, S.M.; Paccagnella, M.L.; Batzel, G.N.; Yin, D.; Pritchard-Jones, K.; Judson, I.; et al. Safety, pharmacokinetics, and preliminary activity of the anti-IGF-1R antibody figitumumab (CP-751,871) in patients with sarcoma and Ewing’s sarcoma: A phase 1 expansion cohort study. Lancet Oncol. 2010, 11, 129–135. [Google Scholar] [CrossRef]
- Juergens, H.; Daw, N.C.; Geoerger, B.; Ferrari, S.; Villarroel, M.; Aerts, I.; Whelan, J.; Dirksen, U.; Hixon, M.L.; Yin, D.; et al. Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J. Clin. Oncol. 2011, 29, 4534–4540. [Google Scholar] [CrossRef] [PubMed]
- Langer, C.J.; Novello, S.; Park, K.; Krzakowski, M.; Karp, D.D.; Mok, T.; Benner, R.J.; Scranton, J.R.; Olszanski, A.J.; Jassem, J. Randomized, phase III trial of first-line figitumumab in combination with paclitaxel and carboplatin versus paclitaxel and carboplatin alone in patients with advanced non-small-cell lung cancer. J. Clin. Oncol. 2014, 32, 2059–2066. [Google Scholar] [CrossRef] [PubMed]
- Tap, W.D.; Demetri, G.; Barnette, P.; Desai, J.; Kavan, P.; Tozer, R.; Benedetto, P.W.; Friberg, G.; Deng, H.; McCaffery, I.; et al. Phase II study of ganitumab, a fully human anti-type-1 insulin-like growth factor receptor antibody, in patients with metastatic Ewing family tumors or desmoplastic small round cell tumors. J. Clin. Oncol. 2012, 30, 1849–1856. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, C.S.; Azevedo, S.; Okusaka, T.; Van Laethem, J.L.; Lipton, L.R.; Riess, H.; Szczylik, C.; Moore, M.J.; Peeters, M.; Bodoky, G.; et al. A phase 3 randomized, double-blind, placebo-controlled trial of ganitumab or placebo in combination with gemcitabine as first-line therapy for metastatic adenocarcinoma of the pancreas: The GAMMA trial. Ann. Oncol. 2015, 26, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Single Patient Expanded Access to Ganitumab for Metastatic Ewing Sarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03029481 (accessed on 24 January 2017).
- Insulin-Like Growth Factor 1 Receptor (IGF-1R) Antibody AMG479 (Ganitumab) in Combination with the SRC Family Kinase (SFK) Inhibitor Dasatinib in People with Embryonal and Alveolar Rhabdomyosarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT03041701 (accessed on 3 February 2017).
- Combination Chemotherapy with or without Ganitumab in Treating Patients with Newly Diagnosed Metastatic Ewing Sarcoma. Available online: https://clinicaltrials.gov/ct2/show/NCT02306161 (accessed on 3 December 2014).
- Sugita, S.; Arai, Y.; Tonooka, A.; Hama, N.; Totoki, Y.; Fujii, Y.; Aoyama, T.; Asanuma, H.; Tsukahara, T.; Kaya, M.; et al. A novel CIC-FOXO4 gene fusion in undifferentiated small round cell sarcoma: A genetically distinct variant of Ewing-like sarcoma. Am. J. Surg. Pathol. 2014, 38, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Sung, Y.S.; Zhang, L.; Singer, S.; Maki, R.G.; Coindre, J.M.; Antonescu, C.R. High prevalence of CIC fusion with double-homeobox (DUX4) transcription factors in EWSR1-negative undifferentiated small blue round cell sarcomas. Genes Chromosomes Cancer 2012, 51, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Stiles, Z.E.; Dickson, P.V.; Glazer, E.S.; Murphy, A.J.; Davidoff, A.M.; Behrman, S.W.; Bishop, M.W.; Martin, M.G.; Deneve, J.L. Desmoplastic small round cell tumor: A nationwide study of a rare sarcoma. J. Surg. Oncol. 2018, 117, 1759–1767. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Lamhamedi-Cherradi, S.E.; Cuglievan, B.; Menegaz, B.A.; Camacho, P.; Huh, W.; Ramamoorthy, V.; Anderson, P.M.; Pollock, R.E.; Lev, D.C.; et al. Multimodality treatment of desmoplastic small round cell tumor: Chemotherapy and complete cytoreductive surgery improve patient survival. Clin. Cancer Res. 2018, 24, 4865–4873. [Google Scholar] [CrossRef] [PubMed]
- Menegaz, B.A.; Cuglievan, B.; Benson, J.; Camacho, P.; Lamhamedi-Cherradi, S.E.; Leung, C.H.; Warneke, C.L.; Huh, W.; Subbiah, V.; Benjamin, R.S.; et al. Clinical activity of pazopanib in patients with advanced desmoplastic small round cell tumor. Oncologist 2018, 23, 360–366. [Google Scholar] [CrossRef] [PubMed]
- De Brachène, A.C.; Demoulin, J.B. FOXO transcription factors in cancer development and therapy. Cell. Mol. Life Sci. 2016, 73, 1159–1172. [Google Scholar] [CrossRef] [PubMed]
- Gallego, S.; Zanetti, I.; Orbach, D.; Ranchère, D.; Shipley, J.; Zin, A.; Bergeron, C.; de Salvo, G.L.; Chisholm, J.; Ferrari, A.; et al. Fusion status in patients with lymph node-positive (N1) alveolar rhabdomyosarcoma is a powerful predictor of prognosis: Experience of the European Paediatric Soft Tissue Sarcoma Study Group (EpSSG). Cancer 2018, 124, 3201–3209. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.; Gordon, T.; McManus, A.; Mapp, T.; Gould, S.; Kelsey, A.; McDowell, H.; Pinkerton, R.; Shipley, J.; Pritchard-Jones, K. UK Children’s Cancer Study Group (UKCCSG) and the UK Cancer Cytogenetics Group. Detection of the PAX3-FKHR fusion gene in paediatric rhabdomyosarcoma: A reproducible predictor of outcome? Br. J. Cancer 2001, 85, 831–835. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, P.H.; Lynch, J.C.; Qualman, S.J.; Tirabosco, R.; Lim, J.F.; Maurer, H.M.; Bridge, J.A.; Crist, W.M.; Triche, T.J.; Barr, F.G. PAX3-FKHR and PAX7-FKHR gene fusions are prognostic indicators in alveolar rhabdomyosarcoma: A report from the children’s oncology group. J. Clin. Oncol. 2002, 20, 2672–2679. [Google Scholar] [CrossRef] [PubMed]
- Missiaglia, E.; Williamson, D.; Chisholm, J.; Wirapati, P.; Pierron, G.; Petel, F.; Concordet, J.P.; Thway, K.; Oberlin, O.; Pritchard-Jones, K.; et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J. Clin. Oncol. 2012, 30, 1670–1677. [Google Scholar] [CrossRef] [PubMed]
- Raney, R.B.; Maurer, H.M.; Anderson, J.R.; Andrassy, R.J.; Donaldson, S.S.; Qualman, S.J.; Wharam, M.D.; Wiener, E.S.; Crist, W.M. The Intergroup Rhabdomyosarcoma Study Group (IRSG): Major lessons from the IRS-I through IRS-IV studies as background for the current IRS-V treatment protocols. Sarcoma 2001, 5, 9–15. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, G.; Jenney, M.; Bergeron, C.; Gallego Melcón, S.; Ferrari, A.; Oberlin, O.; Carli, M.; Stevens, M.; Kelsey, A.; De Paoli, A.; et al. Addition of dose-intensified doxorubicin to standard chemotherapy for rhabdomyosarcoma (EpSSG RMS 2005): A multicentre, open-label, randomised controlled, phase 3 trial. Lancet Oncol. 2018, 19, 1061–1071. [Google Scholar] [CrossRef]
- Walterhouse, D.O.; Pappo, A.S.; Meza, J.L.; Breneman, J.C.; Hayes-Jordan, A.A.; Parham, D.M.; Cripe, T.P.; Anderson, J.R.; Meyer, W.H.; Hawkins, D.S. Shorter-duration therapy using vincristine, dactinomycin, and lower-dose cyclophosphamide with or without radiotherapy for patients with newly diagnosed low-risk rhabdomyosarcoma: A report from the Soft Tissue Sarcoma Committee of the Children’s Oncology Group. J. Clin. Oncol. 2014, 32, 3547–3552. [Google Scholar] [CrossRef] [PubMed]
- Sultan, I.; Qaddoumi, I.; Yaser, S.; Rodriguez-Galindo, C.; Ferrari, A. Comparing adult and pediatric rhabdomyosarcoma in the surveillance, epidemiology and end results program, 1973 to 2005: An analysis of 2600 patients. J. Clin. Oncol. 2009, 27, 3391–3397. [Google Scholar] [CrossRef] [PubMed]
- Weigel, B.J.; Lyden, E.; Anderson, J.R.; Meyer, W.H.; Parham, D.M.; Rodeberg, D.A.; Michalski, J.M.; Hawkins, D.S.; Arndt, C.A. Intensive multiagent therapy, including dose-compressed cycles of ifosfamide/etoposide and vincristine/doxorubicin/cyclophosphamide, irinotecan, and radiation, in patients with high-risk rhabdomyosarcoma: A report from the Children’s Oncology Group. J. Clin. Oncol. 2016, 34, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Yohe, M.E.; Chou, H.C.; Zhang, X.; Marques, J.; Wachtel, M.; Schaefer, B.; Sen, N.; Song, Y.; Gualtieri, A.; et al. PAX3-FOXO1 establishes myogenic super enhancers and confers BET bromodomain vulnerability. Cancer Discov. 2017, 7, 884–899. [Google Scholar] [CrossRef] [PubMed]
- Bid, H.K.; Phelps, D.A.; Xaio, L.; Guttridge, D.C.; Lin, J.; London, C.; Baker, L.H.; Mo, X.; Houghton, P.J. The bromodomain BET inhibitor JQ1 suppresses tumor angiogenesis in models of childhood sarcoma. Mol. Cancer Ther. 2016, 15, 1018–1028. [Google Scholar] [CrossRef] [PubMed]
- Berthon, C.; Raffoux, E.; Thomas, X.; Vey, N.; Gomez-Roca, C.; Yee, K.; Taussig, D.C.; Rezai, K.; Roumier, C.; Herait, P.; et al. Bromodomain inhibitor OTX015 in patients with acute leukaemia: A dose-escalation, phase 1 study. Lancet Haematol. 2016, 3, e186–e195. [Google Scholar] [CrossRef]
- Lacey, A.; Hedrick, E.; Cheng, Y.; Mohankumar, K.; Warren, M.; Safe, S. Interleukin-24 (IL-24) is suppressed by PAX3-FOXO1 and is a novel therapy for rhabdomyosarcoma. Mol. Cancer Ther. 2018. [Google Scholar] [CrossRef] [PubMed]
- Van Gaal, J.C.; Flucke, U.E.; Roeffen, M.H.; de Bont, E.S.; Sleijfer, S.; Mavinkurve-Groothuis, A.M.; Suurmeijer, A.J.; van der Graaf, W.T.; Versleijen-Jonkers, Y.M. Anaplastic lymphoma kinase aberrations in rhabdomyosarcoma: Clinical and prognostic implications. J. Clin. Oncol. 2012, 30, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Van Erp, A.E.M.; Hillebrandt-Roeffen, M.H.S.; van Houdt, L.; Fleuren, E.D.G.; van der Graaf, W.T.A.; Versleijen-Jonkers, Y.M.H. Targeting anaplastic lymphoma kinase (ALK) in rhabdomyosarcoma (RMS) with the second-generation ALK inhibitor ceritinib. Target. Oncol. 2017, 12, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Leahy, M.G.; Aamdal, S.; Rutkowski, P.; Bauer, S.; Richter, S.; Grünwald, V.; Debiec-Rychter, M.; Sciot, R.; et al. The tyrosine kinase inhibitor crizotinib does not have clinically meaningful activity in heavily pre-treated patients with advanced alveolar rhabdomyosarcoma with FOXO rearrangement: European Organisation for Research and Treatment of Cancer phase 2 trial 90101 ‘CREATE’. Eur. J. Cancer 2018, 94, 156–167. [Google Scholar] [CrossRef] [PubMed]
- Argani, P.; Antonescu, C.R.; Illei, P.B.; Lui, M.Y.; Timmons, C.F.; Newbury, R.; Reuter, V.E.; Garvin, A.J.; Perez-Atayde, A.R.; Fletcher, J.A.; et al. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: A distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am. J. Pathol. 2001, 159, 179–192. [Google Scholar] [CrossRef]
- Kuroda, N.; Mikami, S.; Pan, C.C.; Cohen, R.J.; Hes, O.; Michal, M.; Nagashima, Y.; Tanaka, Y.; Inoue, K.; Shuin, T.; et al. Review of renal carcinoma associated with Xp11.2 translocations/TFE3 gene fusions with focus on pathobiological aspect. Histol. Histopathol. 2012, 27, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Lui, M.Y.; Antonescu, C.R.; Krause-Boehm, A.; Meindl, A.; Argani, P.; Healey, J.H.; Ueda, T.; Yoshikawa, H.; Meloni-Ehrig, A.; et al. The der(17)t(X;17)(p11;q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene 2001, 20, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Williams, A.; Bartle, G.; Sumathi, V.P.; Meis, J.M.; Mangham, D.C.; Grimer, R.J.; Kindblom, L.G. Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: Useful diagnostic tools in cases with unusual histological features. Virchows Arch. 2011, 458, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.H.; Brennan, M.F.; Kimmel, M.; Erlandson, R.A.; Garin-Chesa, P.; Flehinger, B.Y. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer 1989, 63, 1–13. [Google Scholar] [CrossRef]
- Sparber-Sauer, M.; Seitz, G.; von Kalle, T.; Vokuhl, C.; Scheer, M.; Münter, M.; Bielack, S.S.; Kazanowska, B.; Ladenstein, R.; Niggli, F.; et al. Alveolar soft-part sarcoma: Primary metastatic disease and metastatic relapse occurring during long-term follow-up: Treatment results of four Cooperative Weichteilsarkom Studiengruppe (CWS) trials and one registry. Pediatr. Blood Cancer 2018, e27405. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Homme, M.; Yamazaki, Y.; Shimizu, R.; Takazawa, Y.; Nakamura, T. Modeling alveolar soft part sarcoma unveils novel mechanisms of metastasis. Cancer Res. 2017, 77, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Pink, D.; Bertz-Lepel, J.; Busemann, C.; Bitz, U.; Reichardt, P. Efficacy of trabectedin in patients with advanced or metastatic alveolar soft-part sarcoma. Onkologie 2012, 35, 249–252. [Google Scholar] [CrossRef] [PubMed]
- Kummar, S.; Allen, D.; Monks, A.; Polley, E.C.; Hose, C.D.; Ivy, S.P.; Turkbey, I.B.; Lawrence, S.; Kinders, R.J.; Choyke, P.; et al. Cediranib for metastatic alveolar soft part sarcoma. J. Clin. Oncol. 2013, 31, 296–302. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Tamborini, E.; Marrari, A.; Brich, S.; Rota, S.A.; Orsenigo, M.; Crippa, F.; Morosi, C.; Gronchi, A.; Pierotti, M.A.; et al. Response to sunitinib malate in advanced alveolar soft part sarcoma. Clin. Cancer Res. 2009, 15, 1096–1104. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Negri, T.; Zaffaroni, N.; Palassini, E.; Morosi, C.; Brich, S.; Conca, E.; Bozzi, F.; Cassinelli, G.; Gronchi, A.; et al. Sunitinib in advanced alveolar soft part sarcoma: Evidence of a direct antitumor effect. Ann. Oncol. 2011, 22, 1682–1690. [Google Scholar] [CrossRef] [PubMed]
- Schöffski, P.; Wozniak, A.; Kasper, B.; Aamdal, S.; Leahy, M.G.; Rutkowski, P.; Bauer, S.; Gelderblom, H.; Italiano, A.; Lindner, L.H.; et al. Activity and safety of crizotinib in patients with alveolar soft part sarcoma with rearrangement of TFE3: European Organization for Research and Treatment of Cancer (EORTC) phase II trial 90101 ‘CREATE’. Ann. Oncol. 2018, 29, 758–765. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Mir, O.; Le Cesne, A.; Vincenzi, B.; Fedenko, A.; Maki, R.G.; Somaiah, N.; Patel, S.; Brahmi, M.; Blay, J.Y.; et al. Activity of pazopanib and trabectedin in advanced alveolar soft part sarcoma. Oncologist 2018, 23, 62–70. [Google Scholar] [CrossRef] [PubMed]
- Lewin, J.; Davidson, S.; Anderson, N.D.; Lau, B.Y.; Kelly, J.; Tabori, U.; Salah, S.; Butler, M.O.; Aung, K.L.; Shlien, A.; et al. Response to immune checkpoint inhibition in two patients with alveolar soft-part sarcoma. Cancer Immunol. Res. 2018, 6, 1001–1007. [Google Scholar] [CrossRef] [PubMed]
- Mouw, K.W.; D’Andrea, A.D. DNA Repair deficiency and immunotherapy response. J. Clin. Oncol. 2018, 36, 1710–1713. [Google Scholar] [CrossRef] [PubMed]
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 blockade in tumors with mismatch-repair deficiency. N. Engl. J. Med. 2015, 372, 2509–2520. [Google Scholar] [CrossRef] [PubMed]
- Stacchiotti, S.; Van Tine, B.A. Synovial sarcoma: Current concepts and future perspectives. J. Clin. Oncol. 2018, 36, 180–187. [Google Scholar] [CrossRef] [PubMed]
- Kawai, A.; Woodruff, J.; Healey, J.H.; Brennan, M.F.; Antonescu, C.R.; Ladanyi, M. SYT-SSX gene fusion as a determinant of morphology and prognosis in synovial sarcoma. N. Engl. J. Med. 1998, 338, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ladanyi, M.; Antonescu, C.R.; Leung, D.H.; Woodruff, J.M.; Kawai, A.; Healey, J.H.; Brennan, M.F.; Bridge, J.A.; Neff, J.R.; Barr, F.G.; et al. Impact of SYT-SSX fusion type on the clinical behavior of synovial sarcoma: A multi-institutional retrospective study of 243 patients. Cancer Res. 2002, 62, 135–140. [Google Scholar] [PubMed]
- Stegmaier, S.; Leuschner, I.; Poremba, C.; Ladenstein, R.; Kazanowska, B.; Ljungman, G.; Scheer, M.; Blank, B.; Bielack, S.; Klingebiel, T.; et al. The prognostic impact of SYT-SSX fusion type and histological grade in pediatric patients with synovial sarcoma treated according to the CWS (Cooperative Weichteilsarkom Studie) trials. Pediatr. Blood Cancer 2017, 64, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, G.; Sambri, A.; Righi, A.; Dei Tos, A.P.; Picci, P.; Donati, D. Histology and grading are important prognostic factors in synovial sarcoma. Eur. J. Surg. Oncol. 2017, 43, 1733–1739. [Google Scholar] [CrossRef] [PubMed]
- Sleijfer, S.; Ouali, M.; van Glabbeke, M.; Krarup-Hansen, A.; Rodenhuis, S.; Le Cesne, A.; Hogendoorn, P.C.; Verweij, J.; Blay, J.Y. Prognostic and predictive factors for outcome to first-line ifosfamide-containing chemotherapy for adult patients with advanced soft tissue sarcomas: An exploratory, retrospective analysis on large series from the European Organization for Research and Treatment of Cancer-Soft Tissue and Bone Sarcoma Group (EORTC-STBSG). Eur. J. Cancer 2010, 46, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Sleijfer, S.; Ray-Coquard, I.; Papai, Z.; Le Cesne, A.; Scurr, M.; Schöffski, P.; Collin, F.; Pandite, L.; Marreaud, S.; De Brauwer, A.; et al. Pazopanib, a multikinase angiogenesis inhibitor, in patients with relapsed or refractory advanced soft tissue sarcoma: A phase II study from the European Organisation for Research and Treatment of Cancer-soft Tissue and Bone Sarcoma group (EORTC study 62043). J. Clin. Oncol. 2009, 27, 3126–3132. [Google Scholar] [CrossRef] [PubMed]
- Kawano, S.; Grassian, A.R.; Tsuda, M.; Knutson, S.K.; Warholic, N.M.; Kuznetsov, G.; Xu, S.; Xiao, Y.; Pollock, R.M.; Smith, J.S.; et al. Preclinical evidence of anti-tumor activity induced by EZH2 inhibition in human models of synovial sarcoma. PLoS ONE 2016, 11, e0158888. [Google Scholar] [CrossRef] [PubMed]
- Italiano, A.; Soria, J.C.; Toulmonde, M.; Michot, J.M.; Lucchesi, C.; Varga, A.; Coindre, J.M.; Blakemore, S.J.; Clawson, A.; Suttle, B.; et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: A first-in-human, open-label, phase 1 study. Lancet Oncol. 2018, 19, 649–659. [Google Scholar] [CrossRef]
- Schoffski, P.; Agulnik, M.; Stacchiotti, S.; Davis, L.E.; Villalobos, V.M.; Italiano, A.; George, S.; Cote, G.M.; Blakemore, S.; Clawson, A.; et al. Phase 2 multicenter study of the EZH2 inhibitor tazemetostat in adults with synovial sarcoma (NCT02601950). J. Clin. Oncol. 2017, 35, 11057. [Google Scholar] [CrossRef]
- Italiano, A. Role of the EZH2 histone methyltransferase as a therapeutic target in cancer. Pharmacol. Ther. 2016, 165, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Merchant, M.S.; Wright, M.; Baird, K.; Wexler, L.H.; Rodriguez-Galindo, C.; Bernstein, D.; Delbrook, C.; Lodish, M.; Bishop, R.; Wolchok, J.D.; et al. Phase I clinical trial of ipilimumab in pediatric patients with advanced solid tumors. Clin. Cancer Res. 2016, 22, 1364–1370. [Google Scholar] [CrossRef] [PubMed]
- Tawbi, H.A.; Burgess, M.; Bolejack, V.; Van Tine, B.A.; Schuetze, S.M.; Hu, J.; D’Angelo, S.; Attia, S.; Riedel, R.F.; Priebat, D.A.; et al. Pembrolizumab in advanced soft-tissue sarcoma and bone sarcoma (SARC028): A multicentre, two-cohort, single-arm, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1493–1501. [Google Scholar] [CrossRef]
- D’Angelo, S.P.; Mahoney, M.R.; Van Tine, B.A.; Atkins, J.; Milhem, M.M.; Jahagirdar, B.N.; Antonescu, C.R.; Horvath, E.; Tap, W.D.; Schwartz, G.K.; et al. Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): Two open-label, non-comparative, randomised, phase 2 trials. Lancet Oncol. 2018, 19, 416–426. [Google Scholar] [CrossRef]
- Dallos, M.; Tap, W.D.; D’Angelo, S.P. Current status of engineered T-cell therapy for synovial sarcoma. Immunotherapy 2016, 8, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, S.P.; Melchiori, L.; Merchant, M.S.; Bernstein, D.; Glod, J.; Kaplan, R.; Grupp, S.; Tap, W.D.; Chagin, K.; Binder, G.K.; et al. Antitumor activity associated with prolonged persistence of adoptively transferred NY-ESO-1 c259T cells in synovial sarcoma. Cancer Discov. 2018, 8, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.T.J.; Thway, K.; Huang, P.H.; Jones, R.L. Clinical and molecular spectrum of liposarcoma. J. Clin. Oncol. 2018, 36, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Panagopoulos, I.; Höglund, M.; Mertens, F.; Mandahl, N.; Mitelman, F.; Aman, P. Fusion of the EWS and CHOP genes in myxoid liposarcoma. Oncogene 1996, 12, 489–494. [Google Scholar] [CrossRef]
- Aman, P.; Panagopoulos, I.; Lassen, C.; Fioretos, T.; Mencinger, M.; Toresson, H.; Höglund, M.; Forster, A.; Rabbitts, T.H.; Ron, D.; et al. Expression patterns of the human sarcoma-associated genes FUS and EWS and the genomic structure of FUS. Genomics 1996, 37, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; Schöffski, P.; Grignani, G.; Blay, J.Y.; Maki, R.G.; Van Tine, B.A.; Alcindor, T.; Jones, R.L.; D’Adamo, D.R.; Guo, M.; et al. Activity of eribulin in patients with advanced liposarcoma demonstrated in a subgroup analysis from a randomized phase III study of eribulin versus dacarbazine. J. Clin. Oncol. 2017, 35, 3433–3439. [Google Scholar] [CrossRef] [PubMed]
- Demetri, G.D.; von Mehren, M.; Jones, R.L.; Hensley, M.L.; Schuetze, S.M.; Staddon, A.; Milhem, M.; Elias, A.; Ganjoo, K.; Tawbi, H.; et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: Results of a phase III randomized multicenter clinical trial. J. Clin. Oncol. 2016, 34, 786–793. [Google Scholar] [CrossRef] [PubMed]
- Gronchi, A.; Ferrari, S.; Quagliuolo, V.; Broto, J.M.; Pousa, A.L.; Grignani, G.; Basso, U.; Blay, J.Y.; Tendero, O.; Beveridge, R.D.; et al. Histotype-tailored neoadjuvant chemotherapy versus standard chemotherapy in patients with high-risk soft-tissue sarcomas (ISG-STS 1001): An international, open-label, randomised, controlled, phase 3, multicentre trial. Lancet Oncol. 2017, 18, 812–822. [Google Scholar] [CrossRef]
- Grosso, F.; Jones, R.L.; Demetri, G.D.; Judson, I.R.; Blay, J.Y.; Le Cesne, A.; Sanfilippo, R.; Casieri, P.; Collini, P.; Dileo, P.; et al. Efficacy of trabectedin (ecteinascidin-743) in advanced pretreated myxoidliposarcomas: A retrospective study. Lancet Oncol. 2007, 8, 595–602. [Google Scholar] [CrossRef]
- Forni, C.; Minuzzo, M.; Virdis, E.; Tamborini, E.; Simone, M.; Tavecchio, M.; Erba, E.; Grosso, F.; Gronchi, A.; Aman, P.; et al. Trabectedin (ET-743) promotes differentiation in myxoid liposarcoma tumors. Mol. Cancer Ther. 2009, 8, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Hantschke, M.; Mentzel, T.; Rütten, A.; Palmedo, G.; Calonje, E.; Lazar, A.J.; Kutzner, H. Cutaneous clear cell sarcoma: A clinicopathologic, immunohistochemical, and molecular analysis of 12 cases emphasizing its distinction from dermal melanoma. Am. J. Surg. Pathol. 2010, 34, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Song, J.S.; Choi, J.; Kim, J.H.; Jang, S.J.; Cho, K.J. Diagnostic utility of EWS break-apart fluorescence in situ hybridization in distinguishing between non-cutaneous melanoma and clear cell sarcoma. Pathol. Int. 2010, 60, 608–613. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Chen, Y.; Cui, T.; Knösel, T.; Zhang, Q.; Geier, C.; Katenkamp, D.; Petersen, I. Identification of biomarkers to distinguish clear cell sarcoma from malignant melanoma. Hum. Pathol. 2012, 43, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Protsenko, S.A.; Semionova, A.I.; Komarov, Y.I.; Aleksakhina, S.N.; Ivantsov, A.O.; Iyevleva, A.G.; Imyanitov, E.N. BRAF-mutated clear cell sarcoma is sensitive to vemurafenib treatment. Investig. New Drugs 2015, 33, 1136–1143. [Google Scholar] [CrossRef] [PubMed]
- Postow, M.A.; Callahan, M.K.; Wolchok, J.D. Immune checkpoint blockade in cancer therapy. J. Clin. Oncol. 2015, 33, 1974–1982. [Google Scholar] [CrossRef] [PubMed]
- Marcrom, S.; De Los Santos, J.F.; Conry, R.M. Complete response of mediastinal clear cell sarcoma to pembrolizumab with radiotherapy. Clin. Sarcoma Res. 2017, 7, 14. [Google Scholar] [CrossRef] [PubMed]
- Lovly, C.M.; Gupta, A.; Lipson, D.; Otto, G.; Brennan, T.; Chung, C.T.; Borinstein, S.C.; Ross, J.S.; Stephens, P.J.; Miller, V.A.; et al. Inflammatory myofibroblastic tumors harbor multiple potentially actionable kinase fusions. Cancer Discov. 2014, 4, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Antonescu, C.R.; Suurmeijer, A.J.; Zhang, L.; Sung, Y.S.; Jungbluth, A.A.; Travis, W.D.; Al-Ahmadie, H.; Fletcher, C.D.; Alaggio, R. Molecular characterization of inflammatory myofibroblastic tumors with frequent ALK and ROS1 gene fusions and rare novel RET rearrangement. Am. J. Surg. Pathol. 2015, 39, 957–967. [Google Scholar] [CrossRef] [PubMed]
- Alassiri, A.H.; Ali, R.H.; Shen, Y.; Lum, A.; Strahlendorf, C.; Deyell, R.; Rassekh, R.; Sorensen, P.H.; Laskin, J.; Marra, M.; et al. ETV6-NTRK3 is expressed in a subset of ALK-negative inflammatory myofibroblastic tumors. Am. J. Surg Pathol 2016, 40, 1051–1061. [Google Scholar] [CrossRef] [PubMed]
- Petridis, A.K.; Hempelmann, R.G.; Hugo, H.H.; Eichmann, T.; Mehdorn, H.M. Metastatic low-grade inflammatory myofibroblastic tumor (IMT) in the central nervous system of a 29-year-old male patient. Clin Neuropathol 2004, 23, 158–166. [Google Scholar] [PubMed]
- Jehangir, M.; Jang, A.; Ur Rehman, I.; Mamoon, N. Synchronous inflammatory myofibroblastic tumor in lung and brain: A case report and review of literature. Cureus 2017, 9, e1183. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.; Kim, C.; Hagstrom, N.; Ferrer, F. Successful preoperative treatment of pediatric bladder inflammatory myofibroblastic tumor with anti-inflammatory therapy. Urology 2007, 70, 372.e13–372.e15. [Google Scholar] [CrossRef] [PubMed]
- Tsuma, Y.; Miyachi, M.; Ouchi, K.; Tsuchiya, K.; Iehara, T.; Naitoh, Y.; Konishi, E.; Yanagisawa, A.; Hosoi, H. Neoadjuvant treatment with cyclooxygenase-2 inhibitor and prednisolone allows conservative surgery for inflammatory myofibroblastic tumor of the bladder. J. Pediatr. Hematol. Oncol. 2016, 38, e283–e285. [Google Scholar] [CrossRef] [PubMed]
- Butrynski, J.E.; D’Adamo, D.R.; Hornick, J.L.; Dal Cin, P.; Antonescu, C.R.; Jhanwar, S.C.; Ladanyi, M.; Capelletti, M.; Rodig, S.J.; Ramaiya, N.; et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N. Engl. J. Med. 2010, 363, 1727–1733. [Google Scholar] [CrossRef] [PubMed]
- Kimbara, S.; Takeda, K.; Fukushima, H.; Inoue, T.; Okada, H.; Shibata, Y.; Katsushima, U.; Tsuya, A.; Tokunaga, S.; Daga, H.; et al. A case report of epithelioid inflammatory myofibroblastic sarcoma with RANBP2-ALK fusion gene treated with the ALK inhibitor, crizotinib. Jpn. J. Clin. Oncol. 2014, 44, 868–871. [Google Scholar] [CrossRef] [PubMed]
- Gaudichon, J.; Jeanne-Pasquier, C.; Deparis, M.; Veyssière, A.; Heyndrickx, M.; Minckes, O.; Orbach, D. Complete and repeated response of a metastatic ALK-rearranged inflammatory myofibroblastic tumor to crizotinib in a teenage girl. J. Pediatr. Hematol. Oncol. 2016, 38, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Saiki, M.; Ohyanagi, F.; Ariyasu, R.; Koyama, J.; Sonoda, T.; Nishikawa, S.; Kitazono, S.; Yanagitani, N.; Horiike, A.; Ninomiya, H.; et al. Dramatic response to alectinib in inflammatory myofibroblastic tumor with anaplastic lymphoma kinase fusion gene. Jpn. J. Clin. Oncol. 2017, 47, 1189–1192. [Google Scholar] [CrossRef] [PubMed]
- Mossé, Y.P.; Lim, M.S.; Voss, S.D.; Wilner, K.; Ruffner, K.; Laliberte, J.; Rolland, D.; Balis, F.M.; Maris, J.M.; Weigel, B.J.; et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: A Children’s Oncology Group phase 1 consortium study. Lancet Oncol. 2013, 14, 472–480. [Google Scholar] [CrossRef]
- Mossé, Y.P.; Voss, S.D.; Lim, M.S.; Rolland, D.; Minard, C.G.; Fox, E.; Adamson, P.; Wilner, K.; Blaney, S.M.; Weigel, B.J. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A Children’s Oncology Group Study. J. Clin. Oncol. 2017, 35, 3215–3221. [Google Scholar] [CrossRef] [PubMed]
- Church, A.J.; Calicchio, M.L.; Nardi, V.; Skalova, A.; Pinto, A.; Dillon, D.A.; Gomez-Fernandez, C.R.; Manoj, N.; Haimes, J.D.; Stahl, J.A.; et al. Recurrent EML4-NTRK3 fusions in infantile fibrosarcoma and congenital mesoblastic nephroma suggest a revised testing strategy. Mod. Pathol. 2018, 31, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Lannon, C.L.; Sorensen, P.H. ETV6-NTRK3: A chimeric protein tyrosine kinase with transformation activity in multiple cell lineages. Semin. Cancer Biol. 2005, 15, 215–223. [Google Scholar] [CrossRef] [PubMed]
- Brennan, M.F.; Antonescu, C.R.; Alektiar, K.M.; Maki, R.G. Management of Soft Tissue Sarcoma, 2nd ed.; Springer: New York, NY, USA, 2016; ISBN 978-3-319-41906-0. [Google Scholar]
- Bailey, J.J.; Schirrmacher, R.; Farrell, K.; Bernard-Gauthier, V. Tropomyosin receptor kinase inhibitors: An updated patent review for 2010–2016—Part, I. Expert Opin. Ther. Pat. 2017, 27, 733–751. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.J.; Schirrmacher, R.; Farrell, K.; Bernard-Gauthier, V. Tropomyosin receptor kinase inhibitors: An updated patent review for 2010–2016—Part, I.I. Expert Opin. Ther. Pat. 2017, 27, 831–849. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; DuBois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Siena, S.; Ou, S.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and antitumor activity of the multitargeted Pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Nagasubramanian, R.; Wei, J.; Gordon, P.; Rastatter, J.C.; Cox, M.C.; Pappo, A. Infantile fibrosarcoma with NTRK3-ETV6 fusion successfully treated with the tropomyosin-related kinase inhibitor LOXO-101. Pediatr. Blood Cancer 2016, 63, 1468–1470. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Drilon, A.; Ou, S.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Sugita, S.; Asanuma, H.; Hasegawa, T. Diagnostic use of fluorescence in situ hybridization in expert review in a phase 2 study of trabectedin monotherapy in patients with advanced, translocation-related sarcoma. Diagn. Pathol. 2016, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.A.; Letai, A.; Fisher, D.E.; Flaherty, K.T. Precision medicine for cancer with next-generation functional diagnostics. Nat. Rev. Cancer 2015, 15, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Lucchesi, C.; Khalifa, E.; Laizet, Y.; Soubeyran, I.; Mathoulin-Pelissier, S.; Chomienne, C.; Italiano, A. Targetable alterations in adult patients with soft-tissue sarcomas: Insights for personalized therapy. JAMA Oncol. 2018, 4, 1398–1404. [Google Scholar] [CrossRef] [PubMed]
- Baslan, T.; Hicks, J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat. Rev. Cancer 2017, 17, 557–569. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; Roy, A.; Xu, L.; Black, J.O.; Coffin, C.M.; Chi, Y.Y.; Tian, J.; Spunt, S.L.; Hawkins, D.S.; Bridge, J.A.; et al. Undifferentiated sarcomas in children harbor clinically relevant oncogenic fusions and gene copy-number alterations: A report from the Children’s Oncology Group. Clin. Cancer Res. 2018, 24, 3888–3897. [Google Scholar] [CrossRef] [PubMed]
- Chudasama, P.; Mughal, S.S.; Sanders, M.A.; Hübschmann, D.; Chung, I.; Deeg, K.I.; Wong, S.H.; Rabe, S.; Hlevnjak, M.; Zapatka, M.; et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat. Commun. 2018, 9, 144. [Google Scholar] [CrossRef] [PubMed]
- Knott, G.J.; Doudna, J.A. CRISPR-Cas guides the future of genetic engineering. Science 2018, 361, 866–869. [Google Scholar] [CrossRef] [PubMed]
- Vanoli, F.; Jasin, M. Generation of chromosomal translocations that lead to conditional fusion protein expression using CRISPR-Cas9 and homology-directed repair. Methods 2017, 121–122, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Spraggon, L.; Martelotto, L.G.; Hmeljak, J.; Hitchman, T.D.; Wang, J.; Wang, L.; Slotkin, E.K.; Fan, P.D.; Reis-Filho, J.S.; Ladanyi, M. Generation of conditional oncogenic chromosomal translocations using CRISPR-Cas9 genomic editing and homology-directed repair. J. Pathol. 2017, 242, 102–112. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Histological Subtype | Chromosomal Translocation | Fusion Gene |
|---|---|---|
| Ewing sarcoma | t(11;22)(q24;q12) | EWSR1-FLI1 |
| t(21;22)(q22;q12) | EWSR1-ERG | |
| t(7;22)(q22;q12) | EWSR1-ETV1 | |
| t(17;22)(q12;q12) | EWSR1-ETV4 | |
| t(2;22)(q33;q12) | EWSR1-FEV | |
| t(16;21)(p11;q22) | FUS-ERG | |
| Ewing sarcoma-like small blue round cell tumor | t(4;19)(q35;q13) | CIC-DUX4 |
| t(X;19)(q13;q13) | CIC-FOXO4 | |
| Desmoplastic small round cell tumor | t(11;22)(p13;q12) | EWSR1-WT1 |
| Alveolar rhabdomyosarcoma | t(2;13)(q35;q14) | PAX3-FOXO1 |
| t(1;13)(p36;q14) | PAX7-FOXO1 | |
| Alveolar soft part sarcoma | t(X;17)(p11;q25) | ASPL-TFE3 |
| Synovial sarcoma | t(X;18)(p11;q11) | SS18-SSX1 |
| t(X;18)(p11;q11) | SS18-SSX2 | |
| t(X;18)(p11;q11) | SS18-SSX4 | |
| Myxoid liposarcoma | t(12;16)(q13;p11) | FUS-CHOP |
| t(12;22)(q13;q12) | EWSR1-CHOP | |
| Clear cell sarcoma | t(12;22)(q13;q12) | EWSR1-ATF1 |
| Inflammatory myofibroblastic tumor | t(1;2)(q21;p23) | TMP3-ALK |
| t(2;19)(p23;p13) | TMP4-ALK | |
| t(2;17)(p23;q23) | CLTC-ALK | |
| Infantile fibrosarcoma | t(12;15)(p13;q25) | ETV6-NTRK3 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nakano, K.; Takahashi, S. Translocation-Related Sarcomas. Int. J. Mol. Sci. 2018, 19, 3784. https://doi.org/10.3390/ijms19123784
Nakano K, Takahashi S. Translocation-Related Sarcomas. International Journal of Molecular Sciences. 2018; 19(12):3784. https://doi.org/10.3390/ijms19123784
Chicago/Turabian StyleNakano, Kenji, and Shunji Takahashi. 2018. "Translocation-Related Sarcomas" International Journal of Molecular Sciences 19, no. 12: 3784. https://doi.org/10.3390/ijms19123784
APA StyleNakano, K., & Takahashi, S. (2018). Translocation-Related Sarcomas. International Journal of Molecular Sciences, 19(12), 3784. https://doi.org/10.3390/ijms19123784